

Abstract
Sulfiredoxin (Srx) catalyzes a novel enzymatic reaction, the reduction of
protein cysteine sulfinic acid,
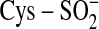 . This reaction is unique to the
typical 2-Cys peroxiredoxins (Prx) and plays a role in peroxide-mediated
signaling by regulating the activity of Prxs. Two mechanistic schemes have
been proposed that differ regarding the first step of the reaction. This step
involves either the direct transfer of the γ-phosphate of ATP to the Prx
molecule or through Srx acting as a phosphorylated intermediary. In an effort
to clarify this step of the Srx reaction, we have determined the 1.8Å
resolution crystal structure of Srx in complex with ATP and Mg2+.
This structure reveals the role of the Mg2+ ion to position the
γ-phosphate toward solvent, thus preventing an in-line attack by the
catalytic residue Cys-99 of Srx. A model of the quaternary complex is
consistent with this proposal. Furthermore, phosphorylation studies on several
site-directed mutants of Srx and Prx, including the Prx-Asp mimic of the
. This reaction is unique to the
typical 2-Cys peroxiredoxins (Prx) and plays a role in peroxide-mediated
signaling by regulating the activity of Prxs. Two mechanistic schemes have
been proposed that differ regarding the first step of the reaction. This step
involves either the direct transfer of the γ-phosphate of ATP to the Prx
molecule or through Srx acting as a phosphorylated intermediary. In an effort
to clarify this step of the Srx reaction, we have determined the 1.8Å
resolution crystal structure of Srx in complex with ATP and Mg2+.
This structure reveals the role of the Mg2+ ion to position the
γ-phosphate toward solvent, thus preventing an in-line attack by the
catalytic residue Cys-99 of Srx. A model of the quaternary complex is
consistent with this proposal. Furthermore, phosphorylation studies on several
site-directed mutants of Srx and Prx, including the Prx-Asp mimic of the
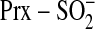 species, support a mechanism
where phosphorylation of
species, support a mechanism
where phosphorylation of 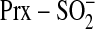 is the
first chemical step.
is the
first chemical step.
The ubiquitous peroxiredoxins (Prxs)2 are thiol-dependent peroxidases that reduce H2O2, peroxynitrite, and alkyl hydroperoxides to water, nitrite, and alcohols, respectively (1). Besides this detoxification role, Prxs can regulate hydrogen peroxide-mediated cell signaling in response to receptor stimulation and oxidative stress (2–5). Humans have six isoforms of Prxs that can be divided into three classes depending on the number and location of Cys residues: 2-Cys, atypical 2-Cys, and 1-Cys (6). Although all Prxs contribute to peroxide homeostasis, the 2-Cys Prxs (PrxI–IV) are unique in that they can be can be post-translationally inactivated by the peroxide substrate and subsequently repaired by an enzyme known as sulfiredoxin (Srx or Npn3) (7–11). Inactivation of Prx is thought to lead to a local increase in peroxide levels and signal potentiation (2–4). Moreover, inactivation appears to endow high molecular weight aggregates of Prx with chaperone activity (12, 13). The action of Srx restores peroxidase activity, thus turning off the peroxide signal and chaperone activity. This novel redox switch has been shown to regulate the transcription of antioxidant genes in Schizosaccharomyces pombe (14, 15). Moreover, the return of human PrxII (hPrxII) to the reduced state can allow cells to renter the cell cycle after H2O2-dependent arrest (16). Recent studies have also illustrated the importance of Srx in synaptic N-methyl-d-aspartic acid receptor activity and antioxidant defenses (17).
Eukaryotic 2-Cys Prxs have evolved structural features that make them
susceptible to elevated levels of peroxides
(4). In the catalytic cycle the
peroxidatic cysteine (Cys-SPH) is oxidized by the peroxide to form
a Cys sulfenic acid (Cys-SPOH). The interaction between the
conserved YF and GGLG structural motifs is thought to impede the attack of the
resolving Cys (Cys-SRH) from the adjacent monomer of the Prx
homodimer on the Cys-SPOH to form a disulfide bond
(Prx-Cys-SP–SR-Cys-Prx)
(18). The resulting catalytic
“pause” permits excess peroxide to further oxidize
Cys-SPOH to the catalytically inactive Cys sulfinic acid
(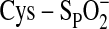 ).
Although a general thiol-disulfide oxidoreductase such as thioredoxin (Trx)
readily returns the disulfide-bonded form of Prx to the active state, Cys
sulfinic acid reduction requires Srx.
).
Although a general thiol-disulfide oxidoreductase such as thioredoxin (Trx)
readily returns the disulfide-bonded form of Prx to the active state, Cys
sulfinic acid reduction requires Srx.
The Srx repair reaction involves ATP, Mg2+, a conserved active
site Cys (Cys-99 in hSrx), and an exogenous thiol reductant, such as
glutathione (GSH) or Trx (10,
19,
20). Conflicting schemes for
the reaction mechanism (Fig. 1)
have been proposed, including which protein is phosphorylated in the first
step (10,
11,
21). In the originally
proposed reaction mechanism (Fig.
1, Path 1),
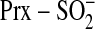 is phosphorylated to generate a
transient, novel sulfinic acid phosphoryl ester
(
is phosphorylated to generate a
transient, novel sulfinic acid phosphoryl ester
(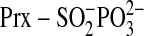 )
intermediate (6). In this
mechanism Srx functions as a phosphotransferase
(Fig. 1, Path 1, Step
1) and a thioltransferase (Path 1, Step 2). Another study
proposed that Cys-99 of hSrx attacks the γ-phosphate of ATP resulting in
the generation of a thiophosphate
(
)
intermediate (6). In this
mechanism Srx functions as a phosphotransferase
(Fig. 1, Path 1, Step
1) and a thioltransferase (Path 1, Step 2). Another study
proposed that Cys-99 of hSrx attacks the γ-phosphate of ATP resulting in
the generation of a thiophosphate
(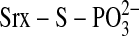 ) intermediate
(Fig. 1, Path 2, Step
1) (7). This intermediate
would then be attacked by the Prx sulfinic moiety to generate the same
) intermediate
(Fig. 1, Path 2, Step
1) (7). This intermediate
would then be attacked by the Prx sulfinic moiety to generate the same
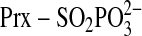 intermediate. It was further proposed that this latter intermediate is
resolved by an external thiol (i.e. GSH,
Fig. 1, Path 2, Step
2) and not Srx-Cys-99 (Path 1, Step 2). In Path 2 Srx functions
exclusively to transfer the phosphate to Prx. Neither of these studies were
able to obtain direct experimental evidence for formation of the sulfinic acid
phosphoryl ester
(
intermediate. It was further proposed that this latter intermediate is
resolved by an external thiol (i.e. GSH,
Fig. 1, Path 2, Step
2) and not Srx-Cys-99 (Path 1, Step 2). In Path 2 Srx functions
exclusively to transfer the phosphate to Prx. Neither of these studies were
able to obtain direct experimental evidence for formation of the sulfinic acid
phosphoryl ester
(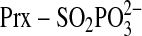 ) nor
was Jeong et al. (21)
able to demonstrate the existence of the proposed thiophosphate
(
) nor
was Jeong et al. (21)
able to demonstrate the existence of the proposed thiophosphate
(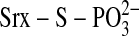 ) intermediate.
) intermediate.
FIGURE 1.

Comparison of the proposed sulfinic acid reduction mechanisms of Srx. Path 1 represents the mechanism originally proposed by Biteau et al. (10). Path 2 incorporates modifications to the reaction pathway as suggested by Jeong et al. (21). Step 1 involves the formation of the sulfinic acid phosphoryl ester intermediate. In Step 2 of the reaction, the addition of a thiol group leads to the formation of different thiosulfinate intermediates. This intermediate is subsequently resolved by additional thiols in Step 3. The thiol RSH could represent GSH or Trx.
In an effort to resolve the uncertainty surrounding the first step of the
repair mechanism, we solved the crystal structure of Srx in complex with
ATP·Mg2+. This allowed us to generate a model of the
quaternary enzyme·substrate complex,
ATP·Mg2+·Srx·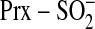 .
Phosphorylation studies on mutant proteins were performed to test this model.
Overall, our results support that
.
Phosphorylation studies on mutant proteins were performed to test this model.
Overall, our results support that
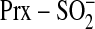 repair proceeds through a
sulfinic acid phosphoryl ester intermediate formed by the direct transfer of
the γ-phosphate from ATP to Prx.
repair proceeds through a
sulfinic acid phosphoryl ester intermediate formed by the direct transfer of
the γ-phosphate from ATP to Prx.
EXPERIMENTAL PROCEDURES
Protein Preparation and Modification—An engineered truncation of hSrx (residues 32–137) was expressed from a pET19 (Novagen) vector derivative containing a PreScission protease (GE Healthcare) cleavage site between Srx and the N-terminal His tag. The protein was expressed in C41(DE3) Escherichia coli cells and purified using nickel affinity and size-exclusion chromatography after the removal of the His tag (20). His-tagged hPrxII was purified in a similar manner as described previously (20, 22). Mutant forms of hSrx (C99S and C99A) and hPrxII (C70S, C172S, and C51D) were generated using the QuikChange site-directed mutagenesis kit from Stratagene, and the proteins were purified as described for the wild-type proteins. Oxidation of PrxII to the sulfinic acid form was performed with excess dithiothreitol and H2O2 as described previously (20). A PD10 desalting column (GE Healthcare) was used to remove excess dithiothreitol and H2O2.
Crystallization—Human SrxET-C99S was crystallized by the vapor diffusion method. Equal volumes of protein (16.6 mg/ml in 20 mm HEPES, pH 7.5, 100 mm NaCl) and well solutions (1 m sodium-potassium phosphate, pH 8.8, 8–13% (±)-2-methyl-2,4-pentanediol (MPD)) were mixed and incubated at 20 °C for a week as sitting drops. The crystal drop solution was slowly changed over the course of a day to a phosphate-free solution containing 1 m sodium malonate, pH 7.5, and 10% MPD. The ligands, 90 mm ATP and 5 mm Mg2+ in the same solution, were slowly added to the crystal drop over an 8-h period and allowed to incubate overnight. A cryoprotectant consisting of the ATP soak solution with 20% ethylene glycol was added to the crystal drop over 2 h before flash-freezing the crystal in nitrogen gas at 100 K.
Data Collection and Structure Determination—A single wavelength (0.979 Å) data set was collected on beamline X4A at NSLS, Brookhaven National Laboratory. Diffraction intensities were integrated using HKL2000 and scaled to 1.8 Å resolution. Cross-validation was performed with 4.9% of the reflections that were set aside. The space group of the crystal was P3221 with unit cell dimensions a = b = 68.5, c = 51.1. Initial phases for the ATP-bound Srx complex were obtained by molecular replacement using PHASER and the apo-Srx structure (Protein Data Bank 1XW3) as the search model (20). The molecular replacement solution was refined with CNS using alternating cycles of simulated annealing and positional and B-factor refinement (23). Model building was performed with COOT (24). Water molecules were visually confirmed within an Fo - Fc map contoured at 3σ and added with COOT. The final cycles of refinement were performed with REFMAC5 (25). The structure was validated using the MOLPROBITY server, which reported 100% of the residues in the Ramachandran favored regions (26). The data collection and refinement statistics for the structure are summarized in Table 1. All structural figures were prepared with the program PyMol (DeLano Scientific).
TABLE 1.
Crystallographic data and refinement statistics
| ATP·Mg2+·SrxET-C99Sa | |
|---|---|
| Data collection | |
| Space group | P3221 |
| Cell dimensions | |
| a, b, c (Å) | 68.5, 68.5, 51.1 |
| α, β, γ (°) | 90, 90, 120 |
| Wavelength (Å) | 0.979 |
| Resolutiona | 38.7 to 1.8 (1.86 to 1.80) |
| Rmerge (%) | 3.8 (29.8) |
| I/σI | 65.6 (7.5) |
| Completeness (%) | 99.8 (100) |
|
Redundancy
|
10.5 (10.5)
|
| Refinement | |
| Resolution | 38.7 to 1.8 |
| Rwork/Rfree (%) | 20.1/23.1 |
| No. of atoms | |
| Protein | 829 |
| Ligands | 33 |
| Water | 68 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.018 |
| Bond angles (°) | 1.77 |
| Average B-factor (Å2) | |
| Protein | 33.2 |
| Ligands | 31.6 |
| Solvent | 37.6 |
| Ramachandran analysis | |
| Favored regions | 100% |
Numbers in parentheses are for the highest resolution shell
γ-32P Phosphorylation—Proteins (Srx variants at 2 μm and PrxII variants at 1 μm) and 1 μm ATP spiked with [γ-32P]ATP (500,000 dpm) were incubated at 37 °C in a reaction mixture containing 25 mm HEPES, pH 7.5, 100 mm NaCl, 1 mm MgCl2. The reactions were quenched by the addition of 5× nonreducing, SDS-PAGE sample-loading buffer containing EDTA (final concentrations in sample, 12 mm Tris, pH 6.8, 5 mm EDTA, 5% glycerol, 0.4% SDS, 0.002% bromphenol blue). The samples were kept on ice until loaded onto 15% SDS-polyacrylamide gels that were also run on ice. The gels were wrapped in plastic wrap and compressed against a phosphorimaging plate and stored overnight at -80 °C. The gels were visualized using a STORM 840 analyzer (GE Healthcare). The data presented are representative of at least three experiments.
RESULTS AND DISCUSSION
Crystal Structure of Srx in Complex with ATP·Mg2+—To understand the structural basis underlying the first step of the repair mechanism, we set out to determine the crystal structure of hSrx (residues 32–137) in complex with the substrate ATP and Mg2+. Potential ATP hydrolysis was prevented by mutating the active site Cys-99 to Ser. Our previous attempts to crystallize Srx in the presence of ATP were unsuccessful, presumably because of the competition with the high concentration of phosphate (1 m) in the crystallization solution (20). Assuming that removal of phosphate buffer was essential for introducing ATP into the protein, the crystals were extensively washed with a solution containing sodium malonate. Excess ATP and Mg2+ were then added and soaked overnight. The ATP·Mg2+·SrxET-C99S structure was determined to 1.8 Å resolution and solved by molecular replacement using wild-type apo-Srx as the search model.
The overall protein structure of the solved complex was essentially identical to our previously determined, wild-type engineered truncation of the hSrx crystal structure (root mean square deviation 0.3 Å). The Srx fold consists of a five-stranded, mixed β-sheet in the center of the protein, two flanking helices (α1 and α2) involved in nucleotide binding, and the C-terminal helix (α3). The electron density around the ATP molecule and the Mg2+ ion was unambiguous (Fig. 2A). The adenosine ring of ATP hydrogen bonds with Ser-64 and Thr-68 located on α1. The phosphate binding pocket of Srx, consisting of His-100 and Arg-101 in the conserved active site motif 96P(G/S)GCHR101, provides hydrogen-bonding interactions with the β- and γ-phosphate groups, respectively. Lys-61 from α1 complements these interactions by hydrogen bonding with the α-phosphate group. Compared with our ADP-bound structure of Srx, the side chain of His-100 has rotated so that the imidazole ring coordinates with the γ-phosphate rather than the β-phosphate of ATP (20). The Mg2+ ion is coordinated by the three phosphate groups of ATP and two waters, W44 and W53 (Fig. 1B). The average Mg2+-oxygen distance in the coordination sphere is 2.2 Å.
FIGURE 2.

Structure of human sulfiredoxin C99S mutant in complex with ATP and Mg2+. A, stereoview of a simulated-annealing omit Fo–Fc electron density map (gray) contoured at 3σ in the active site of Srx (cyan). The carbon atoms of ATP are colored green, and the Mg2+ ion is colored light blue. B, triphosphate group of ATP within the active site of Srx. Hydrogen-bonding interactions are shown by dashed yellow lines. The hydrogen-bonding interactions to the β-phosphate from the backbone of His100 and the side chain of Arg101 are not shown for clarity.
Sulfiredoxin-dependent repair of eukaryotic, 2-Cys Prxs has been suggested
to proceed via two different mechanisms
(Fig. 1)
(10,
21). In the originally
proposed reaction mechanism (Fig.
1, Path 1),
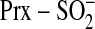 is phosphorylated to generate a
transient sulfinic acid phosphoryl ester
(
is phosphorylated to generate a
transient sulfinic acid phosphoryl ester
(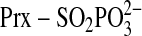 )
intermediate (10). While
testing this mechanism, Jeong et al.
(21) observed that Ser-99
within the enzymatically inactive, C99S hSrx mutant was phosphorylated. This
led the authors to propose that Cys-99 attacks the γ-phosphate of ATP
resulting in the generation of a thiophosphate
(
)
intermediate (10). While
testing this mechanism, Jeong et al.
(21) observed that Ser-99
within the enzymatically inactive, C99S hSrx mutant was phosphorylated. This
led the authors to propose that Cys-99 attacks the γ-phosphate of ATP
resulting in the generation of a thiophosphate
(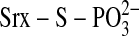 ) intermediate. In the
Srx·ATP·Mg2+ complex
(Fig. 2), there was no
indication of Ser-99 phosphorylation even though the crystal was soaked with
an excess of ATP overnight. The OH group of Ser-99 within this mutant is 4.4
Å away from the phosphorous atom of the γ-phosphate of ATP.
Moreover, as a consequence of its interaction with the Mg2+ ion,
the γ-phosphate projects into solvent parallel to Ser-99. This
observation suggests that Cys-99 of the wild-type enzyme would not be able to
perform an in-line nucleophilic attack on the ATP molecule, the preferred mode
of attack for kinases and other ATP-dependent enzymes (Ref.
27 and references cited
therein). The importance of Mg2+ for the correct positioning of the
γ-phosphate is supported further by the loss of activity upon the
addition of EDTA (10).
) intermediate. In the
Srx·ATP·Mg2+ complex
(Fig. 2), there was no
indication of Ser-99 phosphorylation even though the crystal was soaked with
an excess of ATP overnight. The OH group of Ser-99 within this mutant is 4.4
Å away from the phosphorous atom of the γ-phosphate of ATP.
Moreover, as a consequence of its interaction with the Mg2+ ion,
the γ-phosphate projects into solvent parallel to Ser-99. This
observation suggests that Cys-99 of the wild-type enzyme would not be able to
perform an in-line nucleophilic attack on the ATP molecule, the preferred mode
of attack for kinases and other ATP-dependent enzymes (Ref.
27 and references cited
therein). The importance of Mg2+ for the correct positioning of the
γ-phosphate is supported further by the loss of activity upon the
addition of EDTA (10).
Model of ATP·Mg2+ Bound to the
Srx·Prx Complex—As shown in
Fig. 3A,
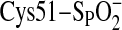 (Csd51) of the hPrxII active site exists in a salt bridge to Arg-127 and is
covered by the GGLG and YF structural motifs. These interactions prevent
access to Csd51 and illustrate the need for dramatic structural changes to
occur before Srx can access and repair Csd51
(4,
22,
28). In the rearrangement
process one of the oxygen atoms of Csd51 could perform an in-line attack on
the ATP molecule bound with the Srx active site. To further explore the
structural basis for the first step of the repair mechanism, we superimposed
the Srx·ATP·Mg2+ complex onto the structure of the
hSrx·hPrxI complex stabilized via a disulfide bond between the active
site Cys residues (supplemental Fig. S1)
(22). The phosphorous atom of
the γ-phosphate of ATP is located 3.1 Å from the S-γ atom of
Prx-Cys-SPH and 3.7 Å from the S-γ atom of Srx-Cys-99.
From this superposition we generated an enzyme-substrate model
(Fig. 3B) by
disrupting the intermolecular disulfide bond and converting the peroxidatic
Cys to the sulfinic acid form. As a result the active site loop was adjusted
slightly to accommodate the changes. In this model one oxygen atom of
(Csd51) of the hPrxII active site exists in a salt bridge to Arg-127 and is
covered by the GGLG and YF structural motifs. These interactions prevent
access to Csd51 and illustrate the need for dramatic structural changes to
occur before Srx can access and repair Csd51
(4,
22,
28). In the rearrangement
process one of the oxygen atoms of Csd51 could perform an in-line attack on
the ATP molecule bound with the Srx active site. To further explore the
structural basis for the first step of the repair mechanism, we superimposed
the Srx·ATP·Mg2+ complex onto the structure of the
hSrx·hPrxI complex stabilized via a disulfide bond between the active
site Cys residues (supplemental Fig. S1)
(22). The phosphorous atom of
the γ-phosphate of ATP is located 3.1 Å from the S-γ atom of
Prx-Cys-SPH and 3.7 Å from the S-γ atom of Srx-Cys-99.
From this superposition we generated an enzyme-substrate model
(Fig. 3B) by
disrupting the intermolecular disulfide bond and converting the peroxidatic
Cys to the sulfinic acid form. As a result the active site loop was adjusted
slightly to accommodate the changes. In this model one oxygen atom of
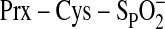 is located 1.7 Å from γ-phosphate atom. The geometric relationship
between the sulfinic acid moiety and ATP suggests that the oxygen atom of
is located 1.7 Å from γ-phosphate atom. The geometric relationship
between the sulfinic acid moiety and ATP suggests that the oxygen atom of
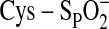 performs a direct in-line attack on the γ-phosphate. In contrast, Cys-99
of Srx points away from the γ-phosphate, making phosphate transfer to
this residue unlikely.
performs a direct in-line attack on the γ-phosphate. In contrast, Cys-99
of Srx points away from the γ-phosphate, making phosphate transfer to
this residue unlikely.
FIGURE 3.

Structural representations of mechanistically relevant species.
A, ribbon diagram of the crystal structure of human PrxII in the
hyperoxidized state 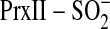 (Protein
Data Bank 1QMV) (28). The
substrate of Srx, Csd51 (dark green) is buried in the active site,
salt-bridged to Arg127 and covered by the YF motif from the adjacent monomer
in the dimer (light green). B, model of the first step of
the reaction. ATP is positioned to be attacked in-line at the
γ-phosphate atom by Csd51 in Prx (dark green). C,
model of the proposed Srx-Prx thiolsulfinate intermediate. This model was
generated by overlaying 2-(phenylmethylsulfanylsulfinyl)ethanol onto the
disulfide bond within the Srx-Prx structure (Protein Data Bank 2RII)
(16).
(Protein
Data Bank 1QMV) (28). The
substrate of Srx, Csd51 (dark green) is buried in the active site,
salt-bridged to Arg127 and covered by the YF motif from the adjacent monomer
in the dimer (light green). B, model of the first step of
the reaction. ATP is positioned to be attacked in-line at the
γ-phosphate atom by Csd51 in Prx (dark green). C,
model of the proposed Srx-Prx thiolsulfinate intermediate. This model was
generated by overlaying 2-(phenylmethylsulfanylsulfinyl)ethanol onto the
disulfide bond within the Srx-Prx structure (Protein Data Bank 2RII)
(16).
Phosphorylated Species in Srx and Prx Variants—To test the enzyme substrate model and to distinguish between the two proposed repair mechanisms, we investigated the phosphorylation status of Srx and Prx. Previous attempts to identify phosphorylated wild-type proteins were unsuccessful suggesting that the phosphoprotein intermediate(s) were labile (10, 21). Because the reaction is thiol-dependent and the presence of additional thiols may inadvertently break down the first reaction intermediate, the reactions were performed without the addition of GSH or Trx. Moreover, all but the active site cysteine residues of PrxII were mutated to serine (C70S and C172S, referred to as hPrxII-C2S). Srx contains only one Cys residue. Furthermore, we envisioned that a reduced temperature would increase the stability of the species during SDS-PAGE analysis. Similar to previous studies with wild-type Srx, these attempts were unsuccessful (Fig. 4A), and no phosphorylated Prx species were observed.
FIGURE 4.

Detection of phospho-protein intermediates of Srx and Prx. A, analysis of Srx and Prx variants within a 30 min reaction at 37 °C containing [γ-32P]-ATP. The reactions were quenched with nonreducing sample buffer containing EDTA and separated on 15% SDS-polyacrylamide gels on ice as described under “Experimental Procedures.” The positions and variants of Srx and PrxII are indicated. The PrxII protein used in these experiments contains the background mutations C70S and C172S, i.e. hPrxII-C2S, to prevent the inadvertent breakdown of phosphorylated intermediates. B, reaction mixtures described in (A) were incubated for the indicated time before analysis. The control samples that did not contain either Srx or Prx were incubated for 120 min. The image in panel 2 was electronically separated only to facilitate alignment of the samples for comparison.
In another approach to potentially stabilize the
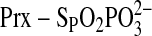 intermediate, we generated reduction inactive variants of hSrx. With the C99S
mutant, the putative thiol attack by Srx to form the thiosulfinate
intermediate (Fig. 1, Path
1, Step 2) would be prevented. We were, however, unable to see
phosphorylation of hPrxII-C2S with this Srx mutant
(Fig. 4A). Instead, we
observed phosphorylation of the C99S mutant as previously described by Jeong
et al. (21). In their
studies Srx C99S, but not Srx C99A, was phosphorylated, which led the authors
to conclude that Srx was phosphorylated at Ser-99 to form a conventional,
stable phosphoserine adduct. Unlike this previous study, we also observed
phosphorylation of Srx C99A and to some extent wild-type Srx. Moreover, in the
absence of
intermediate, we generated reduction inactive variants of hSrx. With the C99S
mutant, the putative thiol attack by Srx to form the thiosulfinate
intermediate (Fig. 1, Path
1, Step 2) would be prevented. We were, however, unable to see
phosphorylation of hPrxII-C2S with this Srx mutant
(Fig. 4A). Instead, we
observed phosphorylation of the C99S mutant as previously described by Jeong
et al. (21). In their
studies Srx C99S, but not Srx C99A, was phosphorylated, which led the authors
to conclude that Srx was phosphorylated at Ser-99 to form a conventional,
stable phosphoserine adduct. Unlike this previous study, we also observed
phosphorylation of Srx C99A and to some extent wild-type Srx. Moreover, in the
absence of 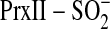 , the Srx variants
were phosphorylated suggesting that the phosphorylation of Srx may be
nonspecific to some degree (Fig.
4B).
, the Srx variants
were phosphorylated suggesting that the phosphorylation of Srx may be
nonspecific to some degree (Fig.
4B).
In the original mechanism proposed for Srx
(Fig. 1), one oxygen atom on
the sulfinic moiety functions as the nucleophile in the attack on the
γ-phosphate of ATP, resulting in the formation of a sulfinic acid
phosphoryl ester intermediate
(10). This mechanism is
reminiscent of the ATP-dependent activation of a carboxyl group by a variety
of enzymes, including glutamine synthetase (GS) and peptide ligases
(29). In GS the
phosphorylation of one oxygen of the carboxyl group of Glu generates a
detectable acyl phosphate intermediate, which in turn is attacked by an
ammonium ion (or amine in other enzymes with similar reactions) to generate
inorganic phosphate (Pi) and Gln. By analogy, we envisioned that
the substitution of the sulfinic acid moiety in
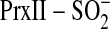 with a carboxyl group
(
with a carboxyl group
(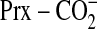 ), i.e. mutation of
Cys-51 to Asp, would stabilize a phosphorylated Prx species. Although this
substitution does alter the nucleophilicity and geometric context of the
attacking oxygen atom, the Asp side chain is structurally the closest analog
of the sulfinic acid moiety. Given the higher pKa value
for a carboxylate versus sulfinic acid (3.9 versus 1.5), we
anticipated that the Asp would be a poorer nucleophile
(30,
31). Therefore, if
phosphorylation of the Asp is observed with WT Srx prior to the Ser residue of
the inactive C99S mutant, then phosphorylation of Prx first would be the most
likely scenario for the WT enyzme. To our knowledge, there is no precedence in
the literature for a thiol attack on a phosphoaspartate, suggesting that the
second step of the Srx reaction will be disabled in this Prx mutant.
), i.e. mutation of
Cys-51 to Asp, would stabilize a phosphorylated Prx species. Although this
substitution does alter the nucleophilicity and geometric context of the
attacking oxygen atom, the Asp side chain is structurally the closest analog
of the sulfinic acid moiety. Given the higher pKa value
for a carboxylate versus sulfinic acid (3.9 versus 1.5), we
anticipated that the Asp would be a poorer nucleophile
(30,
31). Therefore, if
phosphorylation of the Asp is observed with WT Srx prior to the Ser residue of
the inactive C99S mutant, then phosphorylation of Prx first would be the most
likely scenario for the WT enyzme. To our knowledge, there is no precedence in
the literature for a thiol attack on a phosphoaspartate, suggesting that the
second step of the Srx reaction will be disabled in this Prx mutant.
With this approach, incubation of the PrxII-C2S-C51D mutant with wild-type Srx resulted in a stable PrxII phosphoaspartate intermediate after 30 min of incubation, illustrating that hPrxII can be phosphorylated in the active site and that this mutant represents a reasonable mimic of the sulfinic acid substrate (Fig. 4A). To test if Srx could still be phosphorylated in the reaction, we incubated the PrxII-C2S-C51D mutant with the C99S and C99A Srx variants (Fig. 4A). In both cases phosphorylation of PrxII occurred as well as SrxC99S and SrxC99A. In all cases the level of phosphorylation in Prx was greater than the Srx variants.
To further test the validity of the two reaction mechanisms, we monitored
the rate of phosphorylation in Prx versus Srx. In the presence of
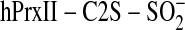 ,
phosphorylation of SrxC99S was observed after 3 min and increased with time
(Fig. 4B, 1st
panel). In contrast, formation of phosphoaspartate in the PrxII-C2S-C51D
mutant was seen within 1 min of incubation with all Srx variants
(Fig. 4B, 2nd
to 4th panels). Phosphorylation of Srx C99S in the presence of
PrxII-C2S-C51D was delayed for 10 min. These data support that PrxII-C51D is
the preferred substrate for phosphorylation. The same pattern was observed
with the C99A Srx mutant. These observations are in agreement with
phosphorylation of Prx being the first step in the reaction mechanism.
Moreover, the phosphorylation of Prx appears to be independent of Cys-99
within Srx.
,
phosphorylation of SrxC99S was observed after 3 min and increased with time
(Fig. 4B, 1st
panel). In contrast, formation of phosphoaspartate in the PrxII-C2S-C51D
mutant was seen within 1 min of incubation with all Srx variants
(Fig. 4B, 2nd
to 4th panels). Phosphorylation of Srx C99S in the presence of
PrxII-C2S-C51D was delayed for 10 min. These data support that PrxII-C51D is
the preferred substrate for phosphorylation. The same pattern was observed
with the C99A Srx mutant. These observations are in agreement with
phosphorylation of Prx being the first step in the reaction mechanism.
Moreover, the phosphorylation of Prx appears to be independent of Cys-99
within Srx.
Structural Models of the Reaction Mechanism—We have
investigated the enzymatic mechanism of sulfinic acid reduction by Srx. Our
studies provide support for the first step of the reaction mechanism proposed
by Biteau et al.
(10). This is demonstrated by
phosphorylation studies of Srx and Prx
(Fig. 4), which show that the
acid form of the active site residue of Prx could be phosphorylated before the
phosphorylation of Srx could be detected. Furthermore, the crystal structure
of Srx in complex with ATP and Mg2+
(Fig. 2) shows that
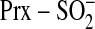 is geometrically favored to
attack ATP over Cys-99 in Srx. This arrangement contrasts with the phosphatase
PTP1B, for example, where the incoming phosphotyrosine is attacked by Cys-215
in an in-line fashion, leading to a phospho-Cys intermediate
(
is geometrically favored to
attack ATP over Cys-99 in Srx. This arrangement contrasts with the phosphatase
PTP1B, for example, where the incoming phosphotyrosine is attacked by Cys-215
in an in-line fashion, leading to a phospho-Cys intermediate
(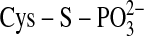 )
(32).
)
(32).
Our recent crystal structure of Srx in complex with Prx (22) and the Srx·ATP·Mg2+ complex presented here provides a structural model of how the repair may occur. Srx accesses the active site of Prx by displacing the YF motif of Prx (Fig. 3A). The binding of the conserved Phe-50 of Prx into a hydrophobic surface pocket of Srx is also thought to result in the unfolding of the Prx active site helix. As a result Csd51 of PrxII reorients to approach the γ-phosphate of ATP for an in-line attack (Fig. 3B). Surprisingly, these conformational changes are dependent on the unfolding and packing of the C-terminal tail of Prx onto the “backside” surface of Srx far from the Prx and Srx active sites (22). This latter interaction is essential for repair to occur.
The second step of the reaction, the thiol-dependent attack on the sulfinic
phosphoryl ester intermediate, is still unproven
(Fig. 1, Paths 1 and
2, Step 2). An external thiol such as GSH could access the phosphoryl
ester from the ATP-binding pocket. Alternatively, GSH could enter
∼120° from the ATP-binding site and attack the sulfinic phosphoryl
ester moiety from the backside. A more compelling scenario is the direct
attack by Cys-99 of Srx on the Prx intermediate. In our quaternary model Csd51
is positioned 1.7 Å from γ-phosphorous atom of ATP. Once the
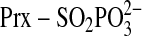 species
has been formed, Cys-99 is in close proximity (∼3 Å) and in a
favorable geometric position to attack the S-γ atom of
species
has been formed, Cys-99 is in close proximity (∼3 Å) and in a
favorable geometric position to attack the S-γ atom of
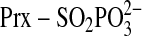 resulting in the formation of a Prx-Srx thiolsulfinate intermediate
(Fig. 3C). This
intermediate could be resolved by the external GSH, resulting in the formation
of a mixed Srx-GSH species (Fig.
1, Path 1, Step 3) or glutathione disulfide (Path 2,
Step 3). Additional experiments are clearly needed to clarify the
reaction beyond Step 1. The identification and temporal appearance of the
thiolsulfinate intermediate(s), Srx-S–S-G and potentially other species
would further help to resolve the unique reaction mechanism of Srx and the
possible cross talk between redox-regulated signaling pathways involving Prxs
and GSH.
resulting in the formation of a Prx-Srx thiolsulfinate intermediate
(Fig. 3C). This
intermediate could be resolved by the external GSH, resulting in the formation
of a mixed Srx-GSH species (Fig.
1, Path 1, Step 3) or glutathione disulfide (Path 2,
Step 3). Additional experiments are clearly needed to clarify the
reaction beyond Step 1. The identification and temporal appearance of the
thiolsulfinate intermediate(s), Srx-S–S-G and potentially other species
would further help to resolve the unique reaction mechanism of Srx and the
possible cross talk between redox-regulated signaling pathways involving Prxs
and GSH.
Supplementary Material
Acknowledgments
We thank the staff of NSLS and beamline X4A for their assistance during data collection and the RapiData course. National Synchrotron Light Source is supported by the United States Department of Energy and National Institutes of Health.
The atomic coordinates and structure factors (code 3CYI) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM072866 (to W. T. L.). This work was also supported by American Heart Association Postdoctoral Fellowship 0725399U (to T. J. J.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: Prx, peroxiredoxin; Csd, Cys sulfinic acid;
Cys-SPH, peroxidatic Cys residue; Cys-SRH, resolving Cys
residue; Cys-SPOH, Cys sulfenic acid;
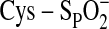 ,
Cys sulfinic acid; GSH, glutathione; Srx, sulfiredoxin; Trx, thioredoxin.
,
Cys sulfinic acid; GSH, glutathione; Srx, sulfiredoxin; Trx, thioredoxin.
References
- 1.Hofmann, B., Hecht, H. J., and Flohé, L. (2002) Biol. Chem. 383 347-364 [DOI] [PubMed] [Google Scholar]
- 2.Rhee, S. G. (2006) Science 312 1882-1883 [DOI] [PubMed] [Google Scholar]
- 3.Rhee, S. G., Kang, S. W., Jeong, W., Chang, T. S., Yang, K. S., and Woo, H. A. (2005) Curr. Opin. Cell Biol. 17 183-189 [DOI] [PubMed] [Google Scholar]
- 4.Wood, Z. A., Poole, L. B., and Karplus, P. A. (2003) Science 300 650-653 [DOI] [PubMed] [Google Scholar]
- 5.Choi, M. H., Lee, I. K., Kim, G. W., Kim, B. U., Han, Y. H., Yu, D. Y., Park, H. S., Kim, K. Y., Lee, J. S., Choi, C., Bae, Y. S., Lee, B. I., Rhee, S. G., and Kang, S. W. (2005) Nature 435 347-353 [DOI] [PubMed] [Google Scholar]
- 6.Rhee, S. G., Kang, S. W., Chang, T. S., Jeong, W., and Kim, K. (2001) IUBMB Life 52 35-41 [DOI] [PubMed] [Google Scholar]
- 7.Woo, H. A., Jeong, W., Chang, T. S., Park, K. J., Park, S. J., Yang, J. S., and Rhee, S. G. (2005) J. Biol. Chem. 280 3125-3128 [DOI] [PubMed] [Google Scholar]
- 8.Woo, H. A., Chae, H. Z., Hwang, S. C., Yang, K. S., Kang, S. W., Kim, K., and Rhee, S. G. (2003) Science 300 653-656 [DOI] [PubMed] [Google Scholar]
- 9.Chevallet, M., Wagner, E., Luche, S., van Dorsselaer, A., Leize-Wagner, E., and Rabilloud, T. (2003) J. Biol. Chem. 278 37146-37153 [DOI] [PubMed] [Google Scholar]
- 10.Biteau, B., Labarre, J., and Toledano, M. B. (2003) Nature 425 980-984 [DOI] [PubMed] [Google Scholar]
- 11.Jönsson, T. J., and Lowther, W. T. (2007) Subcell. Biochem. 44 115-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang, H. H., Lee, K. O., Chi, Y. H., Jung, B. G., Park, S. K., Park, J. H., Lee, J. R., Lee, S. S., Moon, J. C., Yun, J. W., Choi, Y. O., Kim, W. Y., Kang, J. S., Cheong, G. W., Yun, D. J., Rhee, S. G., Cho, M. J., and Lee, S. Y. (2004) Cell 117 625-635 [DOI] [PubMed] [Google Scholar]
- 13.Moon, J. C., Hah, Y. S., Kim, W. Y., Jung, B. G., Jang, H. H., Lee, J. R., Kim, S. Y., Lee, Y. M., Jeon, M. G., Kim, C. W., Cho, M. J., and Lee, S. Y. (2005) J. Biol. Chem. 280 28775-28784 [DOI] [PubMed] [Google Scholar]
- 14.Bozonet, S. M., Findlay, V. J., Day, A. M., Cameron, J., Veal, E. A., and Morgan, B. A. (2005) J. Biol. Chem. 280 23319-23327 [DOI] [PubMed] [Google Scholar]
- 15.Vivancos, A. P., Castillo, E. A., Biteau, B., Nicot, C., Ayte, J., Toledano, M. B., and Hidalgo, E. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 8875-8880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phalen, T. J., Weirather, K., Deming, P. B., Anathy, V., Howe, A. K., van der Vliet, A., Jönsson, T. J., Poole, L. B., and Heintz, N. H. (2006) J. Cell Biol. 175 779-789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papadia, S., Soriano, F. X., Leveille, F., Martel, M. A., Dakin, K. A., Hansen, H. H., Kaindl, A., Sifringer, M., Fowler, J., Stefovska, V., McKenzie, G., Craigon, M., Corriveau, R., Ghazal, P., Horsburgh, K., Yankner, B. A., Wyllie, D. J., Ikonomidou, C., and Hardingham, G. E. (2008) Nat. Neurosci. 11 476-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood, Z. A., Schröder, E., Robin Harris, J., and Poole, L. B. (2003) Trends Biochem. Sci. 28 32-40 [DOI] [PubMed] [Google Scholar]
- 19.Chang, T. S., Jeong, W., Woo, H. A., Lee, S. M., Park, S., and Rhee, S. G. (2004) J. Biol. Chem. 279 50994-51001 [DOI] [PubMed] [Google Scholar]
- 20.Jönsson, T. J., Murray, M. S., Johnson, L. C., Poole, L. B., and Lowther, W. T. (2005) Biochemistry 44 8634-8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeong, W., Park, S. J., Chang, T. S., Lee, D. Y., and Rhee, S. G. (2006) J. Biol. Chem. 281 14400-14407 [DOI] [PubMed] [Google Scholar]
- 22.Jönsson, T. J., Johnson, L. C., and Lowther, W. T. (2008) Nature 451 98-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 24.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 25.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 26.Davis, I. W., Murray, L. W., Richardson, J. S., and Richardson, D. C. (2004) Nucleic Acids Res. 32 W615-W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matte, A., Tari, L. W., and Delbaere, L. T. (1998) Structure (Lond.) 6 413-419 [DOI] [PubMed] [Google Scholar]
- 28.Schröder, E., Littlechild, J. A., Lebedev, A. A., Errington, N., Vagin, A. A., and Isupov, M. N. (2000) Structure (Lond.) 8 605-615 [DOI] [PubMed] [Google Scholar]
- 29.Purich, D. L. (2002) Methods Enzymol. 354 168-177 [DOI] [PubMed] [Google Scholar]
- 30.Palmieri, F., Stipani, I., and Iacobazzi, V. (1979) Biochim. Biophys. Acta 555 531-546 [DOI] [PubMed] [Google Scholar]
- 31.Weast, R. C., Astle, M. J., and Beyer, W. H. (eds) (1983) CRC Handbook of Chemistry and Physics, 64th Ed., p. C719, Chemical Rubber Publishing Co., Boca Raton, FL
- 32.Pannifer, A. D., Flint, A. J., Tonks, N. K., and Barford, D. (1998) J. Biol. Chem. 273 10454-10462 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.