

Abstract
ABCA1-mediated lipid efflux to lipid poor apoA-I results in the gradual lipidation of apoA-I. This leads to the formation of discoidal HDL which are subsequently converted to spherical HDL by the action of LCAT. We have investigated the effect of point mutations and deletions in the carboxy-terminal region of apoA-I on the biogenesis of HDL using adenovirus-mediated gene transfer in apoA-I deficient mice. It was found that the plasma HDL levels were greatly reduced in mice expressing the carboxy-terminal deletion mutants apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)], shown previously to diminish the ABCA1-mediated lipid efflux. The HDL levels were normal in mice expressing the WT apoA-I, the apoA-I[Δ(232-243)] deletion mutant or the apoA-I[E191A/H193A/K195A] point mutant, which promote normal ABCA1-mediated lipid efflux. Electron microscopy and two-dimensional gel electrophoresis showed that the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] mutants formed mainly preβ-HDL particles and few spherical particles enriched in apoE, while WT apoA-I, apoA-I[Δ(232-243)] and apoA-I[E191A/H193A/K195A] formed spherical α-HDL particles. The findings establish that a) deletions that eliminate the 220-231 region of apoA-I prevent the synthesis of α-HDL, but allow the synthesis of preβ-HDL particles in vivo, b) the amino-terminal segment 1-184 of apoA-I can promote synthesis of preβ-HDL type particles in an ABCA1-independent process and c) the charged residues in the 191-195 region of apoA-I do not influence the biogenesis of HDL.
Apolipoprotein A-I (apoA-I) is the major protein component of high density lipoproteins (HDL), and plays an essential role in the biogenesis, structure, function and plasma concentration of HDL (1-5). ApoA-I contains 22- and 11-amino acid repeats (6;7) which based on earlier x-ray crystallography (8) and computer modeling (7) are organized in amphipathic α-helices. Most recently the lipid-free apoA-I has been crystallized in salt buffers containing 500 μM Cr(III)-Tris-acetylacetonate (Cr-acac3) (9). Under the conditions of crystallization, the protein consists of a four helix amino-terminal bundle and two carboxy-terminal helices.
The biogenesis and catabolism of HDL can be considered as a complex pathway that involves several proteins (5). In the early steps of this pathway, apoA-I is secreted mostly lipid-free by the liver and acquires phospholipid and cholesterol via its interactions with the ATP-binding cassette A1 (ABCA1) lipid transporter (2;10;11). Through a series of intermediate steps that are poorly understood, apoA-I is gradually lipidated and proceeds to form discoidal particles which are converted to spherical particles by the action of lecithin:cholesterol acyl transferase (LCAT) (3;12). Both the discoidal and the spherical HDL particles interact functionally with the HDL receptor scavenger receptor class B type I (SR-BI) (4;13;14). They also interact with the ABCG1 transporter (15). The late steps of the HDL pathway involve the transfer of cholesteryl esters to VLDL/LDL for eventual catabolism by the LDL receptor, the hydrolysis of phospholipids and residual triglycerides by the various lipases (lipoprotein lipase, hepatic lipase, and endothelial lipase), and the transfer of phospholipids from VLDL/LDL to HDL by the action of phospholipid transfer protein (16).
In previous studies we used adenovirus-mediated gene transfer of apoA-I mutants to identify steps in the HDL biogenesis pathway where intermediates of the pathway cannot be converted to products and therefore accumulate in plasma (2;17-20). Discrete phenotypes were observed that were characterized by total lack of HDL synthesis, accumulation of discoidal particles of abnormal preβ-/α-HDL ratios and various forms of dislipidemias (2;17-20).
In the present study we analyzed the impact of carboxy-terminal deletion mutants apoA-I[Δ(185-243)], apoA-I[Δ(220-243)] and apoA-I[Δ(232-243)] as well as of the point mutant apoA-I[E191A/H193A/K195A], on the biogenesis of HDL in vivo using adenovirus-mediated gene transfer of apoA-I mutants in apoA-I deficient (apoA-I-/-) mice. The formation of HDL was assessed by FPLC fractionation, electron microscopy (EM) analysis and two-dimensional gel electrophoresis of plasma.
Our findings indicated that carboxy terminal deletions that removes the 220-231 region of apoA-I, prevents the biogenesis of normal α-HDL particles but allows the formation of preβ-HDL particles by processes which appear to be independent of apoA-I/ABCA1 interactions.
Experimental Procedures
Materials
Materials not mentioned in the experimental procedures have been obtained from sources described previously (2;17).
Generation of adenoviruses expressing the wild-type (WT) and the mutant apoA-I forms
The construction of recombinant adenoviruses carrying the genomic sequence for the WT apoA-I, apoA-I[Δ(220-243)] and apoA-I[Δ(232-243)] has been described before (2;19). The adenoviruses expressing apoA-I[Δ(185-243)] and apoA-I[E191A/H193A/K195A] were generated in a similar way. Briefly, the fourth exon of the human apoA-I gene was amplified and mutagenized by polymerase chain reaction, using a set of specific mutagenic primers (185F and 185R for apoA-I[Δ(185-243)] and M32S and M32A for apoA-I[E191A/H193A/K195A]) containing the mutation of interest and a set of flanking universal primers (AINOTF and AISALR) containing the restriction sites NotI and SalI. The sequences of the primers are shown in Table I. The pCA13AIgN vector, which contains a NotI site in intron 3 and a XhoI site in the 3′-end of the apoA-I gene, was used as a template in the amplification reactions (19;21). The DNA fragment containing the mutation of interest was digested with NotI and SalI and sub cloned into the NotI and XhoI sites of the pCA13AIgN vector, thus replacing the WT with the mutated exon 4 sequence. The pCA13-A-I plasmids, containing the 185-243 deletion or the E191A/H193A/K195A point mutation, along with a helper PJM17 adenovirus plasmid were used to generate recombinant adenoviruses as described previously (17;19).
TABLE 1. Oligonucleotide sequence of primers used in PCR amplifications.
| Name | Sequence | Location of sequence |
|---|---|---|
| 185F | 5′-CTC AAG GAG AAC TGAa GGC GCC AGA CTG-3′ | nt 612-638 of apoA-I cDNAb (sense) (amino acids +181 to +189) |
| 185R | 5′-CAG TCT GGC GCC TCA GTT CTC CTT GAG-3′ | nt 638-612 (antisense) (amino acids +189 to +181)c |
| M32S | 5′-CTG GCC GCGd TAC GCC GCC GCG GCC ACC GAG-3′ | nt 638-665 of apoA-I cDNA (sense) (amino acids +189 to +198) |
| M32A | 5′-CTC GGT GGC CGC GGC GGC GTA CGC GGC CAG TCT-3′ | nt 665-633 of apoA-I cDNA (antisense) (amino acids +198 to +188) |
| AINOT F | 5′ CCT CCG CGG ACA GGC GGC CGCe CAG GG 3′ | nt 886-911 of apoA-I genomic sequencef that contains a NotI site (sense), intron 3 of apoA-I gene |
| AISAL R | 5′ A CAT GTC GAC CCC CTT TCA GGG CAC CTG GCC TTG 3′ | ACAT + SalI site + nt 1917-1894 of apoA-I genomic sequence (antisense), at 3′ end of apoA-I gene |
the stop codon is underlined
nucleotide number of the human apoA-I cDNA sequence (57), oligonucleotide position (+) relative to the translation initiation ATG codon
amino acid position (+) refers to the mature plasma apoA-I sequence
mutagenized residues are marked in boldface type and are underlined
the restriction enzyme recognition sites are marked in boldface type
nucleotide number of the human apoA-I genomic sequence (21), oligonucleotide position (+) relative to the translation initiation ATG codon
Animal studies, plasma lipids, apoA-I and apoA-I mRNA levels analyses
ApoA-I-/- (ApoA1tm1Unc) C57BL/6J mice (22) were purchased from Jackson Laboratories (Bar Harbor, ME). The mice were maintained on a 12-h light/dark cycle and standard rodent chow. All procedures performed on the mice were in accordance with National Institutes of Health and institutional guidelines. ApoA-I-/- mice, 6-8 weeks of age, were injected via the tail vein with 1×109 pfu of recombinant adenovirus per animal and the animals sacrificed four days post-injection following a four-hour fast.
The concentration of total cholesterol, free cholesterol, phospholipids and triglycerides of plasma drawn four days post-infection was determined using the Cholesterol CII, Free Cholesterol C, Phospholipids B (Wako Chemicals USA, Inc.) and INFINITY triglycerides (ThermoDMA) reagents, respectively, according to the manufacturer's instructions. The concentration of cholesteryl esters was determined by subtracting the concentration of free cholesterol from the concentration of total cholesterol. Plasma apoA-I levels were measured by turbidimetric immunoassay using the Autokit Apo A1 reagents (Wako Chemicals USA, Inc.), according to the manufacturer's instructions. Hepatic human apoA-I mRNA levels were determined by Northerm blotting as described (2;4).
For FPLC analysis of plasma, 17 μl plasma obtained from mice infected with adenovirus-expressing WT or mutant apoA-I forms were loaded onto a Sepharose 6 PC column (Amersham Biosciences) in a SMART micro FPLC system (Amersham Biosciences) and eluted with phosphate-buffered saline (PBS). A total of 25 fractions of 50 μl volume each were collected for further analysis. The concentration of lipids and apoA-I in the FPLC fractions was determined as described above.
Fractionation of plasma by density gradient ultracentrifugation and electron microscopy analysis of the apoA-I-containing fractions
For this analysis, 300 μl of plasma obtained from adenovirus-infected mice was diluted with saline to a total volume of 0.5 ml. The mixture was adjusted to a density of 1.23 g/ml with KBr and overlayed with 1 ml of KBr solution of d=1.21 g/ml, 2.5 ml of KBr solution of d=1.063 g/ml, 0.5 ml of KBr solution of d=1.019 g/ml, and 0.5 ml of normal saline. The mixture was centrifuged for 22 h in SW55 rotor at 30,000 rpm. Following ultracentrifugation, 0.5 ml fractions were collected from the top for further analyses. The refractive index of the fractions was measured using a refractometer (American Optical Corp.) and it was converted to density for each sample based on a standard curve derived from solutions of known densities. The fractions were dialyzed against ammonium acetate and carbonate buffer (126 mM ammonium acetate, 2.6 mM ammonium carbonate, 0.26 mM EDTA, pH 7.4). Aliquots of the fractions were subjected to SDS-PAGE and the protein bands were visualized by staining with Coomassie Brilliant Blue. The fractions that were obtained from the plasma of mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] mutants were further analyzed by SDS-PAGE and Western blotting. The nitrocellulose membranes were probed with goat polyclonal anti-human apoA-I antibody (Chemicon International) and/or goat polyclonal anti-mouse apoE antibody (Santa Cruz Biotechnology, Inc. Santa Cruz, CA).
For EM analysis, fractions 6-8 that float in the HDL region were dialyzed against ammonium acetate and carbonate buffer. The samples were applied on carbon-coated grids, were stained with sodium phosphotungstate, were visualized in the Phillips CM-120 electron microscope (Phillips Electron Optics, Eindhoven, Netherlands) and photographed as described previously (2). The photomicrographs were taken at 75,000× magnification and enlarged 3 times.
Non-denaturating two-dimensional electrophoresis
The distribution of HDL subfractions in plasma was analyzed by two dimensional electrophoresis as described (23) with some modifications. Briefly, in the first dimension, 1 μL of plasma sample was separated by electrophoresis at 4°C in a 0.75% agarose gel using a 50 mM barbital buffer (pH 8.6, Sigma, St. Louis, MO) until the bromophenol blue marker had migrated 5.5 cm. Agarose gel strips containing the separated lipoproteins were then transferred to a 4% to 20% polyacrylamide gradient gel. Separation in the second dimension was performed at 90 V for 2 to 3 hours at 4°C. The separated proteins were transferred to a nitrocellulose membrane and human apoA-I and mouse apoE were detected by using a goat polyclonal anti-human apoA-I antibody (Chemicon International) and a goat polyclonal anti-mouse apoE antibody (Santa Cruz Biotechnology), respectively.
Agarose gel electrophoresis
Preβ- and α-HDL were separated by 0.7% agarose gel electrophoresis followed by Oil Red O neutral lipid staining (Sigma, St. Louis, MO) according to manufacturer's instructions or Western blotting using a goat polyclonal anti-human apoA-I antibody (Chemicon International).
Cell secretion of WT and mutant apoA-I forms
For assessing the secretion of WT and mutant apoA-I forms, human HTB13 cells (SW 1783, human astrocytoma) grown to 80% confluence in Leibovitz's L-15 medium containing 10% (v/v) fetal bovine serum (FBS) in 100 mm diameter dishes were infected with adenoviruses expressing WT and mutant apoA-I forms at a multiplicity of infection of 20. Twenty-four hours post-infection, the cells were washed twice with PBS and preincubated in serum-free medium for 2 h. Following an additional wash with PBS, fresh serum-free medium was added. After 24 h of incubation, medium was collected and analyzed by SDS-PAGE for apoA-I expression. HTB-13 cells have been chosen in these experiments and for large scale growing in roller bottles because they produce higher yields of apoA-I as compared to Chinese Hamster ovary cells (CHO) and C127 mouse mammary tumour carcinoma cells (ATCC CRC1616).
Cholesterol efflux assay
ABCA1-dependent efflux of [3H]cholesterol to lipid–free apoA-I acceptor was measured using J774 macrophages in which expression of ABCA1 was induced by a cAMP analogue, as described previously (2). The apoA-I forms used as cholesterol acceptors were produced by infection of HTB-13 cells grown in large scale in roller bottles and purification of apoA-I followed as described (3). On day 0 J774 macrophages were plated in 12-well plates at density of 5×105 cells/well in RPMI 1640 with 10 % (v/v) FBS and antibiotics. On day 1 cells were labeled with 1 ml of labeling medium (6 μCi/ml 1,2[3H]cholesterol) for 24 h. Following 24 h of labeling and washing, cells were treated with serum-free medium and equilibrated for 24 h with or without 0.3 mM 8-(4-chlorophenylthio)adenosine 3′:5′-cyclic monophosphate (cpt-cAMP). At the end of the treatment period with cpt-cAMP, cells were washed twice and incubated with 1 ml of RPMI 1640, supplemented with 0.2 % (w/v) bovine serum albumin (BSA), with or without 1 μM WT apoA-I or mutant forms at 37°C. At different time points up to 6 h, 55 μl of medium were collected and clarified by centrifugation in a microcentrifuge for 2 min. The radioactivity in 40 μl of the supernatant was determined by liquid scintillation counting. At the end of the incubation, cells were lysed by 800 μl of lysis buffer (PBS containing 1 % (v/v) Triton X-100) for 30 min at room temperature and radioactivity was measured in 40 μl cell lysate. The percentage of secreted [3H]cholesterol was calculated by dividing the medium-derived counts by the sum of the total counts present in the culture medium and the cell lysate. To calculate the net cpt-cAMP-dependent efflux, the cholesterol efflux of the untreated cells was subtracted from the cholesterol efflux of the cells treated with cpt-cAMP. The total and net cpt-cAMP-dependent efflux of cholesterol was linear over a four-hour period and was defined as 100%.
Results
In vitro studies: ABCA1-mediated efflux of cellular cholesterol
The overall objective of these studies was to assess the importance of domains and residues of the carboxy-terminal region of apoA-I in the ABCA1-mediated lipid efflux and the biogenesis of HDL following the expression of these mutants in apoA-I-/- mice.
Based on the three-dimensional structure of apoA-I the amino-terminal four helix bundle of apoA-I is connected with a loop, consisting of amino acids 188-195, to the two carboxy-terminal helices of apoA-I (9). Previous studies have shown that the carboxy-terminal region of apoA-I is involved in binding to multilamelar phospholipid particles and to HDL particles (3). We have shown previously that the ABCA1-mediated cholesterol efflux to the carboxy-terminal deletion mutants apoA-I[Δ(185-243)] or apoA-I[Δ(220-243)], that lack the 220-231 region, was reduced to 20% and 9% of WT control, respectively (2). However, the ABCA1-mediated cholesterol efflux to the carboxy-terminal deletion mutant apoA-I[Δ(232-243)], that retains the 220-231 region, was normal (2).
In the current study we showed that the cpt-cAMP-dependent (ABCA1-mediated) cholesterol efflux in the presence of apoA-I[E191A/H193A/K195A] acceptor was 92% of the WT control (Figure 1).
FIGURE 1. ABCA1-mediated cholesterol efflux in the presence of WT apoA-I and apoA-I[E191A/H193A/K195A].
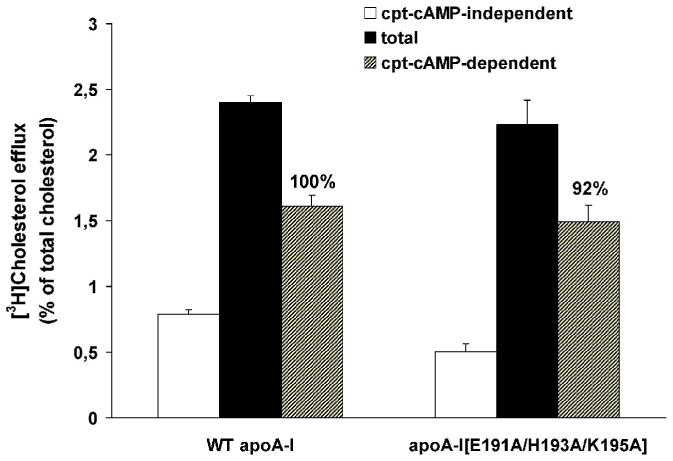
Cells labeled with 6 μCi/ml [3H]cholesterol for 24 h and treated with or without 0.3 mM cpt-cAMP for 24 h were incubated with 1 μM WT apoA-I and apoA-I[E191A/H193A/K195A] for 4 h at 37°C. Media and cells were collected separately and the radioactivity was measured as described in Experimental Procedures. The percent of [3H]cholesterol efflux represents the amount of the radioactivity released in the medium divided by the total radioactivity present in the culture medium and the cell lysate. The percent of the net cpt-cAMP-dependent [3H]cholesterol efflux was calculated as the difference in percent of cholesterol efflux between treated cells (total efflux) and untreated cells (cpt-cAMP independent efflux). Black and white bars show the percent of [3H]cholesterol efflux from cpt-cAMP treated and untreated cells, respectively. Shaded bars show the percent of the net cpt-cAMP-dependent [3H]cholesterol efflux. The numbers on top of the bars represent the cholesterol efflux relative to the WT control set to 100%. Values are the means ± S.D. from three independent experiments performed in duplicate.
In vivo studies: Plasma lipid, apoA-I and hepatic apoA-I mRNA levels following adenovirus mediated-gene transfer in apoA-I-/- mice
To determine the effect of the carboxy-terminal deletions Δ(185-243), Δ(220-243) and Δ(232-243) and the point mutation E191A/H193A/K195A in apoA-I on the biogenesis of HDL we used adenovirus-mediated gene transfer of apoA-I mutants in apoA-I-/- mice. For a typical experiment 4-7 mice were injected with 1×109 pfu of recombinant adenoviruses expressing the WT or the mutants apoA-I forms or of the control adenovirus expressing GFP. Plasma samples and the liver of mice were collected four days post-infection.
Analysis of plasma lipids and apoA-I levels, and hepatic apoA-I mRNA levels showed that apoA-I-/- mice infected with adenoviruses expressing the carboxy-terminal deletion mutants apoA-I[Δ(185-243)] or apoA-I[Δ(220-243)] and apoA-I-/- mice infected with the control adenovirus expressing the green fluorescent protein (apoA-I-/- GFP) had reduced levels of total and esterified cholesterol and decreased cholesteryl ester/ total cholesterol (CE/TC) ratio. In contrast, the apoA-I-/- mice infected with adenoviruses expressing the WT apoA-I, the carboxy-terminal deletion mutant apoA-I[Δ(232-243)] and the carboxy-terminal point mutant apoA-I[E191A/H193A/K195A] had normal levels of total and esterified cholesterol and similar CE/TC ratios (Table 2). The phospholipid levels were normal in mice expressing the WT apoA-I and the apoA-I[Δ(232-243)] and apoA-I[E191A/H193A/K195A] mutants, but were greatly reduced in mice expressing the apoA-I[Δ(185-243)] or apoA-I[Δ(220-243)] mutants as well as in the control mice that express GFP (Table 2). The plasma triglycerides in mice expressing the WT or mutant apoA-I forms were moderately increased as compared to apoA-I-/- GFP, but remained within the normal range (Table 2). The plasma apoA-I levels in mice expressing the apoA-I[Δ(232-243)] and apoA-I[E191A/H193A/K195A] mutants were 40% and 85% of the levels in mice expressing the WT apoA-I, respectively. In contrast, the plasma apoA-I levels were greatly reduced in mice expressing the apoA-I[Δ(220-243)] mutant, and were diminished in mice expressing the apoA-I[Δ(185-243)] mutant (Table 2). The differences in plasma lipid and apoA-I levels do not reflect differences in apoA-I expression, since the relative amounts of apoA-I mRNA were comparable (Table 2). In addition the WT and all apoA-I mutant forms were secreted with the same efficiency into the medium of HTB-13 cells following infection with adenoviruses expressing the WT and mutant apoA-I forms (Figure 2). As seen in Figure 2 the non infected HTB-13 cells do not synthesize apoA-I.
TABLE 2. Comparison of plasma lipids and apoA-I levels and hepatic mRNA levels of apoA-I-/- mice 4 days post-infection with recombinant adenoviruses expressing the WT apoA-I or apoA-I mutants or the control protein GFP.
| Cholesterol
(mg/dl) |
Free cholesterol
(mg/dl) |
Cholesteryl esters
(mg/dl) |
CE/TC | Phospholipids
(mg/dl) |
Triglycerides
(mg/dl) |
ApoA-I
(mg/dl) |
Relative apoA-I mRNA
(%) |
|
|---|---|---|---|---|---|---|---|---|
| WT apoA-I | 116±33 | 36±18 | 81±18 | 0.71±0.08 | 273±52 | 91±6 | 216±36 | 100 |
| apoA-I[Δ(185-243)] | 28±3 | 23±2 | 5±2 | 0.17±0.07 | 69±7 | 53±13 | 0.8±0.1 | 94±17 |
| apoA-I[Δ(220-243)] | 43±4 | 24±1 | 18±3 | 0.42±0.03 | 52±6 | 51±13 | 13±3 | 96±2 |
| apoA-I[Δ(232-243)] | 79±17 | 20±5 | 59±12 | 0.75±0.03 | 142±24 | 68±8 | 87±25 | 100±20 |
| apoA-I[E191A/H193A/K195A] | 113±11 | 30±3 | 83±9 | 0.73±0.03 | 241±35 | 81±30 | 183±32 | 82±13 |
| apoA-I-/- GFP | 27±4 | 13±3 | 14±6 | 0.53±0.09 | 94±13 | 37±9 | - | - |
Values are means ± S.D. (n=4-7).
FIGURE 2. Expression of WT and mutant apoA-I forms in cultures of HTB-13 cells following infection with the corresponding recombinant adenoviruses.
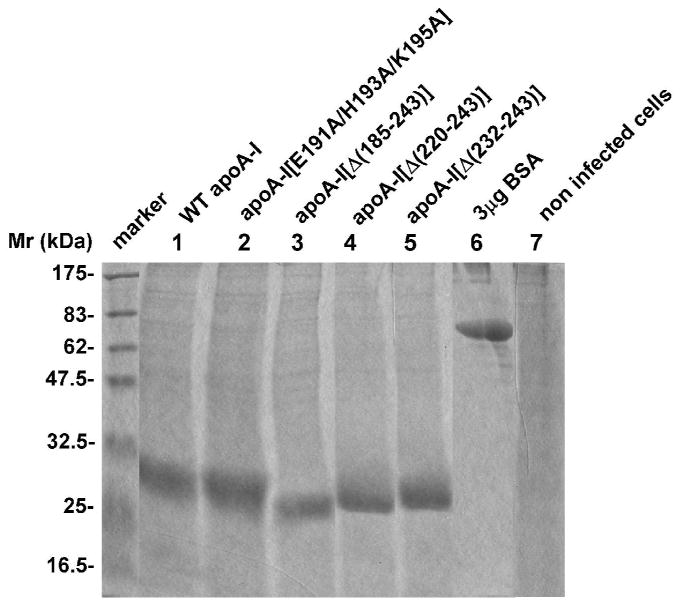
SDS-PAGE analysis of medium obtained from HTB-13 cells in 100 mm dishes infected with adenoviruses expressing the WT and mutant apoA-I forms as described in the Experimental Procedures. An aliquot of 30 μL of serum-free culture medium was analyzed. “Marker” indicates protein markers of different molecular mass, as shown in the figure. Lane 6 contains 3 μg BSA. It was estimated that the infected cultures (5×106 cells) secreted approximately 60-100 μg/ml WT and mutant apoA-I forms over 24 hours of incubation.
FPLC profiles of plasma isolated from mice infected with adenoviruses expressing the WT and the mutant apoA-I forms
FPLC analysis of plasma from apoA-I-/- mice infected with recombinant adenoviruses expressing the apoA-I[Δ(185-243)] or apoA-I[Δ(220-243)] mutants showed that small amounts of cholesterol and phospholipids where detected in the HDL region (Figure 3A, B). The cholesterol and phospholipid distribution and levels in these mice were comparable to those of mice infected with the control adenovirus expressing the GFP (Figure 3A, B). Small amounts of apoA-I were also found in the HDL region in mice infected with the adenovirus expressing the apoA-I[Δ(220-243)] mutant and barely detectable amounts of apoA-I were found in the HDL region in mice infected with the adenovirus expressing the apoA-I[Δ(185-243)] mutant (Figure 3D). The plasma cholesterol, phospholipids and apoA-I in mice expressing the WT apoA-I, apoA-I[Δ(232-243)] or apoA-I[E191A/H193A/K195A] were distributed in the HDL region (Figure 3A, B, D). The cholesterol, phospholipids and apoA-I FPLC profiles of mice expressing the apoA-I[Δ(232-243)] mutant also had a small shoulder at the LDL region (Figure 3A, B, D). The cholesteryl ester profiles in these mice were identical to those of the total cholesterol (data not shown). In all mice infected with the recombinant adenoviruses the plasma triglycerides were distributed in the VLDL region (Figure 3C).
FIGURE 3. FPLC profiles of total cholesterol, phospholipids, triglycerides and apoA-I in plasma of apoA-I-/- mice expressing the WT apoA-I or the carboxy-terminal mutants apoA-I[Δ(185-243)], apoA-I[Δ(220-243)], apoA-I[Δ(232-243)], apoA-I[E191A/H193A/K195A] or the control protein GFP.
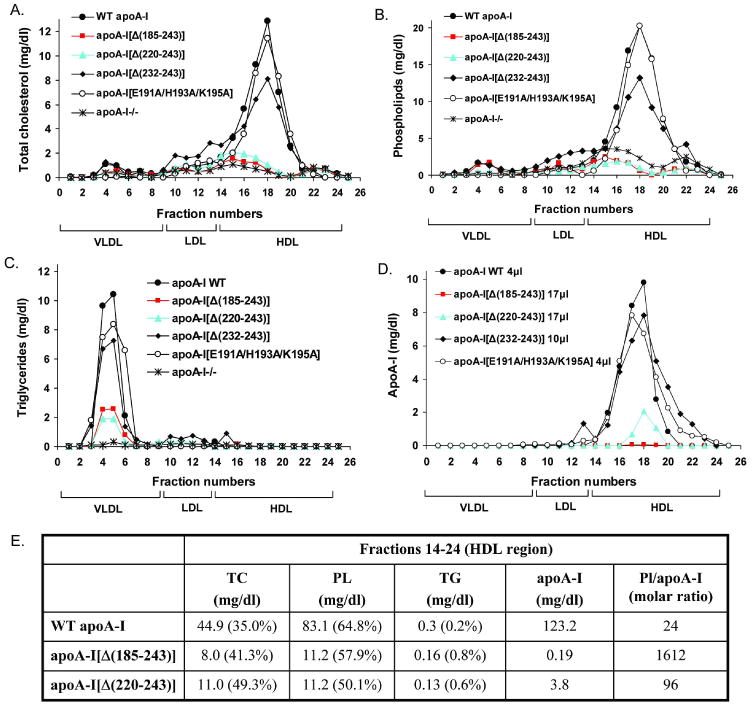
Plasma samples were obtained from mice infected with 1×109 pfu of the recombinant adenoviruses expressing the WT or mutant forms of apoA-I or the control protein GFP four days post-infection. The samples were fractionated by FPLC and then the total cholesterol (A), phospholipids (B), triglycerides (C) and apoA-I (D) levels of each FPLC fraction were determined as described in Experimental Procedures. Panel E, Lipids and apoA-I concentrations from a pool of lipoprotein fractions that correspond to the HDL region (fractions 14-24) expressed as mg/dl. TG, triglycerides; PL, phospholipids; TC, total cholesterol; %, percentage composition relative to the sum of TG, PL and TC values. PL/apoA-I is expressed as molar ratio.
The fractions 14-24, that correspond to the HDL region, obtained from mice expressing the apoA-I[Δ(185-243)] had increased molar ratio of phospholipids/apoA-I compared to that of fractions 14-24 obtained from mice expressing the WT apoA-I (67.2-fold increase) (Figure 3E). An increase in the molar ratio of phospholipids/apoA-I of fractions 14-24 obtained from mice expressing the apoA-I[Δ(220-243)] as compared to mice expressing WT apoA-I (4-fold) was also observed, but this increase was less pronounced than that observed for apoA-I[Δ(185-243)] (Figure 3E). In a previous study it was observed that the HDL isolated by FPLC from ABCA1-deficient mice also had increased molar ratio of phospholipids/apoA-I compared to HDL isolated from control mice (11-fold increase) (24).
Furthermore, based on the lipid composition it was observed an increased percentage of triglycerides in fractions 14-24 obtained from mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] (4- and 3-fold increase, respectively) (Figure 3E). An increased percentage of triglycerides (40-fold increase) was also observed in the HDL isolated from ABCA1 deficient mice (24).
Effect of the carboxy-terminal mutations on the distribution of apoA-I in different densities and the composition of HDL
The fractions obtained following density gradient ultracentrifugation of the plasma of mice expressing the WT or the mutant apoA-I forms or the control protein GFP were analyzed by SDS-PAGE and the protein bands were visualized by staining with Coomassie Brilliant Blue. This analysis showed that in mice infected with adenoviruses expressing the WT apoA-I, the apoA-I[Δ(232-243)] and the apoA-I[E191A/H193A/K195A] mutants, apoA-I was distributed in the HDL2 and HDL3 region (Figure 4A, D, E). In mice expressing the apoA-I[Δ(232-243)] mutant there was a shift in the distribution of apoA-I towards the HDL3 region (Figure 4D). In mice expressing the apoA-I[Δ(220-243)] mutant, the low levels of apoA-I were detected in HDL2, HDL3 and the d>1.21g/ml fractions (Figure 4C). The majority of apoA-I was found in the HDL3 and the lipid poor fractions. In mice expressing the apoA-I[Δ(185-243)] mutant, apoA-I could not be detected by Coomassie Brilliant Blue staining (Figure 4B). Western blotting showed that the majority of apoA-I was found in the d>1.21 g/ml fractions and small quantities were in the HDL3 region (Figure 4G). The low levels of apoA-I detected by SDS-PAGE analysis of the density gradient ultracentrifugation fractions for the carboxy-terminal mutants are consistent with the low plasma apoA-I levels detected by turbidimetric immunoassay (Table 2). In mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] carboxy-terminal deletion mutants the apoE levels were increased and apoE was distributed in the HDL2 region (Figure 4B,C). The apoE levels and distribution in these mice were similar to those observed in mice infected with the control adenovirus that expresses GFP (Figure 4F). The HDL fraction of apoA-I-/- mice was shown previously to contain mainly apoE, as well as apoA-IV, apoA-II and apoCs (25).
FIGURE 4. SDS-PAGE analysis of density gradient ultracentrifugation fractions of plasma of apoA-I-/- mice expressing the WT or mutant forms of apoA-I or the control protein GFP.
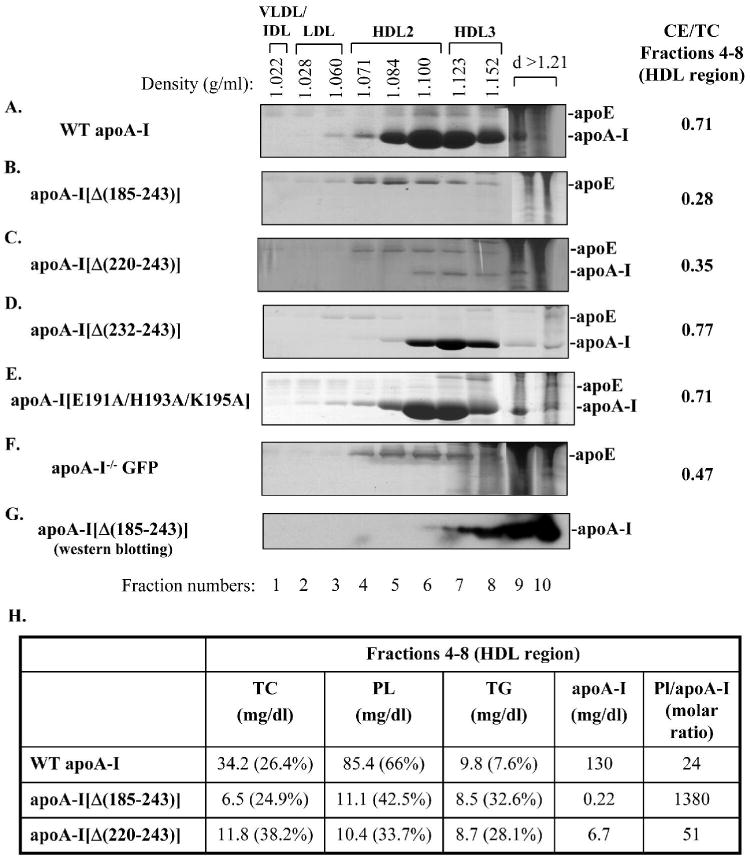
Fractionation of plasma was performed as described in Experimental Procedures. The fractions that were obtained from the plasma of mice expressing the WT apoA-I (A) or the carboxy-terminal mutants apoA-I[Δ(185-243)] (B), apoA-I[Δ(220-243)] (C), apoA-I[Δ(232-243)] (D), apoA-I[E191A/H193A/K195A] (E) or the control protein GFP (F) were subjected to SDS-PAGE and the protein bands were visualized by staining with Coomassie Briliant Blue. On the right side of panels A-F is shown the CE/TC ratio from a pool of lipoprotein fractions that correspond to the HDL region (fractions 4-8). The fractions that were obtained from the plasma of mice expressing the apoA-I[Δ(185-243)] were further analyzed by SDS-PAGE and Western blotting using an anti-human apoA-I antibody (G) as described in Experimental Procedures. The densities of the fractions are indicated on the top of the figure. Panel H, Lipids and apoA-I concentrations from a pool of lipoprotein fractions that correspond to the HDL region (fractions 4-8) expressed as mg/dl. TG, triglycerides; PL, phospholipids; TC, total cholesterol; %, percentage composition relative to the sum of TG, PL and TC values. PL/apoA-I is expressed as molar ratio.
Analysis of the distribution of total cholesterol, cholesteryl ester, free cholesterol, triglycerides and phospholipids following density gradient ultracentrifugation of plasma essentially confirmed the distribution of these lipids to different lipoprotein fractions that were obtained by FPLC fractionation (data not shown). The CE/TC ratio was calculated in the fractions 4-8 that correspond to the HDL region (Figure 4A-F). This analysis showed that the CE/TC ratio in mice infected with the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] carboxy-terminal deletion mutants was lower than that of the mice infected with the GFP-expressing adenovirus and was greatly reduced as compared to the CE/TC ratio of mice infected with adenoviruses expressing the WT apoA-I or the apoA-I[Δ(232-243)] and the apoA-I[E191A/H193A/K195A] mutants (Figure 4A-F). Similar information for the CE/TC ratio of the HDL region for the WT and mutant apoA-I forms was obtained by analysis of the FPLC fractions (data not shown).
Consistent with the lipid composition of the FPLC fractions that correspond to the HDL region, the fractions 4-8 obtained by density gradient ultracentrifugation that also correspond to the HDL region had increased molar ratio of phospholipids/apoA-I (57.5-fold increase) and had an increased percentage of triglycerides (4.3-fold increase) in mice expressing the apoA-I[Δ(185-243)] compared to fractions 4-8 obtained from mice expressing the WT apoA-I (Figure 4H). A less pronounced increase in the molar ratio of phospholipids/apoA-I (2.1-fold) and the percentage of triglycerides (3.7-fold) was observed for fractions 4-8 obtained from mice expressing the apoA-I[Δ(220-243)] as compared to mice expressing the WT apoA-I (Figure 4H).
Effect of the carboxy-terminal mutations on the formation of HDL
Analysis by EM of the HDL fractions 6 and 7 (density 1.100-1.123 g/ml), obtained by density gradient ultracentrifugation (Figure 4A-F), showed that the mice expressing the WT apoA-I and the apoA-I[Δ(232-243)] and apoA-I[E191A/H193A/K195A] mutants formed a large number of spherical HDL particles (Figure 5A, D, E). In contrast the HDL fraction of mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] mutants contained few spherical particles (Figure 5B,C) similar to those seen in control mice infected with the adenovirus expressing GFP (Figure 5F). Similar results were obtained by EM analysis of fraction 8, whereas analysis of d.1.21g/ml fractions did not show the presence of any particles (data not shown).
FIGURE 5. Electron microscopy pictures of the fractions corresponding to the HDL region obtained from the plasma of apoA-I-/- mice expressing the WT or mutant forms of apoA-I or the control protein GFP.

Following density gradient ultracentrifugation the fractions that float to the HDL region obtained from the plasma of mice expressing the WT apoA-I (A) or the carboxy-terminal mutants apoA-I[Δ(185-243)] (B), apoA-I[Δ(220-243)] (C), apoA-I[Δ(232-243)] (D), apoA-I[E191A/H193A/K195A] (E) or the control protein GFP (F) were analyzed by EM. The densities of the fractions used are indicated on the bottom of each picture. The photomicrographs were taken at 75,000× magnification and enlarged 3 times. The fractions that float to the HDL from mice expressing the WT apoA-I or the carboxy-terminal deletion mutants apoA-I[Δ(185-243)], apoA-I[Δ(220-243)] or the control protein GFP were also analyzed by SDS-PAGE and Western blotting using an anti-mouse apoE antibody (G).
The HDL fractions 6 and 7 from mice expressing the WT apoA-I or the carboxy-terminal deletion mutants apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] or the control protein GFP were also analyzed by SDS-PAGE and Western blotting using an anti-mouse apoE antibody. It was found that the apoE levels in the HDL fractions of mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] mutants, as well as in apoA-I-/- mice infected with the adenovirus-expressing GFP were increased compared to the HDL fractions of mice expressing the WT apoA-I (Figure 5G). It has been previously shown that the levels of mouse apoE in plasma as well as in the HDL fraction of apoA-I-/- mice are high and are reduced by expression of WT apoA-I in these mice (26;27).
The carboxy-terminal deletions inhibit the formation of α-HDL particles, but can promote the formation of preβ-HDL particles
Two-dimensional gel electrophoresis of plasma showed that the WT apoA-I and the apoA-I[Δ(232-243)] and apoA-I[E191A/H193A/K195A] mutants formed α-HDL particles and small amounts of preβ-HDL particles (Figure 6A, J, K). In contrast, the apoA-I[Δ(185-243)] mutant formed only preβ-HDL particles and the apoA-I[Δ(220-243)] formed preβ-HDL particles and a very small amount of α-HDL particles (Figure 6D, G). When duplicate blots corresponding to those shown in Figure 6A, D, G, J, K were treated with anti-mouse apoE antibodies, apoE-containing lipoproteins with fast electrophoretic mobility and larger size were detected in the plasma of apoA-I-deficient mice infected with adenoviruses expressing the control protein GFP and those expressing the carboxy-terminal deletion mutants apoA-I[Δ(185-243)], apoA-I[Δ(220-243)] (Figure 6C, E, H). ApoE was not detected in the plasma of mice expressing the WT apoA-I (Figure 6B), as well as those expressing the apoA-I[Δ(232-243)] or the apoA-I[E191A/H193A/K195A] mutants (data not shown). Figures 6F, I show the overlapping of Figures 6D, E and Figures 6G, H, respectively, in order to establish the relative positions of the apoA-I and apoE containing lipoprotein particles. These observations are consistent with previous findings that showed increased apoE in the HDL fraction of apoA-I-/- mice (25) and decreased apoE in the HDL fraction of WT apoA-I overexpressing mice (26;27). The increase in apoE levels in mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] carboxy-terminal deletion mutants may explain the small number of spherical HDL particles observed in the HDL fraction of these mice as well as of apoA-I-/- mice (Figure 5B,C,F).
FIGURE 6. Two dimensional gel electrophoresis analysis of plasma of apoA-I-/- mice expressing the WT or mutant forms of apoA-I or the control protein GFP. Agarose gel electrophoresis analysis of plasma of apoA-I-/- mice expressing the WT apoA-I or the apoA-I[Δ(185-243)] mutant and of purified apoA-I[Δ(185-243)] protein.
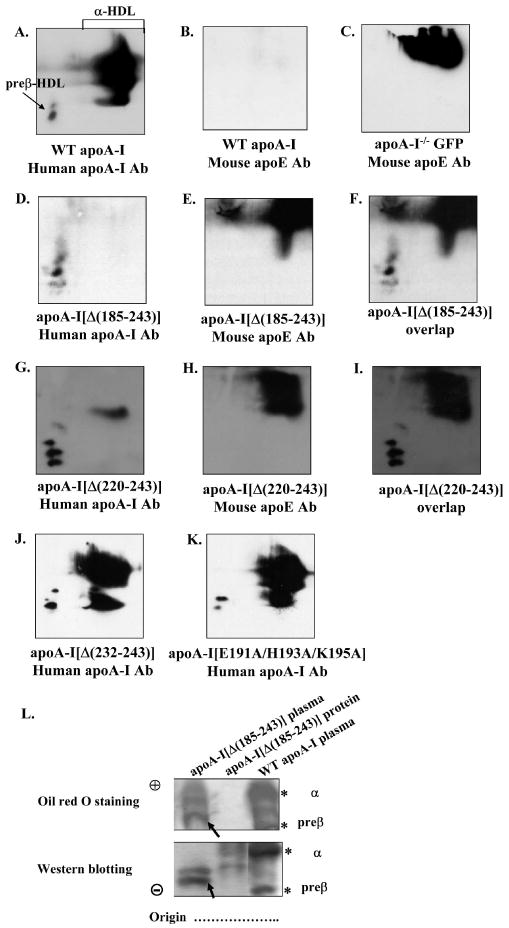
The plasma of mice expressing the WT apoA-I (A, B) or the control protein GFP (C) or the carboxy-terminal mutants apoA-I[Δ(185-243)] (D-F), apoA-I[Δ(220-243)] (G-I), apoA-I[Δ(232-243)] (J), apoA-I[E191A/H193A/K195A] (K) were analyzed by two dimensional gel electrophoresis and Western blotting using anti-human apoA-I antibody (A, D, G, J, K) or anti-mouse apoE antibody (B, E, H), as described in Experimental Procedures. Panels F and I show the overlapping of panels D, E and G, H, respectively. Panel L, The plasma of mice expressing the WT apoA-I or the carboxy-terminal mutant apoA-I[Δ(185-243)] and purified apoA-I[Δ(185-243)] protein were analyzed by 0.7% agarose gel electrophoresis followed by Oil Red O neutral lipid staining or Western blotting using a goat polyclonal anti-human apoA-I antibody as described in Experimental Procedures. The asterisks indicate preβ- and α-HDL that contain WT apoA-I. The apoA-I[Δ(185-243)] mutant formed particles with faster electrophoretic mobility than the preβ-HDL of WT apoA-I as demonstrated by Western blot analysis, that accumulated significant levels of neutral lipid (bands indicated by arrow).
The formation or not of preβ- and α-HDL in mice expressing the WT apoA-I or the apoA-I[Δ(185-243)] mutant was evaluated by agarose gel electrophoresis (Figure 6L). HDL was visualized either by Oil Red O neutral lipid staining or Western blot analysis and detection with an anti-human apoA-I polyclonal antibody. The expression of WT apoA-I was associated with α-migrating and preβ-migrating HDL bands that were detected by with neutral lipid staining. These bands, indicated by an asterisk, corresponded to the position of WT apoA-I as determined by immunoblotting (Figure 6L). Oil Red O-stained bands also appear that migrated in the region between preβ- and α-HDL which did not correspond to an apoA-I immunoreactive band. The apoA-I[Δ(185-243)] mutant gave a band that had faster electrophoretic mobility than preβ-HDL of WT apoA-I. This band, indicated by an arrow, could be stained with Oil Red O and corresponded to the position of apoA-I[Δ(185-24)] as determined by immunoblotting (Figure 6L). Another band of faster electrophoretic mobility was detected by Oil Red O staining which did not correspond to an apoA-I immunoreactive band. In addition, the purified apoA-I[Δ(185-243)] was not stained with Oil Red O and had faster electrophoretic mobility compared to the preβ band containing apoA-I[Δ(185-243)] that was present in the plasma of mice expressing this mutant and was stained by Oil Red O (indicated by an arrow) (Figure 6L).
Discussion
Role of specific domains and residues of the carboxy-terminal region of apoA-I in the biogenesis of HDL
Previous in vitro studies showed that the carboxy-terminal apoA-I deletions that remove the 220-231 region diminished the ABCA1-mediated lipid efflux, whereas the carboxy-terminal (232-243) deletion that retains the 220-231 region does not affect the ABCA1-mediated lipid efflux (2). Chemical cross-linking/immunoprecipitation studies showed that the carboxy-terminal apoA-I deletions that remove the 220-231 region also had diminished ability to be cross-linked to ABCA1 (28).
In this and previous studies we considered HDL biogenesis as a continuous pathway where apoA-I and various participating proteins interact successively to form spherical HDL particles that are biologically active. Prerequisite for the biogenesis of HDL are functional interactions between apoA-I and ABCA1 that promote efflux of cellular phospholipids and cholesterol (2;10;11). Human patients or animal models that lack apoA-I or ABCA1 or have defective forms of ABCA1 fail to form HDL (11;22;29).
Studies in HeLa cells expressing an ABCA1 green fluorescence fusion protein (30;31) and in macrophages (32;33) indicated that following interaction at the cell surface, the apoA-I/ABCA1 complex internalizes, interacts with intracellular lipid pools and is resecreted as a lipidated particle (32;33). Following a similar pathway, apoA-I is transcytosed through endothelial cells and is secreted from the apical surface in a lipid-bound form (34).
Recent data indicate that ABCA1/apoA-I interactions in the liver are essential for the initial lipidation of apoA-I and also determine the subsequent maturation of nascent preβ-HDL to spherical α-HDL particles (35;36). When hepatic ABCA1 is inactivated, preβ-HDL fails to mature to α-HDL and is catabolized rapidly by the kidney, thus resulting in low HDL levels (35;36). Adenovirus-mediated gene transfer of ABCA1 in total or liver-specific knockout mice for ABCA1 restored the HDL cholesterol levels in the liver-specific knockout mice but only partially in the total knockout mice (37). The combined data indicate that the liver is the major site for the initial lipidation of apoA-I, which seems to be the rate-limiting step of HDL biogenesis and the contribution of the peripheral tissues in this process appears to be small. In addition, the ABCA1/apoA-I or ABCA1/preβ-HDL interactions in the peripheral tissues appear to enrich the initially lipidated particle with cholesterol and increase its stability (35-37). However, the fact that liver-specific inactivation of ABCA1 in mice reduces plasma HDL to approximately 17% of the WT control suggests that other proteins produced locally by the liver, such as LCAT, may be crucial for the maturation of HDL (35;36).
A fundamental question that remains is how lipid efflux determined by in vitro assays is co-related to the biogenesis of HDL. Previous adenovirus-mediated gene transfer studies of apoA-I mutants to apoA-I-/- mice showed that a carboxy-terminal deletion (220-243), resulted in low levels of HDL cholesterol and formation of a small number of spherical particles, but no further analysis of the nature of these particles was made (19).
In the current study we examined by adenovirus-mediated gene transfer in apoA-I-/- mice the effect of previously studied carboxy-terminal deletion mutant apoA-I[Δ(220-243)] along with the apoA-I[Δ(185-243)] and apoA-I[Δ(232-243)] deletion mutants, as well as substitutions of charged amino acids in the 188-195 loop on the biogenesis of HDL. Residues His 193 and Lys 195 were shown by x-ray studies to interact with the Cr-acac3 molecules that bridge the amino- and carboxy-terminal regions of apoA-I and therefore support a compact configuration of the two-domain structure of apoA-I in the crystal (9). In addition Glu 191 contributes to a patch of charged residues on the surface of apoA-I that is close to hydrophobic residues of the carboxy-terminal domain (9). The objective was to identify critical domains or residues in the carboxy-terminal segment of apoA-I that are required for the biogenesis of HDL.
The initial parameters determined 4 days post-infection were the plasma lipid levels and the lipid FPLC profile that can initially identify putative defects in the biogenesis of HDL, the distribution of HDL in preβ- and α-HDL subpopulations and the formation of HDL by EM. Hepatic apoA-I mRNA levels were also determined to ensure comparable levels of expression of WT and mutant apoA-I forms in order to interpret the observed phenotypes.
Using the above criteria we have established that the two carboxy-terminal deletion mutants apoA-I[Δ(185-243)] and apoA-I[Δ(220-243),] that lack the 220-231 region, had very low total plasma cholesterol and phospholipid levels, that were comparable to those of the control mice that express the GFP protein and very low HDL levels. The near absence of HDL was corroborated by the low plasma apoA-I levels detected by turbidimetric immunoassay and by density gradient ultracentrifugation analysis of plasma. Since the hepatic apoA-I mRNA levels and the secretion of these deletion apoA-I mutants from cells was normal, the present findings suggest that the observed low HDL levels following adenovirus infection is the result of fast clearance from plasma. Previous studies showed that lipid-free apoA-I or partially lipidated apoA-I forms can be catabolized rapidly in the kidney by the cubulin receptor or other mechanisms (35;38;39).
The carboxy-terminal apoA-I mutants that lack the 220-231 region fail to form α-HDL but they can form preβ-HDL particles by an ABCA1-independent mechanism
Two additional important parameters used to assess biogenesis of HDL particles were the formation of HDL particles as determined by EM and the distribution of HDL in preβ- and α-HDL subpopulations as determined by two-dimensional gel electrophoresis. The EM analysis showed that mutants that lack the 220-231 region fail to promote formation of spherical HDL particles. The small number of spherical HDL particles observed in the plasma of mice expressing the two mutants that lack the 220-231 region or GFP most likely represent apoE-containing HDL particles.
The two-dimensional gel electrophoresis of plasma showed that expression of apoA-I[Δ(185-243)] promoted the formation of preβ-HDL particles but not α-HDL particles. The apoA-I[Δ(220-243)] mutant promoted predominantly the formation of preβ-HDL particles and a few α-HDL particles. The preponderance of preβ-HDL particles in the plasma of mice expressing the two carboxy-terminal mutants that lack the 220-231 region can also explain the low levels of plasma HDL.
The observed phenotypes of the carboxy-terminal mutants that lack the 220-231 region combined with their inability to promote lipid efflux and to cross-link to ABCA1 (2;28) suggest a blockage of the first step in the biogenesis of HDL which involves functional interactions between apoA-I and ABCA1. That is necessary for the correct lipidation of apoA-I and the formation of HDL.
The lack of HDL formation may reflect inability of apoA-I to associate with ABCA1 (28), as well as inability to associate with lipids (3), or both. A recent study of refolding of apoA-I during transition from 5 – 0.45M guanidine HCl using stopped flow circular dichroism showed that deletion of the 186-243 carboxy-terminal segment of apoA-I increases the free energy required for the transition from the native state to a partially unfolded intermediate state (40). It has been proposed that association of apoA-I with lipids requires partial unfolding of apoA-I (40-42). Thus the observed change in the free energy required for the transition from the native state to the partially unfolded intermediate state of apoA-I[Δ(186-243)] may affect the association of apoA-I with lipids and possibly its interactions with ABCA1 that lead to lipid efflux and promote the formation of HDL (40).
In contrast to the properties of the carboxy-terminal deletion mutants that lack the 220 to 231 region, mice expressing the apoA-I[Δ(232-243)] that retains the 220 to 231 region and the point mutant apoA-I[E191A/H193A/K195A] had normal HDL levels. The EM and two-dimensional gel electrophoresis analysis showed formation of spherical HDL and normal preβ- and α-HDL subpopulations. The overall phenotype of mice expressing these mutants was similar to this observed in mice expressing the WT apoA-I. The ability of the apoA-I[Δ(232-243)] and apoA-I[E191A/H193A/K195A] mutants to promote formation of normal HDL particles is consistent with their ability to promote normal ABCA1-mediated lipid efflux in vitro.
Although more rigorous studies may be required to assess the importance of charged residues of the carboxy-terminal region in the biogenesis of HDL the current findings indicate that charged amino acids Glu191, His193 and Lys195 may not be involve in ABCA1/ apoA-I interactions or interactions of the carboxy- and amino-terminal domains of apoA-I that are important for the biogenesis of HDL. Previous studies also showed that substitutions of charged amino acids Glu234, Glu235, Lys238 and Lys239 by Ala did not affect the biogenesis of HDL (19), whereas alteration of hydrophobic residues in the 211- 229 region of apoA-I prevented the maturation of HDL and led to the accumulation of discoidal HDL particles (19).
The amino-terminal 1-184 and 1-219 region of apoA-I can promote formation of preβ-HDL particles in an ABCA1-independent process
Numerous studies have shown that preβ-HDL particles can be formed de novo by an ABCA1-dependent process that leads to the formation of HDL (35-37;43-47). In addition, processes catalyzed by hepatic lipase, CETP and PLTP can generate preβ-HDL from α-HDL particles (48-52). Furthermore, deficiency of apoM inhibits the formation of preβ-HDL (53;54). Previous studies also showed that the plasma of humans with Tangier disease (11;55) and of ABCA1-/- mice (24) contains preβ-HDL but lacks α-HDL particles. In the ABCA1-/- mice, the composition of HDL is abnormal and has increased PL/apoA-I ratio (24). In addition, inhibition of ABCA1 in HepG2 cells and macrophage cultures by glyburide inhibited the formation of α-HDL particles but did not affect the formation of preβ-HDL particles (56). All these findings indicate that some types of preβ-HDL particles can be formed independently of apoA-I/ABCA1 interactions.
Similarly, in this study we show an abnormal PL/apoA-I ratio of the HDL fraction obtained by FPLC or density gradient ultracentrifugation in mice expressing the apoA-I[Δ(185-243)] and apoA-I[Δ(220-243)] mutants. Furthermore, the electrophoretic mobility of the lipid-free apoA-I[Δ(185-243)] is different from that of the lipidated particles formed in mice expressing this carboxy-terminal mutant.
Overall, our studies establish that the 220-231 region of apoA-I is required for functional interactions between apoA-I and ABCA1 that are necessary for the biogenesis of α-HDL particles and the N-terminal domain that lacks the 220-231 region can form preβ-HDL particles in an ABCA1-independent process.
Acknowledgments
We thank Dr Efstratios Stratikos for critical reading and comments and Ms Gayle Forbes for technical assistance.
This work was supported by the Grant of the 6th Framework Programme of the European Union LSHM-CT-2006-037631 (A.C. and V.I.Z.) and the National Institutes of Health Grant HL-48739 (V.I.Z.).
Abbreviations: The abbreviations used are
- ABCA1
ATP-binding cassette transporter A1
- apoA-I
apolipoprotein A-I
- apoA-I-/- mice
apoA-I-deficient mice
- BSA
bovine serum albumin
- CE
cholesteryl ester
- cpt-cAMP
8-(4-chlorophenylthio) adenosine 3′:5′-cyclic monophosphate
- Cr-acac3
Cr(III)-Tris-acetylacetonate
- EM
electron microscopy
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HDL
high density lipoproteins
- LCAT
lecithin cholesterol acyltransferase
- PBS
phosphate-buffered saline
- PL
phospholipids
- TC
total cholesterol
- TG
triglycerides
- WT
wild-type
References
- 1.Zannis VI, Kardassis D, Zanni EE. Genetic mutations affecting human lipoproteins, their receptors, and their enzymes. Adv Hum Genet. 1993;21:145–319. doi: 10.1007/978-1-4615-3010-7_3. [DOI] [PubMed] [Google Scholar]
- 2.Chroni A, Liu T, Gorshkova I, Kan HY, Uehara Y, von Eckardstein A, Zannis VI. The central helices of apoA-I can promote ATP-binding cassette transporter A1 (ABCA1)-mediated lipid efflux. Amino acid residues 220-231 of the wild-type apoA-I are required for lipid efflux in vitro and high density lipoprotein formation in vivo. J Biol Chem. 2003;278:6719–6730. doi: 10.1074/jbc.M205232200. [DOI] [PubMed] [Google Scholar]
- 3.Laccotripe M, Makrides SC, Jonas A, Zannis VI. The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affect its rate of phospholipid binding and its association with high density lipoprotein. J Biol Chem. 1997;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 4.Liu T, Krieger M, Kan HY, Zannis VI. The effects of mutations in helices 4 and 6 of apoA-I on scavenger receptor class B type I (SR-BI)-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted high density lipoprotein and SR-BI is required for efficient lipid transport. J Biol Chem. 2002;277:21576–21584. doi: 10.1074/jbc.M112103200. [DOI] [PubMed] [Google Scholar]
- 5.Zannis VI, Chroni A, Kypreos KE, Kan HY, Cesar TB, Zanni EE, Kardassis D. Probing the pathways of chylomicron and HDL metabolism using adenovirus-mediated gene transfer. Curr Opin Lipidol. 2004;15:151–166. doi: 10.1097/00041433-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Li WH, Tanimura M, Luo CC, Datta S, Chan L. The apolipoprotein multigene family: biosynthesis, structure, structure-function relationships, and evolution. J Lipid Res. 1988;29:245–271. [PubMed] [Google Scholar]
- 7.Nolte RT, Atkinson D. Conformational analysis of apolipoprotein A-I and E-3 based on primary sequence and circular dichroism. Biophys J. 1992;63:1221–1239. doi: 10.1016/S0006-3495(92)81698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy HM. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci U S A. 2006;103:2126–2131. doi: 10.1073/pnas.0506877103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Wang N, Silver DL, Costet P, Tall AR. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J Biol Chem. 2000;275:33053–33058. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- 11.Assmann G, von Eckardstein A, Brewer HB. Familial analphalipoproteinemia: Tangier disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2937–2960. [Google Scholar]
- 12.Soutar AK, Garner CW, Baker HN, Sparrow JT, Jackson RL, Gotto AM, Smith LC. Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: cholesterol acyltransferase. Biochemistry. 1975;14:3057–3064. doi: 10.1021/bi00685a003. [DOI] [PubMed] [Google Scholar]
- 13.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 14.Krieger M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. 2001;108:793–797. doi: 10.1172/JCI14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang XC, Bruce C, Mar J, Lin M, Ji Y, Francone OL, Tall AR. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest. 1999;103:907–914. doi: 10.1172/JCI5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chroni A, Kan HY, Kypreos KE, Gorshkova IN, Shkodrani A, Zannis VI. Substitutions of glutamate 110 and 111 in the middle helix 4 of human apolipoprotein A-I (apoA-I) by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry. 2004;43:10442–10457. doi: 10.1021/bi049782p. [DOI] [PubMed] [Google Scholar]
- 18.Chroni A, Kan HY, Shkodrani A, Liu T, Zannis VI. Deletions of Helices 2 and 3 of Human ApoA-I Are Associated with Severe Dyslipidemia following Adenovirus-Mediated Gene Transfer in ApoA-I-Deficient Mice. Biochemistry. 2005;44:4108–4117. doi: 10.1021/bi047998l. [DOI] [PubMed] [Google Scholar]
- 19.Reardon CA, Kan HY, Cabana V, Blachowicz L, Lukens JR, Wu Q, Liadaki K, Getz GS, Zannis VI. In vivo studies of HDL assembly and metabolism using adenovirus-mediated transfer of ApoA-I mutants in ApoA-I-deficient mice. Biochemistry. 2001;40:13670–13680. doi: 10.1021/bi011451e. [DOI] [PubMed] [Google Scholar]
- 20.Chroni A, Duka A, Kan HY, Liu T, Zannis VI. Point mutations in apolipoprotein A-I mimic the phenotype observed in patients with classical lecithin:cholesterol acyltransferase deficiency. Biochemistry. 2005;44:14353–14366. doi: 10.1021/bi050962o. [DOI] [PubMed] [Google Scholar]
- 21.Roghani A, Zannis VI. Alterations of the glutamine residues of human apolipoprotein AI propeptide by in vitro mutagenesis. Characterization of the normal and mutant protein forms. Biochemistry. 1988;27:7428–7435. doi: 10.1021/bi00419a038. [DOI] [PubMed] [Google Scholar]
- 22.Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci U S A. 1992;89:7134–7138. doi: 10.1073/pnas.89.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fielding CJ, Fielding PE. Two-dimensional nondenaturing electrophoresis of lipoproteins: applications to high-density lipoprotein speciation. Methods Enzymol. 1996;263:251–259. doi: 10.1016/s0076-6879(96)63017-1. [DOI] [PubMed] [Google Scholar]
- 24.Francone OL, Subbaiah PV, van Tol A, Royer L, Haghpassand M. Abnormal phospholipid composition impairs HDL biogenesis and maturation in mice lacking Abca1. Biochemistry. 2003;42:8569–8578. doi: 10.1021/bi034540v. [DOI] [PubMed] [Google Scholar]
- 25.Plump AS, Azrolan N, Odaka H, Wu L, Jiang X, Tall A, Eisenberg S, Breslow JL. ApoA-I knockout mice: characterization of HDL metabolism in homozygotes and identification of a post-RNA mechanism of apoA-I up-regulation in heterozygotes. J Lipid Res. 1997;38:1033–1047. [PubMed] [Google Scholar]
- 26.Scott BR, McManus DC, Franklin V, McKenzie AG, Neville T, Sparks DL, Marcel YL. The N-terminal globular domain and the first class A amphipathic helix of apolipoprotein A-I are important for lecithin:cholesterol acyltransferase activation and the maturation of high density lipoprotein in vivo. J Biol Chem. 2001;276:48716–48724. doi: 10.1074/jbc.M106265200. [DOI] [PubMed] [Google Scholar]
- 27.McManus DC, Scott BR, Frank PG, Franklin V, Schultz JR, Marcel YL. Distinct central amphipathic alpha-helices in apolipoprotein A-I contribute to the in vivo maturation of high density lipoprotein by either activating lecithin-cholesterol acyltransferase or binding lipids. J Biol Chem. 2000;275:5043–5051. doi: 10.1074/jbc.275.7.5043. [DOI] [PubMed] [Google Scholar]
- 28.Chroni A, Liu T, Fitzgerald ML, Freeman MW, Zannis VI. Cross-linking and lipid efflux properties of apoA-I mutants suggest direct association between apoA-I helices and ABCA1. Biochemistry. 2004;43:2126–2139. doi: 10.1021/bi035813p. [DOI] [PubMed] [Google Scholar]
- 29.Matsunaga T, Hiasa Y, Yanagi H, Maeda T, Hattori N, Yamakawa K, Yamanouchi Y, Tanaka I, Obara T, Hamaguchi H. Apolipoprotein A-I deficiency due to a codon 84 nonsense mutation of the apolipoprotein A-I gene. Proc Natl Acad Sci U S A. 1991;88:2793–2797. doi: 10.1073/pnas.88.7.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neufeld EB, Demosky SJ, Jr, Stonik JA, Combs C, Remaley AT, Duverger N, Santamarina-Fojo S, Brewer HB., Jr The ABCA1 transporter functions on the basolateral surface of hepatocytes. Biochem Biophys Res Commun. 2002;297:974–979. doi: 10.1016/s0006-291x(02)02274-x. [DOI] [PubMed] [Google Scholar]
- 31.Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB., Jr Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi Y, Smith JD. Cholesterol efflux to apolipoprotein AI involves endocytosis and resecretion in a calcium-dependent pathway. Proc Natl Acad Sci U S A. 1999;96:11358–11363. doi: 10.1073/pnas.96.20.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith JD, Waelde C, Horwitz A, Zheng P. Evaluation of the role of phosphatidylserine translocase activity in ABCA1-mediated lipid efflux. J Biol Chem. 2002;277:17797–17803. doi: 10.1074/jbc.M201594200. [DOI] [PubMed] [Google Scholar]
- 34.Rohrer L, Cavelier C, Fuchs S, Schluter MA, Volker W, von Eckardstein A. Binding, internalization and transport of apolipoprotein A-I by vascular endothelial cells. Biochim Biophys Acta. 2006;1761:186–194. doi: 10.1016/j.bbalip.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singaraja RR, Stahmer B, Brundert M, Merkel M, Heeren J, Bissada N, Kang M, Timmins JM, Ramakrishnan R, Parks JS, Hayden MR, Rinninger F. Hepatic ATP-binding cassette transporter A1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler Thromb Vasc Biol. 2006;26:1821–1827. doi: 10.1161/01.ATV.0000229219.13757.a2. [DOI] [PubMed] [Google Scholar]
- 37.Singaraja RR, Van Eck M, Bissada N, Zimetti F, Collins HL, Hildebrand RB, Hayden A, Brunham LR, Kang MH, Fruchart JC, van Berkel TJ, Parks JS, Staels B, Rothblat GH, Fievet C, Hayden MR. Both Hepatic and Extrahepatic ABCA1 Have Discrete and Essential Functions in the Maintenance of Plasma High-Density Lipoprotein Cholesterol Levels In Vivo. Circulation. 2006 doi: 10.1161/CIRCULATIONAHA.106.621433. [DOI] [PubMed] [Google Scholar]
- 38.Kozyraki R, Fyfe J, Kristiansen M, Gerdes C, Jacobsen C, Cui S, Christensen EI, Aminoff M, de la CA, Krahe R, Verroust PJ, Moestrup SK. The intrinsic factor-vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein A-I receptor facilitating endocytosis of high-density lipoprotein. Nat Med. 1999;5:656–661. doi: 10.1038/9504. [DOI] [PubMed] [Google Scholar]
- 39.Hammad SM, Stefansson S, Twal WO, Drake CJ, Fleming P, Remaley A, Brewer HB, Jr, Argraves WS. Cubilin, the endocytic receptor for intrinsic factor-vitamin B(12) complex, mediates high-density lipoprotein holoparticle endocytosis. Proc Natl Acad Sci U S A. 1999;96:10158–10163. doi: 10.1073/pnas.96.18.10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gross E, Peng DQ, Hazen SL, Smith JD. A novel folding intermediate state for apolipoprotein A-I: role of the amino and carboxy termini. Biophys J. 2006;90:1362–1370. doi: 10.1529/biophysj.105.075069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc Natl Acad Sci U S A. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banuelos S, Muga A. Binding of molten globule-like conformations to lipid bilayers. Structure of native and partially folded alpha-lactalbumin bound to model membranes. J Biol Chem. 1995;270:29910–29915. doi: 10.1074/jbc.270.50.29910. [DOI] [PubMed] [Google Scholar]
- 43.Castro GR, Fielding CJ. Early incorporation of cell-derived cholesterol into pre-beta-migrating high-density lipoprotein. Biochemistry. 1988;27:25–29. doi: 10.1021/bi00401a005. [DOI] [PubMed] [Google Scholar]
- 44.Forte TM, Goth-Goldstein R, Nordhausen RW, McCall MR. Apolipoprotein A-I-cell membrane interaction: extracellular assembly of heterogeneous nascent HDL particles. J Lipid Res. 1993;34:317–324. [PubMed] [Google Scholar]
- 45.Forte TM, Bielicki JK, Goth-Goldstein R, Selmek J, McCall MR. Recruitment of cell phospholipids and cholesterol by apolipoproteins A-II and A-I: formation of nascent apolipoprotein-specific HDL that differ in size, phospholipid composition, and reactivity with LCAT. J Lipid Res. 1995;36:148–157. [PubMed] [Google Scholar]
- 46.Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Chau P, Nakamura Y, Fielding CJ, Fielding PE. Mechanism of prebeta-HDL formation and activation. Biochemistry. 2006;45:3981–3987. doi: 10.1021/bi052535g. [DOI] [PubMed] [Google Scholar]
- 48.Clay MA, Newnham HH, Barter PJ. Hepatic lipase promotes a loss of apolipoprotein A-I from triglyceride-enriched human high density lipoproteins during incubation in vitro. Arterioscler Thromb. 1991;11:415–422. doi: 10.1161/01.atv.11.2.415. [DOI] [PubMed] [Google Scholar]
- 49.Hennessy LK, Kunitake ST, Kane JP. Apolipoprotein A-I-containing lipoproteins, with or without apolipoprotein A-II, as progenitors of pre-beta high-density lipoprotein particles. Biochemistry. 1993;32:5759–5765. doi: 10.1021/bi00073a006. [DOI] [PubMed] [Google Scholar]
- 50.Liang HQ, Rye KA, Barter PJ. Dissociation of lipid-free apolipoprotein A-I from high density lipoproteins. J Lipid Res. 1994;35:1187–1199. [PubMed] [Google Scholar]
- 51.von Eckardstein A, Jauhiainen M, Huang Y, Metso J, Langer C, Pussinen P, Wu S, Ehnholm C, Assmann G. Phospholipid transfer protein mediated conversion of high density lipoproteins generates pre beta 1-HDL. Biochim Biophys Acta. 1996;1301:255–262. doi: 10.1016/0005-2760(96)00050-1. [DOI] [PubMed] [Google Scholar]
- 52.Settasatian N, Duong M, Curtiss LK, Ehnholm C, Jauhiainen M, Huuskonen J, Rye KA. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J Biol Chem. 2001;276:26898–26905. doi: 10.1074/jbc.M010708200. [DOI] [PubMed] [Google Scholar]
- 53.Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med. 2005;11:418–422. doi: 10.1038/nm1211. [DOI] [PubMed] [Google Scholar]
- 54.Xu N, Dahlback B. A novel human apolipoprotein (apoM) J Biol Chem. 1999;274:31286–31290. doi: 10.1074/jbc.274.44.31286. [DOI] [PubMed] [Google Scholar]
- 55.Asztalos BF, Brousseau ME, McNamara JR, Horvath KV, Roheim PS, Schaefer EJ. Subpopulations of high density lipoproteins in homozygous and heterozygous Tangier disease. Atherosclerosis. 2001;156:217–225. doi: 10.1016/s0021-9150(00)00643-2. [DOI] [PubMed] [Google Scholar]
- 56.Krimbou L, Hajj HH, Blain S, Rashid S, Denis M, Marcil M, Genest J. Biogenesis and speciation of nascent apoA-I-containing particles in various cell lines. J Lipid Res. 2005;46:1668–1677. doi: 10.1194/jlr.M500038-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Karathanasis SK, Salmon E, Haddad IA, Zannis VI. In: Biochemistry and Biology of Plasma Proteins. Scanu AM, Spector AA, editors. Marcel Dekker, Inc.; New York, NY: 1985. pp. 474–493. [Google Scholar]