

Abstract
Three phenotypically related genetic syndromes and their lesions (LKB1, PTEN, and TSC1/2) are identified as frequently altered in lung cancer. LKB1, a kinase inactivated in 30% of lung cancers, is discussed in this review. Loss of LKB1 regulation often coincident with KRAS activation allows for unchecked growth and the metabolic capacity to accommodate the proliferation.
Keywords: LKB1, STK11, Peutz-Jeghers syndrome, review, lung cancer, heritable tumour syndromes
Heritable tumour syndromes and lung cancer
Heritable tumour syndromes have provided invaluable insights into human disease. These focal perversions of normal growth and development shine as a beacon on particular genes and pathways in a manner, which, once deciphered, can impact far beyond the heritable syndrome itself. Just as a jigsaw puzzle assembled in parts suddenly comes together when a key piece is recognised, the genetic syndrome can bring together vastly disparate biologic knowledge to yield a bit of clarity where none existed before. Although the field of lung cancer has lacked such a fortuitous discovery to date, a series of recent events appears likely to open a new era in our understanding of the disease. Three genetic syndromes, Peutz–Jeghers syndrome (PJS), Cowden's disease (CD), and tuberous sclerosis (TS), all sharing parallel phenotypes primarily of non-malignant and non-pulmonary disease, have crystallised a common pathway, which is activated in a significant proportion of all human lung cancers. Key pieces have been identified and joined, such as nutrient metabolism, proliferative signalling, and anti-apoptosis pathways. The puzzle of lung tumour genesis, invasion, and metastasis is somewhat more complete.
LKB1 and PJS
The central figure in the current discussion is the gene LKB1. Its discovery as well as an appreciation of its biology is dominated by its causative role in the autosomal-dominant inherited PJS. First described in the 1920s by Johannes Peutz, an association with cancer was made by Harold Jeghers in the 1940s (Peutz, 1921; Jeghers et al, 1949). The syndrome includes an overgrowth of differentiated tissues called hamartomas, primarily polyps in the gastrointestinal tract (GI), as well as abnormal pigmentation of the mucous membranes and skin. Validating Dr Jegher's observations, large cohorts have clarified the risk and spectrum of malignancies in PJS, including a cumulative cancer risk of 60% by the age 60 years (Hearle et al, 2006). Of primary concern for PJS patients are tumours of GI origin, although breast, gynaecologic, pancreas, and lung have been reported.
The pattern of inheritance of PJS, as well as the high frequency of cancer in affected, indicated that a tumour suppressor gene was likely the causative agent. In 1997, Hemminki et al (1997) identified the chromosome region 19p13.3 as the relevant cytoband. A year later a single gene called serine/threonine kinase 11 (STK11) was identified as the culprit (Hemminki et al, 1998; Jenne et al, 1998). The transcript for STK11 had previously been reported in an unmapped fashion by the more commonly used gene alias LKB1. The identification of a tumour suppressor and its genomic coordinates inspired a flurry of tumour resequencing surveys of LKB1 in a range of tissues. Table 1 reviews a sample of the largest of these reports in the area of lung cancer, and includes associated epigenetic findings. Although infrequently mutated in spontaneous tumours, LKB1 point mutations were seen in a pattern mirroring those seen in association with PJS, including cervix, gastrointestinal, and pancreas (Avizienyte et al, 1999; Sanchez-Cespedes, 2007). A range of atypical PJS tumours was also identified, including prostate and melanoma. Surprisingly, the highest mutation rate, hovering around 25% in multiple studies, was in lung adenocarcinomas, an established but uncommon PJS-associated tumour (Virmani et al, 1998; Sanchez-Cespedes et al, 2001) (Table 1).
Table 1. Summary results of studies reporting LKB1 mutation and other related events in lung cancer.
| Reference | Year | Finding | Method | Comments/observations |
|---|---|---|---|---|
| Virmani et al (1998) | 1998 | 10/12 LOH in NSCLC cell lines and 3/9 in SmCC cell lines in 19p13.2 | Allelotyping by PCR | Lung cancer cell lines used to define common, specific known, and novel deletions in NSCLC and SmCC cell lines |
| Sobottka et al (2000) | 2000 | 7/12 (58%) patients with LOH at 19p in brain metastases of lung cancer patients, whereas glioma LKB1 LOH was <20% | Microsatellite PCR analysis and genomic sequencing | None of brain metastases showed LKB1 mutations by direct genomic sequencing |
| Sanchez-Cespedes et al (2002) | 2002 | 5/20 of primary AC, 0/12 primary SCC; and 2/9 of lung cancer cell lines with mutation of LKB1 | Microsatellite PCR, direct sequencing, long-range PCR | 0/12 mutations in SCC |
| 1/20 tumor with LKB1 promoter hypermethylation | Methylation-specific PCR | |||
| Ghaffar et al (2003) | 2003 | 9/35 loss of LKB1 protein in AC, 10/96 loss in AAH (7/33 loss in high-grade AAH; 3/63 loss in low grade; 9/35 invasive tumors) | Immunohistochemistry | LKB1 protein loss from AAH correlated with severe dysplasia, suggesting loss as essential to transition from pre- to malignant tumor. Also showed strong positive association between biallelic gene inactivation and negative protein expression |
| Fernandez et al (2004) | 2004 | 5/19 lung AD with point mutations of LKB1 | Direct sequencing, long-range PCR | Frequent observation of concurrent KRAS (4/11) and p53 (6/11) in LKB1 mutant samples. Performed microarray analysis with several genes dependent on LKB1 status |
| 0/19 samples with promoter methylation of LKB1 | Methylation-specific PCR | |||
| 10/18 of samples with LOH at 19p | Fluorescent in situ hybridization | |||
| Carretero et al (2004) | 2004 | 6/11 AD contained LKB1 deletion or truncating mutant, 0/11 SCC | Allelotyping by PCR and sequencing | Several AD lines with LKB1 mutant also have KRAS mutant |
| Matsumoto et al (2007) | 2007 | Lung cancer cell lines with mutations in LKB1: 13/31 AD, 1/2 ADSCC, 3/11 SCC, 3/7 LCC, and 1/19 SmCC | RT-PCR, sequencing and genomic sequencing | Strong positive correlation with KRAS status, not correlation with p53 status or EGFR mutation status |
| 8/70 lung cancer cell lines demonstrated shortened LKB1 transcripts, 9/70 demonstrated absent LKB1 transcript | Two cell lines with absent gene expression did not demonstrate mutation, suggesting alternate gene silencing method. The 9/70 with loss of LKB1 had deletions in promoter or exon 1 | |||
| Primary tumors: 1/106 stage 1 tumor with LKB1 mutation, 3/24 stages I–III. 7/91 male smokers, 0/64 female non-smokers | Small numbers limit interpretation for KRAS and EGFR associations, appears to be associated with poorly differentiated tumors | |||
| 3/25 brain metastases of lung cancer patients with LKB1 mutation | ||||
| Ji et al (2007) | 2007 | Primary tumors: 19/80 AD, 6/42 SCC, 1/7 LCC, and 1/4 ADSCC, respectively, with single copy alterations of LKB1; 8/80 AD, 2/42 SCC with homozygous mutations | Direct sequencing, multiplex ligation-dependent probe amplification | SCC, AD, SCC, LCC, and AD: frequent loss of a single copy of LKB1, concomitant p53, and KRAS mutations found frequently |
| Onozato et al (2007) | 2007 | Cell lines with mutations in LKB1: 3/8 AD, 1/6 SCC, 1/3 LCC, and 0/5 SmCC | Direct sequencing | Lung cancer cell lines examined |
| 3/100 primary tumors with LKB1 mutations in Japanese cohort 3/33 male smokers | Direct sequencing | LKB1 mutation rare in Japanese lung cancer population. All mutations were in male smokers with moderately or poorly differentiated tumors |
ADSCC=adenosquamous carcinoma; AD=adenocarcinoma; AAH=atypical adenomatous hyperplasia; LCC=large cell carcinoma; LOH=loss of heterozygosity; NSCLC=non-small-cell lung cancer; PCR=polymerase chain reaction; SmCC=small cell carcinoma; SCC=squamous cell carcinoma.
LKB1 loss in cancer
The LKB1 locus had been previously indicated as important in lung cancer, although admittedly without the laser focus of a known genomic syndrome and target gene. Indeed, 19p13.3 has been repeatedly indicated for its high frequency of loss of heterozygosity (LOH), often above 50%, in lung cancer by chromosomal analysis (Sobottka et al, 2000). Interestingly, the frequency of LOH for many tumours, including lung cancer, continues to be significantly higher than the reported rate of point mutations. The discrepancy between the reported rate of mutation and LOH raises a number of questions, including the accuracy of the reported mutation studies, the potential for other mechanisms of gene silencing (such as, promoter methylation or other epigenetic event), or the possibility that a second gene target lies in the region. In fact, careful attention to resequencing is crucial, and techniques that detect large homozygous deletions are required to fully characterize tumour LKB1 loss (Ji et al, 2007). Yet, thorough sequencing alone does not fully account for the LOH mutation frequency discrepancy, prompting investigators to pursue other mechanisms of gene silencing. Promoter methylation accounts for a small fraction of repressed LKB1 expression cases, but this is unlikely a major contributing mechanism (Esteller et al, 2000; Trojan et al, 2000). Other mechanisms of gene silencing, including microRNA, might contribute to some of the gap but these remain under investigation. Perhaps, the most interesting possibility is that LKB1 haploinsufficiency itself could be tumorigenic, as we have recently shown in animal models (Ji et al, 2007). At this time, however, we cannot exclude the possibility that there is another gene target in the 19p13.3 region that could account for additional cases of LOH, such as BRG1 (Medina et al, 2008). In contrast to the challenges of detecting functional LKB1 gene dosage described above, two authors have reported that immunohistochemical assessment of LKB1 protein status as a straightforward proxy for both biallelic loss and promoter methylation (Ghaffar et al, 2003; Fenton et al, 2006). Taken together, information on genetic status of LKB1 garnered through examining PJS patients and screening multiple tumour types has pointed to LKB1 as an important mediator in the development of cancer.
Frequent LOH at 19p13.3 points only indiscriminately to the importance of the LKB1 locus in lung cancer, as many regions have chromosomal alterations in this disease. Similarly, the dramatic clinical histories of a minority of PJS patients include lung cancer, but not at such a high rate as to suggest the prevalence of LKB1 alteration in this tumour (Hirano et al, 2002; Estrada Trigueros et al, 2005; von Herbay et al, 2005). Yet, cancers in a few young patients with PJS, indeed, suggest a role as a potential early carcinogenic event. Ghaffar et al (2003) have bolstered this possibility by detecting frequent LKB1 loss in the progression of the premalignant lesion atypical adenomatous hyperplasia (AAH) towards frank invasive. Developmentally, LKB1 is expressed in foetal lung and ubiquitously expressed in adult bronchial mucosa, underscoring its importance in normal lung physiology and development (Luukko et al, 1999). Step by step, pieces of a puzzle assemble to document both the importance of LKB1 to the normal lung and its high frequency of aberrancy in cancer. Yet, the reader might still rightfully wonder why does a gene that primarily causes genetic disease of the gut appear to be mutated so early and often in sporadic cases of lung cancer?
LKB1 expression, regulation, and action
Like individual pieces of a jigsaw puzzle, the various facets of LKB1 need further consideration before they reveal a coherent picture. The functional domains of LKB1's 10 exons (9 coding) include a central catalytic domain, nuclear localisation signal, putative cytoplasmic retention signal, and a prenylation motif (Alessi et al, 2006). Localised to the nucleus, it is transported to the cytosol and activated by binding two associated proteins STE20-related adaptor (STRAD), a catalytically inactive kinase-like protein, and an armadillo repeat-containing protein named mouse protein 25 (MO25). The complex is 100-fold more active as a kinase than LKB1 alone. LKB1 also associates with a chaperone complex consisting of HSP90 and Cdc37, which likely aids in stabilisation of LKB1 protein and assembly of the STRAD–MO25 complex. LKB1 is phosphorylated on several sites, either activating or inhibiting, by several kinases, including p90 ribosomal S6 kinase (RSK), protein kinase A (PKA), or ataxia-telengiectasia-mutated (ATM) kinase. Despite the apparently rich regulatory aspects of LKB1, however, no mutation hot spots have emerged.
LKB1 functions as a tumour suppressor that regulates cell polarity, differentiation, and metastasis as well as responds to energy status to regulate cell metabolism (Figure 1) (see Alessi et al, 2006; and references within). One function of LKB1 directly linked to lung cancer is the phosphorylation, ubiquitination, and the degradation of polyomavirus enhancer activator 3 (PEA3). Mutant LKB1 increases the stability of PEA3, which, in turn, increases transcription of genes involved in metastasis, including COX-2, MMP-9, and MMP-14 (Upadhyay et al, 2006; Cowden Dahl et al, 2007). Interestingly, COX-2 is commonly elevated in many disease states, including PJS and lung cancer (Achiwa et al, 1999; Rossi et al, 2002; McGarrity et al, 2003). Indeed, COX-2 inhibition decreased polyps in a small study of PJS patients and a large study of patients with familial adenomatous polyposis (Udd et al, 2004; Bertagnolli et al, 2006). Finally, increased COX-2 is associated with decreased survival in patients with lung adenocarcinoma (Achiwa et al, 1999). Although LKB1 clearly regulates proteins such as PEA3, current understanding is that its primary action is through the target 5′-AMP-activated protein kinase (AMPK) (Towler and Hardie, 2007). AMPK is a master regulator of metabolism that orchestrates efficient energy production with minimal waste in times of energy stress. It is a serine/threonine kinase activated by metabolic signals and stressors, including exercise, starvation, hypoxia, and ischaemia. In short, consumption of ATP, decreased ATP production, and/or increased concentration of AMP activate AMPK to alter numerous metabolic and proliferative pathways (Figure 1). Downstream of LKB1, AMPK tightly regulates the metabolic status of cells. For example, when carbon sources are scarce, AMPK ensures ATP generation by activating catabolic pathways such as glycolysis and fatty acid oxidation, as well as upregulates mitochondrial biogenesis. In parallel, it switches off ATP-consuming anabolic pathways, including fatty acid, cholesterol, glycogen, and protein synthesis. Of relevance to this review, AMP binding to AMPK only causes a fivefold increase in activity, while the AMP-induced conformational change in AMPK primes it as a substrate for LKB1, allowing LKB1 phosphorylation to induce activation by 100-fold (Alessi et al, 2006). Therefore, AMPK action and LKB1 activity are tightly coordinated to sense energy stress and attempt to provide cells with fuel for rapid energy production, while limiting growth and energy loss when nutrients are scarce.
Figure 1.
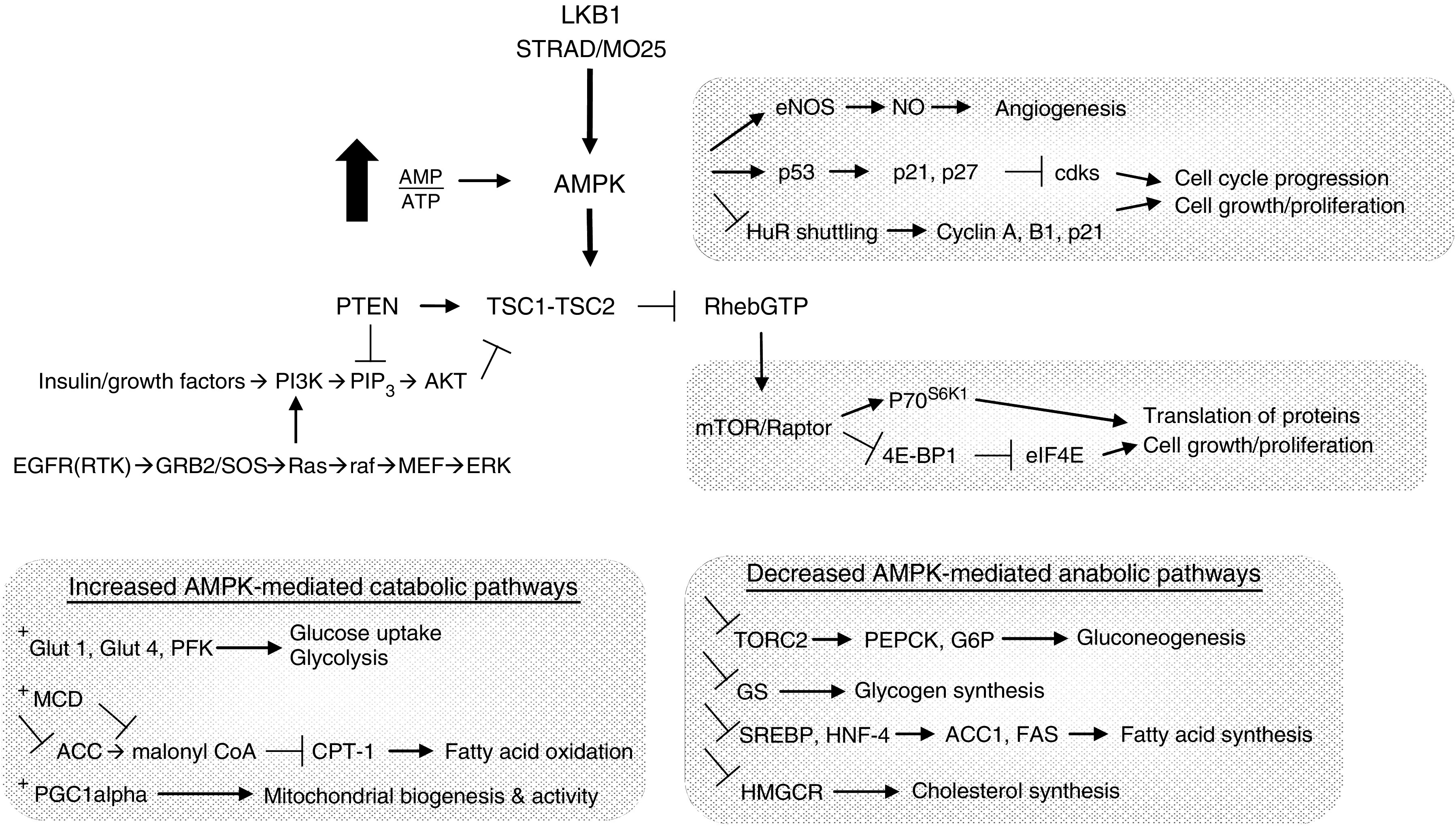
Energy sentinels in proliferation: LKB1 and AMPK. LKB1 is activated by binding STRAD and MO25. AMPK is a master regulator of metabolism, which is activated by LKB1 phosphorylation when the AMP/ATP ratio is high. AMPK drives catabolic pathways such as glucose uptake, glycolysis, mitochondrial proliferation, and fatty acid oxidation to generate ATP in times of stress. It also blunts ATP-consuming anabolic pathways including fatty acid, cholesterol, glycogen, and protein synthesis. The latter role of inhibiting protein synthesis through TSC1/2 and mTOR underlie AMPK's role in cell growth and proliferation. However, LKB1–AMPK regulation of fuel availability and the metabolic status is also central to proliferation. In fact, the LKB1 signal that cells should halt division when sufficient nutrients are not present overrides growth factor signalling through EGFR, for example. In sum, LKB1 and AMPK act together to sense energy stress to provide cells with nutrients for rapid energy production, while limiting growth. The sentinel activity of LKB1 and AMPK is lost in cancer, which allows unlimited proliferation despite metabolic cues to the contrary.
LKB1 proliferation, metastasis, and metabolism
A central target for AMPK's control of proliferation is the mammalian target of rapamycin (mTOR) kinase, which regulates numerous downstream targets, such as amino acid transporters, VEGF, p70 ribosomal protein S6 kinase 1 (S6K), and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) pathways, among others, to increase the translation of proteins and cell growth (Figure 1). In fact, AMPK acts on mTOR through phosphorylating and activating tumour suppressor tuberous sclerosis complex-2 (TSC2), an upstream negative regulator of mTOR. AMPK activation also blocks cell cycle progression from G1 to S phase through phosphorylation and accumulation of the tumour suppressor p53 and the downstream cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p27. Other cell cycle regulation occurs through the alteration of cytoplasmic/nuclear ratios of RNA-binding protein human antigen R (HuR), thereby reducing the ability to stabilise mRNAs encoding cyclins (Wang et al, 2002). Finally, hypoxia-induced AMPK activation of endothelial NOS (eNOS) drives angiogenesis. The relative importance of each mechanism discussed remains to be determined. In sum, LKB1 inactivation in cancer releases the AMPK-mediated breaks on energy waste and permits proliferation. The capacity to sense a nutrient deficit is a function of healthy cells that is lost in proliferating cells, which highlights a distinctly separate but parallel piece of the cancer puzzle.
Hamartomatous syndromes, mTOR, and lung cancer
Decoding the regulatory networks of LKB1, particularly an interaction with the AMPK–mTOR axis, crystallises a link between a genetic disease with little pulmonary implications and a gene frequently altered in lung cancer. Clearly, mTOR is a lung cancer target; yet, the jumble of alternate pathways raises doubts as to whether this is the primary mechanism (Figure 1). Quite remarkably, upstream of the mTOR pathway, however, is the invocation of three genes and two autosomal-dominant inherited syndromes with phenotypes remarkably similar to PJS, including PTEN (CD) and TSC1/TSC2 (tuberous sclerosis, TS). Like PJS, both CD and TS manifest themselves as primary hamartomatous diseases. Patients with CD demonstrate hamartomas of the skin, mucous membranes, breast, and thyroid, with 85% of patients showing PTEN germline mutation. Like PJS, an increased risk of tumours is seen, although again lung cancer is not prominent. The PTEN phosphatase controls the levels of PIP3 induced by growth factor activation of PI3 kinase, and acts to negatively regulate mTOR activity. Patients with TS do not have malignancy as their primary concern, but instead hamartomas of the skin, brain, kidney, skin, lungs, and other tissues (Crino et al, 2006). As in CD, the TS complex acts as a negative regulator of mTOR. In summary, the related phenotypes of PJS, CD, and TS are joined by shared regulation of the mTOR signalling cascade.
Consideration of these syndromes begs the question, is there a broader lung cancer malignant phenotype equivalent to that shared by germline PJS, CD, and TS? In fact, mutations of PTEN have been reported in many tumours, including 4–8% of non-small-cell lung cancers (NSCLC) (Forbes et al, 2006). PTEN protein expression is lost in more than 25% of NSCLC tumours with evidence of epigenetic silencing at work as well (Luukko et al, 1999; Marsit et al, 2005). Additionally, PTEN is co-expressed with LKB1 in foetal lung development. Similarly, both the TSC1 locus (9q34) and the TSC2 locus (16p) are frequent targets of LOH in both lung adenocarcinoma and the pre-invasive lung lesion AAH (Takamochi et al, 2001, 2004). Although the frequency with which these events occur in concert remains unknown, the conclusion must be that targets coalescing on the mTOR pathway defined in large part by dissecting the disease of germline mutations of LKB1, PTEN, and TSC1/2 appear to be frequently altered in lung cancer, suggesting a broad and important contribution of this specific set of interacting proteins to the disease.
Turning to animal models: LKB1 and KRAS
Recent years have seen progress similar to that we document for LKB1 in a number of other lung cancer genes and cancer networks, such as EGFR, KRAS, p53, and CDKN2A (including both transcripts: p16Ink4a and INK4a/ARF). As we suggest above, it is expected that the alteration of only one gene in a pathway may be sufficient for tumorigenesis. As such, an understanding of the relationships between aberrations may help clarify the minimal set of events necessary to cause cancer. To this end, we documented several interactions between LKB1 and other genes commonly altered in lung cancer in experimental animal models (Ji et al, 2007). As predicted from the natural human experiment of PJS, mice deficient in LKB1 do not get lung tumours. The alteration of other lung cancer genes in combination with LKB1, however, did produce tumours of a striking phenotype. First, we observed that homozygous loss of LKB1 in combination with KRAS resulted in an aggressive tumour phenotype with high tumour multiplicity, short survival, and frequent metastases well above those of KRAS alone. The KRAS/LKB1 double knockout was in fact the most aggressive phenotype of all tumours considered in the study, and had an additional feature that had not previously been reported in a mouse model: squamous cell carcinoma histology. Additionally, it was noted that even hemizygous loss of LKB1 in combination with KRAS resulted in a more aggressive phenotype than in KRAS mutation alone.
The clinical relevance of LKB1 alteration remains an area of active investigation, although preliminary results appear likely to confirm findings of the animal models, including the frequent co-mutation of LKB1 inactivation and activating mutations of KRAS (Table 1). As a mirror opposite to the established positive association of EGFR gene mutation with females and nonsmokers, we detect a positive association of LKB1 inactivation with both smoking and male gender. Other interesting reports, including prevalence of brain metastasis, associations with poorly differentiated tumours, the importance of the hemizygous LKB1 state, and apparent independence with p53 mutation, remain to be confirmed. Certainly, we hope for clinical associations of this type on which to base important patient management decisions. In broader terms, one has to note that many of the tumours associated with PJS, including GI and gynaecologic malignancies are also those where KRAS is a frequent target of mutation. The question remains, is the LKB1–KRAS connection part of a broad phenotype of interconnecting cancer pathways? We propose that perhaps KRAS activation and LKB1 loss permit liberation from cell cycle and energy status checkpoints, respectively, which allow for a metabolic advantage.
mTOR and KRAS in the clinic
Paramount in the LKB1 story is the focus now thrust upon very specific cancer pathways, several of which have attractive therapeutic targets. At the top of the list is the mTOR pathway where three Food and Drug Administration (FDA)-approved inhibitors are currently available, such as rapamycin, everolimus, and temsirolimus, with more in development, including the recently described deforolimus (Cohen, 2008). Inhibitors of mTOR have already been investigated in lung cancer, with clear evidence of anticancer activity both as single agents and in combination with cytotoxic chemotherapy and radiation (Milton et al, 2007; Sarkaria et al, 2007; Mita et al, 2008). Interestingly, a recent report in the New England Journal of Medicine documented a striking therapeutic effect of mTOR-targeted therapy by sirolimus to angiomyolipomas, which are lesions attributable to TSC dysfunction in patients with TS (Bissler et al, 2008). Patients with angiomyolipomas, including the pulmonary form lymphangioleiomyomatosis, almost uniformly experienced reduction in the size of their tumours with this therapy. The efficacy of mTOR-targeted therapy in this benign tumour of the lung, marked by one aspect of mTOR activation, certainly is of interest due to its parallel biology in lung cancer.
Although activity in these early studies has been documented, there is little evidence that this will be of a broader spectrum than for other classes of lung cancer therapeutics. Therefore, there is an incentive to define biomarkers of response to therapy, such as mutation status or, perhaps, more encompassing markers such as those indicating AMPK activity. There is increasing evidence that broad patterns of tumour behaviour can be captured as general phenotypes using profiling techniques such as gene expression arrays. Specifically, the squamoid gene expression subtype of lung adenocarcinoma is known to have higher rates of KRAS mutation and demonstrates gene expression correlated with LKB1 inactivation (Hayes et al, 2006). It is also interesting to note that as LKB1 mutation appears to pair with KRAS activation, specific combinations of therapy targeting parallel pathways might be appropriate. Indeed, initial work on this similar line has begun, including targeting downstream elements of RAS signalling (MEK) and mTOR simultaneously (Legrier et al, 2007).
Conclusion
Three phenotypically related genetic syndromes, including PJS, CD, and TS, are united by careful alignment of their mechanisms acting through the mTOR pathway. The lesions responsible for the syndromes, LKB1, PTEN, and TSC1/2, are identified as frequently altered in lung cancer, suggesting that they comprise elements of a common lung caner phenotype. Taken together, when the phenotype of LKB1 mutation is examined in the setting of known alterations of lung cancer, an interesting association of KRAS dependence appears, with specific clinical and treatment potential. Like all good puzzles, the pieces are starting to fall into place.
References
- Achiwa H, Yatabe Y, Hida T, Kuroishi T, Kozaki K, Nakamura S, Ogawa M, Sugiura T, Mitsudomi T, Takahashi T (1999) Prognostic significance of elevated cyclooxygenase 2 expression in primary, resected lung adenocarcinomas. Clin Cancer Res 5: 1001–1005 [PubMed] [Google Scholar]
- Alessi DR, Sakamoto K, Bayascas JR (2006) LKB1-dependent signaling pathways. Annu Rev Biochem 75: 137–163 [DOI] [PubMed] [Google Scholar]
- Avizienyte E, Loukola A, Roth S, Hemminki A, Tarkkanen M, Salovaara R, Arola J, Butzow R, Husgafvel-Pursiainen K, Kokkola A, Jarvinen H, Aaltonen LA (1999) LKB1 somatic mutations in sporadic tumors. Am J Pathol 154: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET (2006) Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355: 873–884 [DOI] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN (2008) Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358: 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero J, Medina PP, Pio R, Montuenga LM, Sanchez-Cespedes M (2004) Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene 23: 4037–4040 [DOI] [PubMed] [Google Scholar]
- Cohen EE (2008) mTOR: the mammalian target of replication. J Clin Oncol 26: 348–349 [DOI] [PubMed] [Google Scholar]
- Cowden Dahl KD, Zeineldin R, Hudson LG (2007) PEA3 is necessary for optimal epidermal growth factor receptor-stimulated matrix metalloproteinase expression and invasion of ovarian tumor cells. Mol Cancer Res 5: 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355: 1345–1356 [DOI] [PubMed] [Google Scholar]
- Esteller M, Avizienyte E, Corn PG, Lothe RA, Baylin SB, Aaltonen LA, Herman JG (2000) Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz–Jeghers syndrome. Oncogene 19: 164–168 [DOI] [PubMed] [Google Scholar]
- Estrada Trigueros G, Lopez-Encuentra A, Garcia Quero C (2005) Non-small cell bronchogenic carcinoma and Peutz–Jeghers syndrome. Arch Bronconeumol 41: 296. [DOI] [PubMed] [Google Scholar]
- Fenton H, Carlile B, Montgomery EA, Carraway H, Herman J, Sahin F, Su GH, Argani P (2006) LKB1 protein expression in human breast cancer. Appl Immunohistochem Mol Morphol 14: 146–153 [DOI] [PubMed] [Google Scholar]
- Fernandez P, Carretero J, Medina PP, Jimenez AI, Rodriguez-Perales S, Paz MF, Cigudosa JC, Esteller M, Lombardia L, Morente M, Sanchez-Verde L, Sotelo T, Sanchez-Cespedes M (2004) Distinctive gene expression of human lung adenocarcinomas carrying LKB1 mutations. Oncogene 23: 5084–5091 [DOI] [PubMed] [Google Scholar]
- Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, Stratton MR (2006) Cosmic 2005. Br J Cancer 94: 318–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffar H, Sahin F, Sanchez-Cepedes M, Su GH, Zahurak M, Sidransky D, Westra WH (2003) LKB1 protein expression in the evolution of glandular neoplasia of the lung. Clin Cancer Res 9: 2998–3003 [PubMed] [Google Scholar]
- Hayes DN, Monti S, Parmigiani G, Gilks CB, Naoki K, Bhattacharjee A, Socinski MA, Perou C, Meyerson M (2006) Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol 24: 5079–5090 [DOI] [PubMed] [Google Scholar]
- Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, Trimbath JD, Giardiello FM, Gruber SB, Offerhaus GJ, de Rooij FW, Wilson JH, Hansmann A, Moslein G, Royer-Pokora B, Vogel T, Phillips RK, Spigelman AD, Houlston RS, Mehenni H, Resta N, Park JG, Miyaki M, Guanti G, Costanza MC, Lim W, Olschwang S, Keller JJ, Westerman AM, Menko FH, Boardman LA, Scott RJ, Trimbath J, Giardiello FM, Gruber SB, Gille JJ, Offerhaus GJ, de Rooij FW, Wilson JH, Spigelman AD, Phillips RK, Houlston RS (2006) Frequency and spectrum of cancers in the Peutz–Jeghers syndrome. Cancer risks in LKB1 germline mutation carriers. Relative frequency and morphology of cancers in STK11 mutation carriers. Clin Cancer Res 12: 3209–3215 [DOI] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA (1998) A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature 391: 184–187 [DOI] [PubMed] [Google Scholar]
- Hemminki A, Tomlinson I, Markie D, Jarvinen H, Sistonen P, Bjorkqvist AM, Knuutila S, Salovaara R, Bodmer W, Shibata D, de la Chapelle A, Aaltonen LA (1997) Localization of a susceptibility locus for Peutz–Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet 15: 87–90 [DOI] [PubMed] [Google Scholar]
- Hirano S, Takiguchi Y, Igari H, Hiroshima K, Shingyoji M, Watanabe R, Moriya T, Tanabe N, Tatsumi K, Kuriyama T (2002) A case of pulmonary adenocarcinoma accompanied by superior vena caval thrombosis in a patient with Peutz–Jeghers syndrome. Jpn J Clin Oncol 32: 307–309 [DOI] [PubMed] [Google Scholar]
- Jeghers H, Mc KV, Katz KH (1949) Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med 241: 993, illust; passim [DOI] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M (1998) Peutz–Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18: 38–43 [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, Bao L, Contreras CM, Li D, Chen L, Krishnamurthy J, Koivunen J, Chirieac LR, Padera RF, Bronson RT, Lindeman NI, Christiani DC, Lin X, Shapiro GI, Janne PA, Johnson BE, Meyerson M, Kwiatkowski DJ, Castrillon DH, Bardeesy N, Sharpless NE, Wong KK (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature 448: 807–810 [DOI] [PubMed] [Google Scholar]
- Legrier ME, Yang CP, Yan HG, Lopez-Barcons L, Keller SM, Perez-Soler R, Horwitz SB, McDaid HM (2007) Targeting protein translation in human non small cell lung cancer via combined MEK and mammalian target of rapamycin suppression. Cancer Res 67: 11300–11308 [DOI] [PubMed] [Google Scholar]
- Luukko K, Ylikorkala A, Tiainen M, Makela TP (1999) Expression of LKB1 and PTEN tumor suppressor genes during mouse embryonic development. Mech Dev 83: 187–190 [DOI] [PubMed] [Google Scholar]
- Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, Kelsey KT (2005) PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol 36: 768–776 [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna JD, Yokota J (2007) Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 26: 5911–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarrity TJ, Peiffer LP, Amos CI, Frazier ML, Ward MG, Howett MK (2003) Overexpression of cyclooxygenase 2 in hamartomatous polyps of Peutz–Jeghers syndrome. Am J Gastroenterol 98: 671–678 [DOI] [PubMed] [Google Scholar]
- Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M (2008) Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 29: 617–622 [DOI] [PubMed] [Google Scholar]
- Milton DT, Riely GJ, Azzoli CG, Gomez JE, Heelan RT, Kris MG, Krug LM, Pao W, Pizzo B, Rizvi NA, Miller VA (2007) Phase 1 trial of everolimus and gefitinib in patients with advanced nonsmall-cell lung cancer. Cancer 110: 599–605 [DOI] [PubMed] [Google Scholar]
- Mita MM, Mita AC, Chu QS, Rowinsky EK, Fetterly GJ, Goldston M, Patnaik A, Mathews L, Ricart AD, Mays T, Knowles H, Rivera VM, Kreisberg J, Bedrosian CL, Tolcher AW (2008) Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol 26: 361–367 [DOI] [PubMed] [Google Scholar]
- Onozato R, Kosaka T, Achiwa H, Kuwano H, Takahashi T, Yatabe Y, Mitsudomi T (2007) LKB1 gene mutations in Japanese lung cancer patients. Cancer Sci 98: 1747–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peutz J (1921) Over een zeer merkwaardige, gecombineerde familiaire polyposis van de slijmliezen van den tractus intestinalis met die van de neuskeelholte en gepaard met eigenaardige pigmentaties van huid-en slijmvliezen. Ned Maandschr v Gen 10: 134–146 [Google Scholar]
- Rossi DJ, Ylikorkala A, Korsisaari N, Salovaara R, Luukko K, Launonen V, Henkemeyer M, Ristimaki A, Aaltonen LA, Makela TP (2002) Induction of cyclooxygenase-2 in a mouse model of Peutz–Jeghers polyposis. Proc Natl Acad Sci USA 99: 12327–12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Cespedes M (2007) A role for LKB1 gene in human cancer beyond the Peutz–Jeghers syndrome. Oncogene 26: 7825–7832 [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, Rosell R, Monzo M, Wu L, Westra WH, Yang SC, Jen J, Sidransky D (2001) Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res 61: 1309–1313 [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D (2002) Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res 62: 3659–3662 [PubMed] [Google Scholar]
- Sarkaria JN, Schwingler P, Schild SE, Grogan PT, Mladek AC, Mandrekar SJ, Tan AD, Kobayashi T, Marks RS, Kita H, Miller RC, Limper AH, Leof EB (2007) Phase I trial of sirolimus combined with radiation and cisplatin in non-small cell lung cancer. J Thorac Oncol 2: 751–757 [DOI] [PubMed] [Google Scholar]
- Sobottka SB, Haase M, Fitze G, Hahn M, Schackert HK, Schackert G (2000) Frequent loss of heterozygosity at the 19p13.3 locus without LKB1/STK11 mutations in human carcinoma metastases to the brain. J Neurooncol 49: 187–195 [DOI] [PubMed] [Google Scholar]
- Takamochi K, Ogura T, Suzuki K, Kawasaki H, Kurashima Y, Yokose T, Ochiai A, Nagai K, Nishiwaki Y, Esumi H (2001) Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol 159: 1941–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamochi K, Ogura T, Yokose T, Ochiai A, Nagai K, Nishiwaki Y, Suzuki K, Esumi H (2004) Molecular analysis of the TSC1 gene in adenocarcinoma of the lung. Lung Cancer 46: 271–281 [DOI] [PubMed] [Google Scholar]
- Towler MC, Hardie DG (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100: 328–341 [DOI] [PubMed] [Google Scholar]
- Trojan J, Brieger A, Raedle J, Esteller M, Zeuzem S (2000) 5′-CpG island methylation of the LKB1/STK11 promoter and allelic loss at chromosome 19p13.3 in sporadic colorectal cancer. Gut 47: 272–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd L, Katajisto P, Rossi DJ, Lepisto A, Lahesmaa AM, Ylikorkala A, Jarvinen HJ, Ristimaki AP, Makela TP (2004) Suppression of Peutz–Jeghers polyposis by inhibition of cyclooxygenase-2. Gastroenterology 127: 1030–1037 [DOI] [PubMed] [Google Scholar]
- Upadhyay S, Liu C, Chatterjee A, Hoque MO, Kim MS, Engles J, Westra W, Trink B, Ratovitski E, Sidransky D (2006) LKB1/STK11 suppresses cyclooxygenase-2 induction and cellular invasion through PEA3 in lung cancer. Cancer Res 66: 7870–7879 [DOI] [PubMed] [Google Scholar]
- Virmani AK, Fong KM, Kodagoda D, McIntire D, Hung J, Tonk V, Minna JD, Gazdar AF (1998) Allelotyping demonstrates common and distinct patterns of chromosomal loss in human lung cancer types. Genes Chromosomes Cancer 21: 308–319 [DOI] [PubMed] [Google Scholar]
- von Herbay A, Arens N, Friedl W, Vogt-Moykopf I, Kayser K, Muller KM, Back W (2005) Bronchioloalveolar carcinoma: a new cancer in Peutz–Jeghers syndrome. Lung Cancer 47: 283–288 [DOI] [PubMed] [Google Scholar]
- Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M (2002) AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol 22: 3425–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]