

The lycopodium family of alkaloids has garnered considerable attention over the years because of their wide-ranging biological activity and structural complexity.1 The parent member of this family, lycopodine (1), was isolated 125 years ago by Bödeker (Figure 1).2 Beneficial medicinal properties, such as antipyretic3 and anticholinesterase activity,4 have been attributed to lycopodine and other lycopodium alkaloids. To date, seven racemic total syntheses and two racemic formal syntheses of 1 have been reported.5 Herein, we report the first enantioselective total synthesis of 1. Our retrosynthetic strategy is shown in Figure 1. Key to this strategy is the diastereoselective intramolecular Michael addition of 4 and the Heathcock-inspired5d Mannich cyclization to form tricycle 2.
Figure 1.
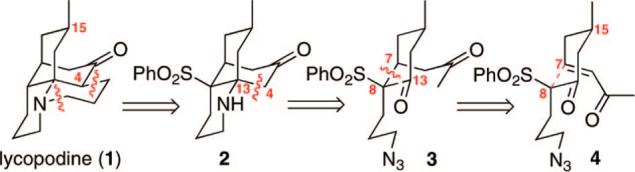
Retrosynthetic strategy for lycopodine.
The synthesis commenced with the known ester 6,6 which is readily accessible in two steps from commercially available acyl sultam 5 (Scheme 1). Next, one-pot treatment of commercially available 1,4-dibromobutane (7) with NaSO2Ph in DMF followed by NaN3 and H2O generated the sulfone 8 in reasonable yield. Double deprotonation of 8 with lithium tetramethylpiperidide (LiTMP) followed by addition of the chiral ester 6 yielded keto sulfone 9 in 74% yield. It should be noted that use of lithium diisopropylamide or n-BuLi resulted in a significant reduction in the yield of this transformation. We have previously observed the superior performance of LiTMP during our synthetic efforts toward the bisspiroketal azaspiracid-1.7 Next, we turned our attention to the cross metathesis of 9 and 3-penten-2-one (10). The presence of the azide moiety as well as an internal nucleophile (the C8 keto sulfone moiety) and Michael acceptor (the C5–C7 enone) within product 4 imparted unique challenges for the cross metathesis. We were gratified to find that use of the highly active second-generation Grubbs–Hoveyda (GH-II) catalyst8 did generate the desired product 4 in good yield (63%, 88% on the basis of recovered starting material). Also key to this metathesis was the specific choice of the enone 10.9 Replacement of 10 with methyl vinyl ketone led to a significant reduction in the efficiency of the transformation.
Scheme 1.
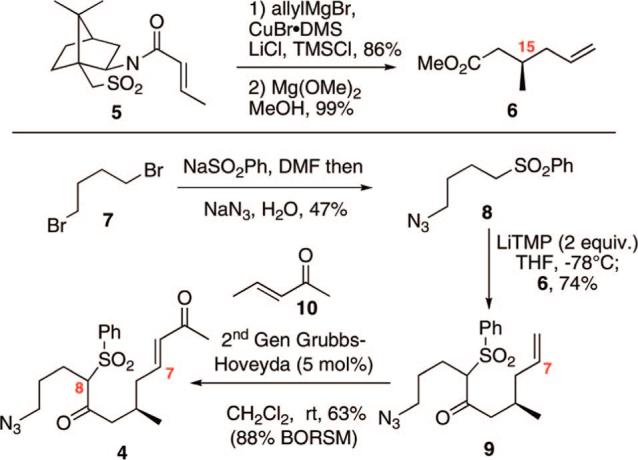
Synthesis of the Keto Sulfone Subunit
Our attention then turned to the key intramolecular Michael addition of keto sulfone 4 (Scheme 2). We had initially hypothesized that the C15 stereogenic center would disrupt the desired stereochemical outcome at the C7 and C8 positions by placing the larger phenyl sulfone and methyl ketone in pseudoequatorial positions in the transition state 11 shown. Consequently, our strategy had intended to explore a suitable organocatalyst for facilitating this reaction while overriding what we perceived to be the favored pathway. Prior to embarking on this investigation, we felt it would be prudent to confirm the inherent stereochemical preference in the system. Treatment of keto sulfone 4 with i-Pr2NH in a mixed-solvent system (4:1 i-PrOH/dichloromethane) at room temperature led to clean conversion to a single product that crystallized out of the reaction in 89% yield. Interestingly, X-ray crystallographic analysis revealed that the desired stereochemical outcome 3 had instead been observed. One possible explanation for this fortuitous diastereoselectivity may be a 1,2-steric interaction between the large substituents at C7 and C8 that forces them to adopt a pseudoaxial conformation in the transition state, a feature that would disfavor transition state 11. Conversion of the methyl ketone 3 into the Mannich cyclization precursor 14 was accomplished in one pot via Staudinger reduction followed by tert-butyldimethylsilyl (TBS) enol ether formation.
Scheme 2.
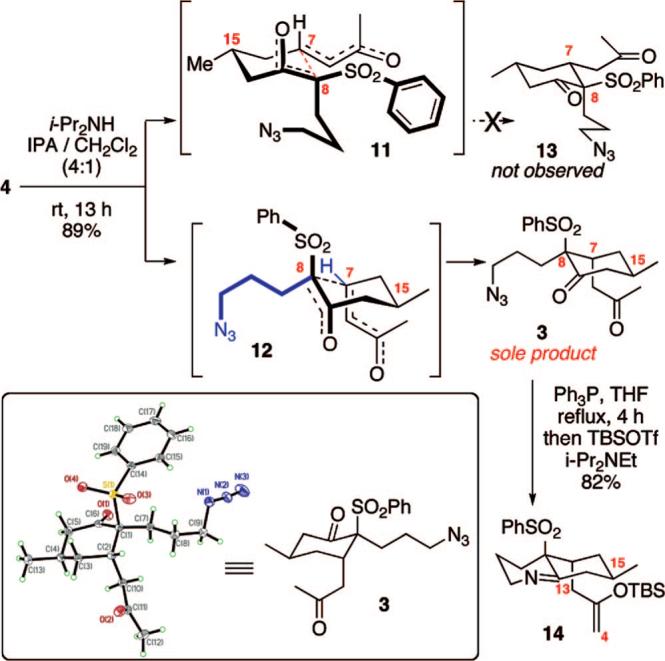
Diastereoselective Intramolecular Michael Addition and ORTEP Representation of Keto Sulfone 3
We next turned to the key Mannich cyclization step (Scheme 3). To this end, treatment of silyl enol ether 14 with Zn(OTf)2(96 °C, ClCH2CH2Cl, 16 h) generated a new compound. One might have expected product 2 to be formed; however, X-ray crystallographic analysis established that the rearranged tricycle 19 had been constructed. Product 19 was the result of a net 1,3-rearrangement of the sulfone moiety from the expected C8 position to the C14 position. While a limited number of examples of 1,3-rearrangements of allylic sulfones have appeared in the literature,10 we believe this result is the first example of an R-sulfonyl imine undergoing such a shift. This rearrangement likely occurs via initial complexation of the imine nitrogen followed by imine/enamine interconversion to form intermediate 16. Next, the 1,3-transposition of the sulfonyl moiety might occur via either heterolytic cleavage of the C–S bond to a tight ion pair11 or homolytic cleavage followed by recombination at C14 to produce the axial sulfone 17. An alternate mechanism would invoke a 2,3-sigmatropic rearrangement to the sulfinate ester followed by reorganization to the sulfone 18.12,13 Subsequent conversion to the C13,14 enamine followed by reprotonation would generate the penultimate intermediate 18. Final Mannich cyclization would yield the tricycle 19. Presumably, this net 1,3-shift of the phenyl sulfone moiety generates a more reactive intermediate for the key Mannich cyclization. Completion of a formal synthesis of lycopodine was accomplished by desulfurization using Na/Hg amalgam in good yield to give the known tricycle 20.5e Attempts to follow Schumann's outlined route for alkylation of 20 with 3-bromopropan-1-ol gave disappointing results.5e Fortunately, alkylation with the 3-iodo variant proved effective (68% yield over 2 steps). The final cyclization was performed using a slight modification of Heathcock's original conditions [t-BuOK (6 equiv), Ph2CO (18 equiv), PhH, 110 °C, sealed tube, 50 min].5d These improved conditions suppressed a retro-Michael pathway that competitively produced the tricycle 20. Finally, reduction using Stryker's reagent14 yielded 1, which matched the reported spectral data.5f,15 Importantly, comparison of the optical rotation {[α]D = −23.2° (c = 0.22, 100% EtOH)} with the literature value16 {[α]D = −24.5° (c = 1.10, 100% EtOH)} allowed us to confirm the assigned absolute configuration of 1.
Scheme 3.
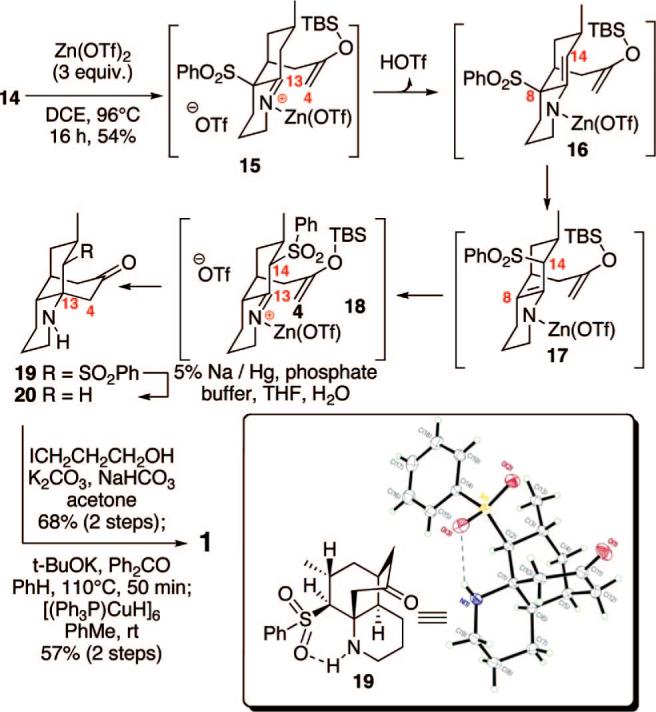
Total Synthesis of Lycopodine
In conclusion, we have completed the first enantioselective total synthesis of lycopodine. This approach should open the door to accessing other lycopodium alkaloids. Further synthetic studies will be reported in due course.
Supplementary Material
Acknowledgment
Financial support was provided by the National Institutes of Health (GM63723). The authors would like to thank Professor Clayton Heathcock (UC-Berkeley) for providing an authentic sample of 1 and Professor Max Deinzer and Dr. Jeff Morré (OSU) for mass spectral data. Finally, the authors are grateful to Professor James D. White (OSU), Professor Paul R. Blakemore (OSU), and Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for helpful discussions.
Footnotes
Supporting Information Available: Complete experimental procedures, including 1H and 13C spectra, for all of the new compounds, and crystallographic data and CIF files for 3 and 19. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Kobayashi J, Morita H. Alkaloids. 2005;61:1–57. doi: 10.1016/s1099-4831(05)61001-2. [DOI] [PubMed] [Google Scholar]; b Ma X, Gang DR. Nat. Prod. Rep. 2004;21:752–772. doi: 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]
- 2.a Bödeker K. Justus Liebigs Ann. Chem. 1881;208:363. [Google Scholar]; b Achmatowicsz O, Uzieblo W. Rocz. Chem. 1938;18:88–95. [Google Scholar]; c Ayer WA, Iverach GG. Tetrahedron Lett. 1962:87–92. doi: 10.1016/s0040-4039(00)72888-7. [DOI] [PubMed] [Google Scholar]; d Rogers D, Quick A, Hague M. Acta Crystallogr. 1974;B30:552–553. [Google Scholar]; e Hague M, Rogers D. J. Chem. Soc., Perkin Trans. 1975;2:93–98. [Google Scholar]
- 3.Nikonorow M. Acta Pol. Pharm. 1939;3:23–56. [Google Scholar]
- 4.Ortega MG, Agnese AM, Cabrera JL. Phytomedicine. 2004;11:539–543. doi: 10.1016/j.phymed.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 5.a Stork G, Kretchmer RA, Schlessinger RH. J. Am. Chem. Soc. 1968;90:1647–1648. doi: 10.1021/ja01008a042. [DOI] [PubMed] [Google Scholar]; b Ayer WA, Bowman WR, Joseph TC, Smith P. J. Am. Chem. Soc. 1968;90:1648–1650. doi: 10.1021/ja01008a043. [DOI] [PubMed] [Google Scholar]; c Kim S, Bando Y, Horii Z. Tetrahedron Lett. 1978:2293–2294. [Google Scholar]; d Heathcock CH, Kleinman EF, Binkley ES. J. Am. Chem. Soc. 1982;104:1054–1068. [Google Scholar]; e Schumann D, Mueller HJ, Naumann A. Liebigs Ann. Chem. 1982:1700–1705. [Google Scholar]; f Kraus GA, Hon YS. Heterocycles. 1987;25:377–386. [Google Scholar]; g Grieco PA, Dai Y. J. Am. Chem. Soc. 1998;120:5128–5129. [Google Scholar]; i Mori M, Hori K, Akashi M, Hori M, Sato Y, Nishida M. Angew. Chem., Int. Ed. 1998;37:637–638. doi: 10.1002/(SICI)1521-3773(19980316)37:5<636::AID-ANIE636>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; For formal syntheses of lycopodine, see: Padwa A, Brodney MA, Jr., Sheehan SM. J. Org. Chem. 1997;62:78–87. doi: 10.1021/jo960829p..
- 6.Boulet SL, Paquette LA. Synthesis. 2002:895–900. [Google Scholar]
- 7.Zhou X-T, Carter RG. Angew. Chem., Int. Ed. 2006;45:1787–1790. doi: 10.1002/anie.200503733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. [Google Scholar]
- 9.We purchased 10 (70% pure) from Aldrich Flavors and Fragrances (Aldrich cat. no. W341703, 25 g, $92). The impurity (mesityl oxide) did not affect the performance of the cross metathesis. Alternate sources of 10 were significantly higher in cost and less pure.
- 10.a Padwa A, Bullock WH, Dyszlewski AD. Tetrahedron Lett. 1987;28:3193–3196. [Google Scholar]; b Ogura K, Iihama T, Kiuchi S, Kajiki T, Koshikawa O, Takahashi K, Iida H. J. Org. Chem. 1986;51:700–705. [Google Scholar]; c Lin P, Whitman GH. J. Chem. Soc., Chem. Commun. 1983:1102–1103. [Google Scholar]; d Baechler RD, Bentley P, Deuring L, Fisk S. Tetrahedron Lett. 1982;23:2269–2272. [Google Scholar]; For Pd-catalyzed methods, see: Jagusch T, Gais H-J, Bondarev O. J. Org. Chem. 2004;69:2731–2736. doi: 10.1021/jo030340d., and references cited within.
- 11.The heterolytic cleavage would appear unlikely given the tethered nucleophile in the form of the silyl enol ether.
- 12.Hatanaka N, Ozaki O, Matsumoto M. Tetrahedron Lett. 1986;27:3169–3172. [Google Scholar]; Also see: Knight DJ, Whitman GH, Williams JG. J. Chem. Soc., Perkin Trans. 1987;1:2149–2152..
- 13.A fourth option, involving formation of an intermediate 1,1-dioxothietane followed by ring opening, also exists.
- 14.Mahoney WS, Stryker JM. J. Am. Chem. Soc. 1989;111:8818–8823. [Google Scholar]
- 15.Nakashima TT, Singer PP, Browne LM, Ayer WA. Can. J. Chem. 1975;53:1936–1942. [Google Scholar]
- 16.Douglas B, Lewis DG, Marion L. Can. J. Chem. 1953;31:272–276. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.