

Abstract
Werner syndrome is an inherited disease displaying a premature aging phenotype. The gene mutated in Werner syndrome encodes both a 3′ → 5′ DNA helicase and a 3′ → 5′ DNA exonuclease. Both WRN helicase and exonuclease preferentially utilize DNA substrates containing alternate secondary structures. By virtue of its ability to resolve such DNA structures, WRN is postulated to prevent the stalling and collapse of replication forks that encounter damaged DNA. Using electron microscopy, we visualized the binding of full-length WRN to DNA templates containing replication forks and Holliday junctions, intermediates observed during DNA replication and recombination, respectively. We show that both wild-type WRN and a helicase-defective mutant bind with exceptionally high specificity (>1000-fold) to DNA secondary structures at the replication fork and at Holliday junctions. Little or no binding is observed elsewhere on the DNA molecules. Calculations of the molecular weight of full-length WRN revealed that, in solution, WRN exists predominantly as a dimer. However, WRN bound to DNA is larger; the mass is consistent with that of a tetramer.
Helicases are motor proteins that utilize the energy derived from nucleotide hydrolysis to disrupt double-stranded or multistranded nucleic acids. Their importance can be gleaned from the following facts: (a) human cells possess up to 25 DNA helicases (1) and perhaps an equal number of RNA helicases; (b) they are essential for numerous cellular transactions, including DNA replication, repair, and recombination, as well as RNA transcription, maturation, and translation. Helicases are recognized by the presence of conserved signature sequence motifs. All helicases possess sequences homologous to the Walker A and B boxes that are characteristic of NTP binding and/or hydrolyzing enzymes (2). They do not display sequence specificity for unwinding, but exhibit preference for the type and structure of the nucleic acid substrate, and can be classified as those that preferentially unwind DNA or RNA. DNA helicases facilitate strand separation either passively, by binding and trapping ssDNA2 that arises during transient breathing of paired DNA, or by an active mode wherein they actively destabilize paired DNA in addition to binding the released ssDNA (2). Unwinding is generally directional, proceeding either 3′ → 5′ or 5′ → 3′ on the translocating strand.
The Werner syndrome protein belongs to the RecQ family of evolutionarily conserved 3′ → 5′ DNA helicases (3). The prototype of this family is Escherichia coli RecQ. Prokaryotes and lower eukaryotes generally have one RecQ member; however, higher eukaryotes possess multiple members. For example, five homologs, designated RecQ1 or RecQL, RecQ2 or BLM, RecQ3 or WRN, RecQ4 or RTS, and RecQ5a/b, have been identified in human cells. All RecQ members share a conserved helicase core and one or two additional C-terminal domains, the RQC (RecQ C-terminal) and HRDC (helicase and RNaseD C-terminal) domains. These domains bind, respectively, proteins and DNA. Eukaryotic RecQ helicases have, in addition, further N- and C-terminal extensions that are involved in protein-protein interactions and postulated to lend unique functional characteristics to each helicase (see Refs. 4, 5 and references therein for a summary of RecQ helicases).
RecQ DNA helicases are noteworthy for several reasons. 1) In addition to unwinding duplex DNA, they unwind DNA containing noncanonical structures such as forks, bubbles, Holliday junctions/D-loops, and tetraplexes (6, 7). In fact, in many instances, they prefer these substrates to standard duplex DNA. 2) Mutations in three RecQ helicases, BLM, WRN, and RTS, are manifested by the rare genomic instability disorders, Bloom syndrome, Werner syndrome, and Rothmund-Thomson syndrome/RAPADILINO/Baller-Gerold syndrome, respectively (4). Werner syndrome, for example, is an autosomal recessive disorder characterized by features of premature aging and a high incidence of uncommon cancers (3). The fact that these disorders are rare emphasizes the importance of these helicases in cellular metabolism. The emerging hypothesis is that RecQ helicases, by virtue of their ability to resolve alternate DNA structures, prevent the stalling and collapse of replication forks that encounter damaged DNA and thus help in maintaining genome integrity (8).
WRN is a unique member of the RecQ helicase family. In addition to exhibiting unwinding activity, WRN also encodes a 3′ → 5′ DNA exonuclease (9). WRN exonuclease excises 3′-terminal single mismatches but does not degrade ssDNA (10), and similar to the helicase, WRN exonuclease hydrolyzes noncanonical DNA substrates (11). The overlapping substrate preference suggests that the helicase and exonuclease may function coordinately; in fact, this has been reported to occur with a model DNA replication fork structure in vitro (12). However, the opposing polarity of the helicase and exonuclease activities raises questions on how this might occur mechanistically.
In this study, we present initial studies on the visualization of WRN binding to model DNA substrates encountered during DNA replication and recombination by electron microscopy (EM). We show that WRN binds at the junction of a replication fork and at Holliday junction structures. Moreover, binding to junction DNA is highly specific because little or no WRN binding is visualized at other sites along these substrates. Furthermore, using apoferritin as a size reference, EM analyses reveal that full-length WRN is predominantly a dimer in solution. However, WRN assembles into a larger complex composed of four monomers upon binding to DNA, consistent with the quaternary structure of WRN being a tetramer.
EXPERIMENTAL PROCEDURES
Wild-type and K577M WRN Purification—WRN was purified as described by successive steps of DEAE-cellulose, phosphocellulose, and Ni2+-affinity chromatography (9). The purified protein exhibits both 3′ → 5′ DNA helicase and exonuclease activities and was visualized as a single polypeptide with a molecular mass of ∼165 kDa on SDS-polyacrylamide gels. Purified WRN was stored at –80 °C in buffer containing 20 mm Tris-HCl (pH 7.9), 0.5 m NaCl, 1 mm EDTA, 0.05% Igepal CA-630, 25% glycerol, 250 mm imidazole, 100 μg/ml bovine serum albumin, and 10 μg/ml protease inhibitors (leupeptin, aprotinin, pepstatin, and phenylmethylsulfonyl fluoride), or the same buffer lacking bovine serum albumin.
DNA Templates—Replication forks and Holliday junctions were synthesized as described previously (13, 14). Briefly, the Holliday junction was synthesized by annealing four oligonucleotides together resulting in a small four-way Holliday junction with AGCC-3′ overhangs. These junctions were transformed into larger Holliday junction templates suitable for visualization with EM by ligation of four 575-bp double strand DNA arms onto the junction. Templates containing all four arms, referred to as Holliday junction, were gel-purified prior to use in binding reactions. Replication fork templates were prepared from a 3.4-kb circular DNA template containing a G-less cassette with a nick site at one end. The construct was nicked with N.BbvCIA followed by strand displacement with Klenow (exo-) in the absence of dCTP. This resulted in a circular template with a 400-bp single strand arm. A primer was annealed to the 3′ end of the displaced single strand arm, and the complementary strand was extended by Klenow fragment of DNA polymerase, resulting in a 3.4-kb circular template with a 400-bp double strand arm.
Protein-DNA Binding Reactions and Preparation of Samples for EM—Wild-type or K577M WRN (0.8 μg/ml) was incubated with DNA (3 μg/ml) on ice in 30 μl of binding buffer containing 40 mm Tris-HCl (pH 7.6), 4 mm MgCl2, 5 mm dithiothreitol, and 1 mm ATPγS for 20 min. Protein-DNA complexes were fixed with 0.6% glutaraldehyde for 5 min at room temperature; excess glutaraldehyde and binding buffer components were removed by chromatography through a 2-ml column of 2% agarose beads (Agarose Bead Technology, Colna, Portugal) equilibrated with 10 mm Tris-HCl (pH 7.6) and 0.1 mm EDTA.
Samples of WRN or apoferritin (used as a size marker) were prepared for EM by dilution to 10 μg/ml in 20 mm HEPES (pH 7.8) buffer; 1 μl of this dilution was mixed with 1 μl of 1.2% glutaraldehyde. Following 10 min of incubation at 0 °C, the sample was further diluted by addition of 8 μl of water. Preparation for EM followed promptly.
Electron Microscopy—Cross-linked free or DNA-bound protein samples were individually mixed with a buffer containing 2.5 mm spermidine and incubated on glow-charged carbon foil grids for 3 min (15). For each shadowcasting, an apoferritin standard was prepared side-by-side. Samples were washed with a series of water/ethanol washes, air-dried, and rotary shadowcast with tungsten at 1 × 10–6 torr. Samples were analyzed using an FEI Tecnai 12 transmission electron microscope (FEI Inc., Hillsboro, OR) at 40 kV, and images were captured on a Gatan Ultrascan 4000 slow-scan CCD camera and supporting software (Gatan Inc., Pleasanton, CA). Size analysis was performed with ImageQuant 5.1 software (GE Healthcare). Image size and contrast were adjusted using Adobe Photoshop (San Jose, CA). Further image manipulations were performed using Corel Photo Paint version 12 (Fremont, CA).
Mass Analysis by Electron Microscopy—Using EM, fields of free or DNA-bound proteins were captured on digital micrographs. The projected protein surface areas were measured using ImageQuant 5.1 software. The numbers of particles traced for size analysis of each sample are given in Table 1. All measured areas were normalized to the same magnification and resolution by using Microsoft Excel 2007 (Redmond, CA).
TABLE 1.
Number of particles traced for size analysis
The following abbreviations are used: WT, wild-type; RF, replication fork DNA; HJ, Holliday junction DNA.
| Sample | No. of particles analyzed |
|---|---|
| Free WRN | |
| WT-WRN | 200 |
| K577M-WRN | 123 |
|
Apoferritin |
139 |
| DNA-bound WRN | |
| WT-WRN: RF | 20 |
| K577M-WRN: RF | 43 |
| WT-WRN: HJ | 28 |
| K577M-WRN: HJ | 16 |
To determine molecular weights of free and DNA-bound WRN, projected areas measured by ImageQuant were transferred into GraphPad Prism version 5. In addition to calculating the means and medians of these areas, a histogram of each data set was built and fitted with a Gaussian distribution equation to determine the peak values (see supplemental Fig. 1). We further calculated the means ± S.E. for the projected areas and determined the 95% confidence intervals for each Gaussian peak value. Finally, using apoferritin as the standard, and Equation 1,
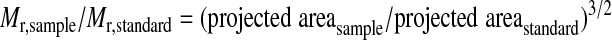 |
(Eq. 1) |
the projected areas of the samples were converted to molecular masses in kDa (16). The calculated molecular masses were similar when the peak projected area from the Gaussian curve or the median and mean projected areas were used. Therefore, for simplicity, we only report the molecular weights calculated from the projected area from the peak of the Gaussian curve (Table 2). Yet, although determining the actual oligomeric state of free and bound WRN and K577M WRN, molecular weights from means and medians were also taken into consideration.
TABLE 2.
Oligomeric states of free and DNA-bound WT-WRN
The following abbreviations are used: RF, replication fork DNA; HJ, Holliday junction DNA; WT, wild-type.
| Sample | Projected area | Calculated Mr | 95% confidence intervals for Mr | Estimated oligomeric State |
|---|---|---|---|---|
| Pixels | ||||
| WT-WRN (free) | 2392 | 301 | 294-307 | Dimer |
| WT-WRN bound to RF | 4103 | 577 | 515-641 | Tetramer |
| WT-WRN bound to HJ | 4097 | 576 | 561-590 | Tetramer |
RESULTS
Visualization of WRN Binding to DNA Containing Alternate Secondary Structures—The binding and localization of WRN on DNAs containing replication forks and Holliday junctions were visualized by electron microscopy. WRN was incubated with DNA, fixed, and adsorbed onto carbon-supporting grids, and then shadowcast with tungsten (Fig. 1A). We observed that WRN bound specifically to the 4-way junction in the Holliday junction DNAs. Scoring 359 DNA molecules at random (three independent experiments) revealed that 35 ± 12% of the DNA molecules contained WRN protein localized at the 4-way junction. The remaining DNA molecules either contained WRN bound at sites other than the 4-way junction, for example, along the DNA arms or at the DNA ends (4 ± 4%), or lacked protein (61 ± 12%) (Fig. 3A). Thus, of the protein-bound fraction, 89 ± 5% of the DNA molecules contained WRN at the 4-way junction. These data indicate a remarkably high preference of WRN for binding alternate secondary structures relative to the much longer regions of duplex B-form DNA.
FIGURE 1.

Binding of WRN to Holliday junction DNA. Wild-type (A) or K577M (B) WRN was incubated with Holliday junction templates, mounted onto carbon-coated copper grids, and rotary shadowcast with tungsten for visualization by EM. Images are shown in reverse contrast. Bar is equivalent to 50 nm.
FIGURE 3.

Quantitative analysis of WRN binding to Holliday junctions and replication forks. Large numbers of DNA molecules were surveyed to determine protein-free and protein-bound fractions. The protein-bound fractions were scored for specific and nonspecific binding (defined respectively, as junction binding or binding elsewhere on the DNA). A and C, percent Holliday junction molecules bound by wild-type (n = 359) and K577M-WRN (n = 527), respectively. B and D, percent replication forks bound by wild-type (n = 230) and K577M-WRN (n = 237), respectively. Error bars represent ± S.D.
As observed with the Holliday junction DNA template, the binding of WRN to large replication fork templates also showed a high specificity for the fork junction. Of 230 protein-DNA complexes scored in two independent experiments, 45 ± 9% of the DNA templates showed WRN bound at the replication fork junction (Fig. 2A and Fig. 3B). The remaining DNA molecules either contained WRN bound to the DNA arms or along the circular portion of the DNA template (9 ± 3%) or had no protein bound (46 ± 12%) (Fig. 3B). This corresponds to 83 ± 19% of the protein-DNA complexes containing WRN specifically at the fork junction. Given the large number of potential DNA-binding sites for WRN on both of these DNA templates, the binding specificity for replication fork and Holliday junctions is highly significant.
FIGURE 2.

Binding of WRN to replication fork DNA. Wild-type (A) or K577M (B) WRN was incubated with replication fork DNA templates and processed for EM as described in the legend to Fig. 1. Images are shown in reverse contrast. Bar is equivalent to 100 nm.
Binding of Helicase-defective, K577M WRN to Holliday Junctions and Replication Forks—K577M WRN is a single amino acid variant that lacks helicase activity; this substitution, however, does not abolish its exonuclease activity or alter its ability to bind DNA (17). Analysis of binding and localization of the mutant protein to the DNA templates by EM confirmed this observation. K577M WRN, like wild-type WRN, was also observed to bind as a large protein complex with an exceptionally high preference for the 4-way junction (Fig. 1B); 93 ± 13% of protein-DNA complexes contained K577M WRN at the 4-way junction. Overall 49 ± 10% of DNA contained a WRN complex at the 4-way junction (n = 257, two independent experiments) (Fig. 3C). The remaining DNA molecules were free of protein (47 ± 11%) or contained WRN bound at other sites on the DNA template (4 ± 2%) (Fig. 3C). Similar results were obtained with the replication fork template, with 68 ± 19% of protein-bound DNA templates containing K577M WRN at the fork junction (Figs. 2 and 3). Overall, 54 ± 5% of the templates showed a large WRN complex at the fork junction; 8 ± 2% showed protein bound elsewhere on the DNA template, and 38 ± 7% of the DNA was protein-free (n = 237; two independent experiments) (Fig. 3).
Mass Analysis of WRN in Solution and Bound to DNA—The ability to clearly visualize WRN by EM allowed us to determine the mass and oligomeric state of WRN, either free in solution or bound to replication fork and Holliday junction DNA templates. The fact that EM requires nanogram amounts of protein circumvents the inability to produce sufficient quantities of full-length WRN protein required for more traditional methods of mass analysis, i.e. ultracentrifugation.
As in most molecular mass determination methods, mass analysis by EM assumes that WRN is a globular protein. In this study, we utilized apoferritin, a large, stable globular protein complex with a mass of 443 kDa (Fig. 4K), because WRN is visually similar in size and shape to apoferritin (Fig. 4), i.e. globular without any central “holes” indicative of a donut shape protein. We can use the projected area of apoferritin to predict the mass of WRN free in solution and bound to DNA. This approach has been utilized previously to determine the oligomeric states of other DNA-binding proteins by this laboratory (16, 18–20). Apoferritin standards were cross-linked in parallel and mounted with WRN samples on separate EM grids followed by side-by-side shadowcasting. This controls for differences in metal shadowcasting and fixation of samples. Following EM imaging, digital image analysis was carried out to measure projected areas of apoferritin and WRN particles (Fig. 5 and supplemental Fig. 1). Using the molecular size of apoferritin as a reference, projected areas of WRN were converted to molecular weights; the calculated molecular sizes were used to estimate the oligomeric form of WRN (Table 2 and “Experimental Procedures”).
FIGURE 4.

Size analysis of free and DNA-bound WRN proteins. Wild-type (top panel) and K577M (middle panel) WRN were bound to Holliday junctions (A, B, E, and F) and replication forks (C, D, G, and H) and were mounted side-by-side with apoferritin (K) on separate grids for EM analysis along with unbound wild-type (I) and K577M-WRN (J). The projected areas of wild-type and K577M-WRN were measured from EM images and used to estimate the oligomeric forms of free and DNA-bound WRN as described under “Experimental Procedures.”
FIGURE 5.

Distribution of WT-WRN and K577M-WRN areas measured by EM. Projected areas of free and DNA-bound wild-type and K577M-WRN and apoferritin were calculated from EM images as described under “Experimental Procedures.” RF and HJ refer to replication fork DNA and Holliday junction DNA, respectively.
The projected area of WRN in solution, 2392 pixels, translates to a calculated molecular mass of 301 kDa. Assuming that WRN, like apoferritin, is a globular protein, the molecular mass is ∼2-fold greater than that of a WRN monomer, suggesting that free WRN is most likely a dimer (see Table 2 for confidence intervals). On the other hand, the projected area of WRN bound to either Holliday junction or replication fork DNA is 4097 and 4103 pixels, respectively. Therefore, the calculated mass of WRN bound to Holliday junction DNA is 576 kDa and the replication fork DNA is 577 kDa, which are four times greater than that of monomeric WRN. It should be noted that these latter estimates include a small amount of mass associated with the DNA bound within the WRN particles. This can be expected to add some, but not a significant, amount of additional mass to these estimates. These values are consistent with the quaternary structure of DNA-bound WRN being a tetramer (Table 2). Our analysis of the oligomerization state of K577M WRN, however, did not reveal distinct multimeric forms like wild-type WRN. Free K577M WRN appears to be in equilibrium between dimeric and trimeric forms, and DNA-bound K577M may be a trimer-tetramer equilibrium (Fig. 5). Therefore, a single Lys → Met amino acid substitution could alter the oligomerization state, or could change the shape of WRN, resulting in different calculated masses than wild-type WRN. Importantly, there is a clear increase in the multimeric state of DNA-bound WRN and K577M WRN relative to its unbound form.
DISCUSSION
We report our findings on the localization of WRN on model DNA substrates, including replication forks and Holliday junctions, by electron microscopy. Both substrates contain single strand-double strand DNA junctions that are resolved by WRN helicase and exonuclease (7, 11). We show that WRN binds at the junction of a replication fork and at the intersection of the four duplex DNA arms in the Holliday junction substrate. The binding shows exceptionally high specificity for structured DNA as little or no WRN localizes to other regions on these DNA substrates. Of note, the Holliday junction DNA substrate comprises four 575-bp DNA duplexes, and the replication fork is created within a 3.4-kb circular, duplex DNA. There are large numbers of potential sites available for DNA binding by WRN, yet >80% of protein-bound DNA templates have WRN bound selectively at junction structures. This translates to a >1000-fold preference of WRN for binding alternate secondary structures. A helicase-deficient variant of WRN, K577M, also exhibits similar preferential binding to junction DNA, indicating that DNA substrate binding preference is independent of unwinding activity.
Binding of WRN and other RecQ helicases to alternate DNA structures has been demonstrated by electrophoretic mobility shift assays (11). Competitive inhibition studies using electrophoretic mobility shift assays have yielded Kd values in the nanomolar range for binding to structured DNA (21). However, visualization of binding by EM has thus far been lacking. The EM images in this study provide a first look at DNA binding by a full-length RecQ helicase. They reveal that WRN recognizes and binds predominantly to structured components of DNA substrates, rather than being localized at duplex regions or at free ends.
The directionality of WRN helicase and WRN exonuclease on duplex B-form DNA necessitates the requirement for a 3′-tail and a 3′-recessed end, respectively. Neither unwinding nor exonucleolytic degradation can occur on blunt-ended duplex DNA. However, the presence of single-strandedness in the form of nicks, gaps, or alternate secondary structures (such as those present in the substrates analyzed) within blunt-ended double strand DNA promotes both helicase and exonuclease activities (7, 11). How then, for example, does WRN bound at a 4-way junction exonucleolytically digest DNA at 3′ ends? It is conceivable that the DNA assumes a folded conformation rendering the ends accessible to junction-bound WRN. Although we did not observe looping of DNA ends into the junctions, this could occur in vivo through the interaction of WRN with one or more proteins and/or following ATP hydrolysis. Alternatively, if the junction is closely apposed to the ends, one of the DNA-bound subunits of WRN (see below) could directly contact the 3′ terminus and catalyze its degradation. Likewise, unwinding could initiate internally and proceed in a manner distinct from that believed to occur on canonical B-form duplex DNA. The EM analyses provide a platform for investigating alternate possibilities for the mode of action of WRN helicase and exonuclease.
Previous attempts at determining the oligomeric state of full-length WRN by glycerol gradient centrifugation were equivocal because of low resolution of the procedure. The ability to visualize single molecules of WRN using amounts that are several orders of magnitude lower than required for traditional methods (e.g. size exclusion chromatography or analytical ultracentrifugation) prompted us to calculate molecular sizes using EM. In these analyses, it was apparent that the size of WRN, both in solution and bound to DNA, was larger than that expected of a globular monomeric protein. Using apoferritin as a standard, size analysis revealed that four monomers of WRN are bound to DNA, consistent with the quaternary structure being a tetramer in the DNA-bound state. Considering that wild-type WRN appears dimeric in solution, this suggests that either a change in the oligomeric state is required for, or acquired upon, DNA binding or that two dimers are needed to bind replication fork and Holliday junction DNA templates. Regardless, the oligomeric nature of WRN suggests that coordinate action of WRN helicase and exonuclease can occur via active sites residing on different monomers.
Quaternary structures of RecQ DNA helicases have been reported using different approaches, including size exclusion chromatography, pre-steady state enzyme kinetics, and electron microscopy. However, the reports are conflicting. For example, E. coli RecQ was reported to be monomeric by stopped flow kinetics and biophysical techniques (22, 23). However, the observation that RecQ helicase activity is cooperative with ATP concentration, yielding a Hill coefficient of 3.3 ± 0.3, led Harmon and Kowalczykowski (24) to postulate that the active form of RecQ must have at least three interacting ATP-binding subunits. Likewise, the RecQ helicase core of BLM was reported to be an active monomer (25), whereas an N-terminal fragment was shown to form hexamers and dodecamers (26), and the full-length protein in solution was visualized as tetramers and hexamers by electron microscopy (27). Consistent with our data that full-length WRN is multimeric, Huang et al. (28) and Xue et al. (29) used size exclusion chromatography to demonstrate that, in solution, an N-terminal fragment and full-length WRN, respectively, are trimeric. Furthermore, using atomic force microscopy, Xue et al. (29) reported that the N-terminal exonuclease domain of WRN exists in a trimer-hexamer equilibrium that shifts to hexamers upon the addition of DNA. In contrast, pre-steady state kinetic determinations have suggested that the functional unit of WRN in unwinding DNA is a monomer (30).
Some of these discrepancies can be explained by the use of partial fragments of WRN instead of the full-length protein that we employed, by differences in the approaches used by different investigators, and perhaps, by nucleotide hydrolysis or its lack thereof, as in our EM analyses. Precedence for nucleotides regulating the quaternary structure of RecQ helicases comes from a recent report that ATP destabilizes higher order multimeric forms of human RecQL (31). This raises an interesting possibility that the oligomeric form assumed by WRN could dictate whether it functions predominantly as a helicase or an exonuclease. WRN interacts with a number of proteins that participate in DNA replication and recombination (5). In vivo, these interactions could also influence the equilibrium between the different oligomeric states of WRN and potentially modulate the helicase and exonuclease activities of WRN.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants F32 GM077900 (to S. A. C.), GM31819 (to J. D. G.), and ES013773 (to J. D. G.), a Glenn foundation award (to J. D. G.), and Genome Network Project Grant PO1 CA077852 (to L. A. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are: ss, single-stranded; ATPγS, adenosine 5′-O-(thiotriphosphate).
References
- 1.Tuteja, N., and Tuteja, R. (2004) Eur. J. Biochem. 2711835 –1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singleton, M. R., Dillingham, M. S., and Wigley, D. B. (2007) Annu. Rev. Biochem. 7623 –50 [DOI] [PubMed] [Google Scholar]
- 3.Ozgenc, A., and Loeb, L. A. (2005) Mutat. Res. 577237 –251 [DOI] [PubMed] [Google Scholar]
- 4.Hanada, K., and Hickson, I. D. (2007) Cell. Mol. Life Sci. 642306 –2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamath-Loeb, A. S., Fry, M., and Loeb, L. A. (2006) DNA Helicases and Human Disease, pp.435 –460, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 6.Fry, M., and Loeb, L. A. (1999) J. Biol. Chem. 27412797 –12802 [DOI] [PubMed] [Google Scholar]
- 7.Mohaghegh, P., Karow, J. K., Brosh, R. M., Jr., Bohr, V. A., and Hickson, I. D. (2001) Nucleic Acids Res. 292843 –2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu, L. (2007) DNA Repair 6936 –944 [DOI] [PubMed] [Google Scholar]
- 9.Shen, J. C., Gray, M. D., Oshima, J., Kamath-Loeb, A. S., Fry, M., and Loeb, L. A. (1998) J. Biol. Chem. 27334139 –34144 [DOI] [PubMed] [Google Scholar]
- 10.Kamath-Loeb, A. S., Shen, J. C., Loeb, L. A., and Fry, M. (1998) J. Biol. Chem. 27334145 –34150 [DOI] [PubMed] [Google Scholar]
- 11.Shen, J. C., and Loeb, L. A. (2000) Nucleic Acids Res. 283260 –3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Opresko, P. L., Laine, J. P., Brosh, R. M., Jr., Seidman, M. M., and Bohr, V. A. (2001) J. Biol. Chem. 27644677 –44687 [DOI] [PubMed] [Google Scholar]
- 13.Lee, S., Elenbaas, B., Levine, A., and Griffith, J. (1995) Cell 811013 –1020 [DOI] [PubMed] [Google Scholar]
- 14.Subramanian, D., and Griffith, J. D. (2005) J. Biol. Chem. 28042568 –42572 [DOI] [PubMed] [Google Scholar]
- 15.Griffith, J. D., and Christiansen, G. (1978) Annu. Rev. Biophys. Bioeng. 719 –35 [DOI] [PubMed] [Google Scholar]
- 16.Griffith, J. D., Makhov, A., Zawel, L., and Reinberg, D. (1995) J. Mol. Biol. 246576 –584 [DOI] [PubMed] [Google Scholar]
- 17.Gray, M. D., Shen, J. C., Kamath-Loeb, A. S., Blank, A., Sopher, B. L., Martin, G. M., Oshima, J., and Loeb, L. A. (1997) Nat. Genet. 17100 –103 [DOI] [PubMed] [Google Scholar]
- 18.Allen, D. J., Makhov, A., Grilley, M., Taylor, J., Thresher, R., Modrich, P., and Griffith, J. D. (1997) EMBO J. 164467 –4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianchi, A., Smith, S., Chong, L., Elias, P., and de Lange, T. (1997) EMBO J. 161785 –1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fouche, N., Moon, I. K., Keppler, B. R., Griffith, J. D., and Jarstfer, M. B. (2006) Biochemistry 459624 –9631 [DOI] [PubMed] [Google Scholar]
- 21.Huber, M. D., Lee, D. C., and Maizels, N. (2002) Nucleic Acids Res. 303954 –3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu, H. Q., Deprez, E., Zhang, A. H., Tauc, P., Ladjimi, M. M., Brochon, J. C., Auclair, C., and Xi, X. G. (2003) J. Biol. Chem. 27834925 –34933 [DOI] [PubMed] [Google Scholar]
- 23.Zhang, X. D., Dou, S. X., Xie, P., Hu, J. S., Wang, P. Y., and Xi, X. G. (2006) J. Biol. Chem. 28112655 –12663 [DOI] [PubMed] [Google Scholar]
- 24.Harmon, F. G., and Kowalczykowski, S. C. (2001) J. Biol. Chem. 276232 –243 [DOI] [PubMed] [Google Scholar]
- 25.Janscak, P., Garcia, P. L., Hamburger, F., Makuta, Y., Shiraishi, K., Imai, Y., Ikeda, H., and Bickle, T. A. (2003) J. Mol. Biol. 33029 –42 [DOI] [PubMed] [Google Scholar]
- 26.Beresten, S. F., Stan, R., van Brabant, A. J., Ye, T., Naureckiene, S., and Ellis, N. A. (1999) Protein Expression Purif. 17239 –248 [DOI] [PubMed] [Google Scholar]
- 27.Karow, J. K., Newman, R. H., Freemont, P. S., and Hickson, I. D. (1999) Curr. Biol. 9597 –600 [DOI] [PubMed] [Google Scholar]
- 28.Huang, S., Beresten, S., Li, B., Oshima, J., Ellis, N. A., and Campisi, J. (2000) Nucleic Acids Res. 282396 –2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue, Y., Ratcliff, G. C., Wang, H., Davis-Searles, P. R., Gray, M. D., Erie, D. A., and Redinbo, M. R. (2002) Biochemistry 412901 –2912 [DOI] [PubMed] [Google Scholar]
- 30.Choudhary, S., Sommers, J. A., and Brosh, R. M., Jr. (2004) J. Biol. Chem. 27934603 –34613 [DOI] [PubMed] [Google Scholar]
- 31.Muzzolini, L., Beuron, F., Patwardhan, A., Popuri, V., Cui, S., Niccolini, B., Rappas, M., Freemont, P. S., and Vindigni, A. (2007) PLoS Biol. 5 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.