

Abstract
Recent computer simulations of the cysteine nucleophilic attack on propanal in human mitochondrial Aldehyde Dehydrogenase (ALDH2) yielded an unexpected result; the chemically reasonable formation of a dead-end cysteine-cofactor adduct when NAD+ was in the “hydride transfer” position. More recently, this adduct found independent crystallographic support in work on formyltetrahydrofolate dehydrogenase, work which further found evidence of the same adduct on re-examination of deposited electron densities of ALDH2. Although the experimental data showed this adduct was reversible, several mechanistic questions arise from the fact that it forms at all. Here, we present results from further Quantum Mechanical/Molecular Mechanical (QM/MM) simulations toward understanding the mechanistic implications of adduct formation. These simulations revealed that formation of the oxyanion thiohemiacetal intermediate only when the nicotinamide ring of NAD+ is oriented away from the active site, contrary to prior arguments. In contrast, and in seeming paradox, when NAD is oriented to receive the hydride, disassociation of the oxyanion intermediate to form the dead-end adduct is more thermodynamically-favored than maintaining the oxyanion intermediate necessary for catalysis to proceed. However, this disassociation to the adduct could be avoided through proton transfer from the enzyme to the intermediate. Our results continue to indicate that the unlikely source of this proton is the Cys302 main chain amide.
The overall mechanism of aldehyde dehydrogenase (E.C. 1.2.1.3) shown in Figure 1 involves 1) nucleophilic attack on the aldehyde to form a thiohemiacetal intermediate from which 2) the hydride is transferred to NAD+, to yield NADH and the thioester which 3) hydrolyses to yield the product carboxylic acid followed by 4) release of the cofactor. Specific atomic level details about each of these steps have been obtained from x-ray crystallography and NMR. The side chain of the conserved Asn169 (ALDH2 numbering) and the main chain amide of Cys302 are positioned to form hydrogen bonds to a negatively charged thiohemiacetal intermediate structure. This configuration is often called the “oxyanion hole”.
Figure 1.
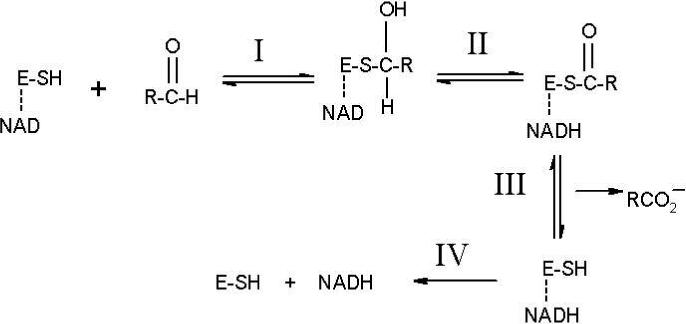
Traditional scheme for the catalytic cycle of ALDH. E-SH denotes the enzyme with Cys302 as the nucleophile and the line to NAD indicates that it is non-covalently bound prior to reacting the substrate. Reaction I: Nucleophilic activation and attack on the substrate by Cys302 to give a thiohemiacetal intermediate. Reaction II: Hydride transfer from the intermediate to NAD to form NADH and a thioester. Reaction III: Hydrolysis of the thioester with release of the carboxylic acid. Reaction IV: Release of NADH.
In the human mitochondrial ALDH2 structure (1), the Asn169 Nδ-to-crotonaldehyde carbonyl oxygen is 4.1 Å while the main chain amide nitrogen of Cys302 is 3.8 Å away. Three sequentially adjacent cysteines are located in the active site; the central member, Cys302, represents the reactive nucleophile (2). (The flanking cysteines are not highly conserved). Cys302 is positioned to react stereospecifically with the aldehyde to form a pro-R tetrahedral intermediate. Crystal structures and NMR experiments show multiple conformations of the NAD nicotinamide and phosphate groups that link to the adenine (1,3) shown in Figure 2. One NAD conformation shows a close approach of the nicotinamide to Cys302 and would be positioned to receive a hydride from the thiohemiacetal intermediate. Hydride transfer is pro-R stereospecific (3,4). The other NAD+ conformation shows the nicotinamide more removed from the active site and is likely the position of the coenzyme during thioester hydrolysis. In this conformation, Glu268 acts as a general base to abstract a proton from the water molecule to form the reactive hydroxide anion that hydrolyzes the thioester intermediate (5).
Figure 2.
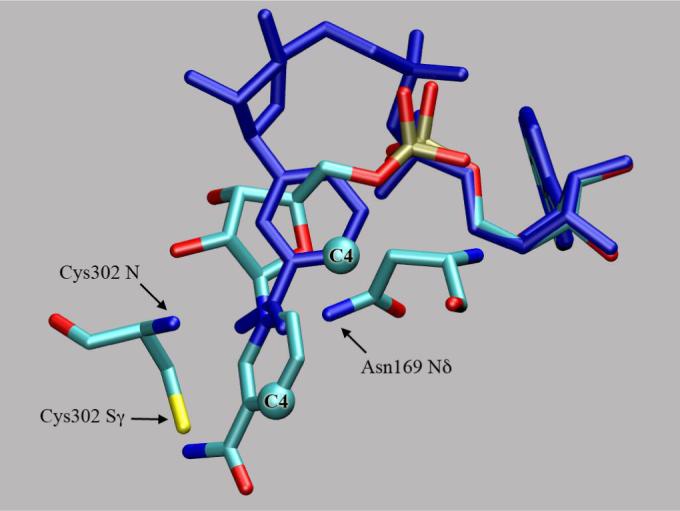
Structural superposition of NAD in hydride transfer (HTX) and hydrolysis (HYOH, atoms in blue) positions. The C4 of the nicotinamide moiety moves 4.6 Å. Asn169 and Cys302 form the “oxyanion” hole.
Despite this wealth of knowledge, several important aspects remain undetermined. Most of these center around the first steps in the catalytic cycle. For example, it is unknown whether Cys302 is stabilized in ionized (thiolate) form or whether a general base is needed to activate it (deprotonate) for reaction with aldehydes. The activation could either occur prior to substrate binding or be induced by it. No conclusive evidence has been put forth on this matter. Furthermore, the coenzyme has been proposed to be in position to help stabilize the thiolate (6,7). Without knowledge of the detailed atomic level structure, such a proposal appears reasonable given that the nicotinamide moiety of NAD+ has a formal positive charge. Yet, no atomic level description has been proposed for this stabilization. It has been known since 1963 that thiols can react with nicotinamides to form C-4 addition compounds (8). On this basis, stabilization of a free thiolate in direct contact with the NAD+ cofactor should not be possible. Re-examination of the crystal structures of two distinct members of the ALDH superfamily, formyltetrahydrofolate dehydrogenase and mitochondrial ALDH2 (Cα rmsd 1.36Å), now confirms that this direct interaction actually leads to a cysteine-cofactor adduct, one which is further confirmed by a broad absorption maximum around 319nm (9). The adduct bond would have to break before the catalytic cycle could proceed. Indeed, that study revealed that this bond is reversible by soaking the crystals in aldehydes. Unfortunately, this result does not indicate whether nucleophilic attack on aldehydes by Cys302 is favored over addition to NAD+. There have been several other reports on the formation of covalent NAD adducts with substrates (10-13) but none between the nicotinamide and an active site nucleophile before the report by Tsybovsky et al. (9).
The protonation state of the thiohemiacetal intermediate in ALDH has only been discussed in a few instances (14). The assumption may be that hydride transfer proceeds from the intermediate in oxyanion form and that no proton transfer coupling is required. Yet in many cases of hydride transfer in enzymes, proton transfer is a vital component to describing the reaction (15,16). The lack of discussion in this area is most likely related to the fact that until now (and in the absence of simulations) the H-bonding potentials of the Asn169 side chain and the main chain amide of Cys302 have generally been considered sufficient to account for stabilization of the catalytic intermediate leading to catalysis without full proton transfer. This is a critical distinction that we investigate in our studies. Asparaginyl side chains and main chain amides are rarely invoked to act as proton donors due to their high pKas and because of the mistaken notion that they would be protonating an intermediate that was more aldehyde-like versus a fully formed oxyanion intermediate. It is important to remember that the thiohemiacetals have been shown to have a pKa in the 12−14 range (17), a range that could presumably be much higher in the relatively solvent-shielded active site. Since any proton donation to the intermediate must be transferred back during the subsequent hydride transfer reaction to form the thioester, this may either not have been seen as a critical step in the catalytic cycle or to have presented a grave theoretical problem if the proton donor were to be one of the more conventional sources with a pKa closer to neutrality. Instead, having an amide as a potential donor puts donor and acceptor on relatively equivalent pKa levels, as should be the case if reversibility is crucial.
Obtaining detailed atomic-level information on intermediates and transition states in most if not all enzymes is extremely difficult (18). In principle, the structures presented by Perez-Miller for the human mitochondrial ALDH2 enzyme can be used to examine certain assumptions about the ALDH mechanism using hybrid Quantum Mechanical/Molecular Mechanical (QM/MM) potential energy functions (19-21). Recognition that in the right configuration a cysteine-cofactor adduct can be formed, as will be shown, is a critical feature of the mechanism. Our initial results indicated this adduct as a possibility and we presented preliminary aspects of it at the 13th (biannual) International Workshop on Carbonyl Metabolizing Enzymes in July 2006 (22). Subsequent crystallography (9) verifies that our modeling of this aspect was correct and motivated our efforts to understand the mechanistic implications of this adduct. We used QM/MM and MM simulations in past studies to understand the binding of benzaldehyde to ALDH3 (23, 24), the formation of the thiohemiacetal intermediate, and to distinguish between different mechanisms for activating the cysteine nucleophile (24). In addition, in ALDH3 we determined a novel mechanism for stabilizing the oxyanion intermediate supported by a “second-sphere” interaction with a lysine residue that is strictly conserved in sequence only within class 3 ALDHs and succinic semialdehyde dehydrogenases (25).
Methods
Ab initio and semiempirical QM calculations of active site ALDH models
Gas phase models of possible ALDH enzymatic reactions were examined for the validation of the PM3 method (26) used in the QM/MM simulations described below. Model reactions used in these studies were defined as 1) between methylthiolate and nicotinamide to produce a neutral adduct and 2) between methylthiolate and fluroacetaldehyde to produce a thiohemiacetal. The fluorine substitution was necessary to produce a product with a stationary point in these model reactions (but not in subsequent QM/MM simulations). Potential energy scans were performed for the reaction of methylthiolate with acetaldehyde using the MP2/6−31+G(d,p) and PM3 methods varying the sulfur-carbonyl carbon distance; no minima were found in either case.
The proton affinities of the thiohemiacetal oxyanion molecule and a negatively charged dimethylamide were also calculated. In addition, a section of the ALDH active site was constructed that included the cysteine side chain and main chain along with acetaldehyde to determine the enthalpic difference between two proton locations. All of these product structures are shown in Figure 3. The reactants and products were first optimized at the MP2/6−31+G(d,p) level and converted to enthalpies by applying the un-scaled thermal corrections derived at this level. The final values reported are energy values taken from MP2/6−311++G(2df,2p) optimizations with the thermal corrections from the smaller basis set MP2 calculation. Basis set superposition error (BSSE) was accounted for by employing the counterpoise method (27). These calculations were performed with Gaussian03 (28). The PM3 results were taken by optimizing the same molecules using Dynamo version 2.2 (29, 30). Other semiempirical molecular orbital methods were examined as part of this study but were not as accurate.
Figure 3.

Product structures with bond lengths from MP2/6−311++G(2df,2p) in red and PM3 in black for A) fluoro-thiohemiacetal B) protonated fluoro-thiohemiacetal C) methylthiolate-nicotinamide adduct D and E) Model ALDH active site structures with differing proton locations.
Construction of the ALDH2 molecular models
The starting models for the QM/MM simulations were based on two crystal structures of human mitochondrial ALDH2 (PDB codes: 1O01 and 1O04 (1)). One of these (1O01) contains crotonaldehyde in the active site with the carbonyl oxygen placed in the oxyanion hole made up of the side chain of Asn169 and the main chain amide of Cys302. The nicotinamide ring of the NAD+ cofactor is in the “hydrolysis” (HY) position, one of two orientations differing by ca. 4.6 Å (Figure 2). The other structure (1O04) is a Cys302Ser mutant and has NAD+ in the hydride transfer (HT) position. Since these structures overlap to a great extent except for the position of the cofactor and surrounding counterions and water molecules, then ternary complexes can be constructed with NAD either in the hydrolysis or HT position with only slight modifications. The position of crotonaldehyde was used to construct a ternary complex of propanal. In all systems, the positions of the Mg2+ and Na+ ions identified by Perez-Miller and Hurley (1) were retained as well as the positions of oxygen atoms from water molecules surrounding the active site.
All residues were kept at their canonical protonation state except for Cys302 which is reasonable to model as a thiolate anion given that we were modeling its reaction with propanal and NAD+. The possible general bases (Glu268 and Glu399) that conceivably could activate a neutral Cys302 are relatively far removed from the chemistry described in this report and therefore will have little effect. The tautomeric state of all histidine residues, although similarly far removed, was visually examined and adjusted accordingly depending on the location of proton acceptors. Otherwise, hydrogen atoms were added to the enzyme, cofactor, substrate and oxygen atoms from water based on internal coordinates in CHARMM (31) using the HBUILD algorithm (32). The entire tetrameric ALDH2 system was then solvated with TIP3P (33) water molecules. The entire solvated system consisted of ca. 99K atoms. A 500 step steepest-descent minimization of the system was then performed with CHARMM to relax the positions of the hydrogen atoms and to place the propanal substrate securely in the oxyanion hole defined previously with Cys302 poised to attack the carbonyl carbon of propanal. Equilibration of the system for MM simulations usually involves simulating the system at room temperature for several picoseconds (ps). This procedure is probably not ideal for our system since we are examining the relevance of the reported crystallographic atomic coordinates to the enzymatic mechanism. Equilibration procedures more appropriate to QM/MM simulations are given below. The QM/MM simulations were focused on the β subunit using a stochastic boundary approach (34). The system used for running QM/MM simulations included the entire tetrameric enzyme, all NAD+ molecules and counterions in close contact, water molecules from the x-ray and all added water molecules within a 35 Å sphere centered on the Sγ atom of Cys302. A representation of the system is shown in Figure 4.
Figure 4.

Tetrameric human mitochondrial ALDH structure (left) with the chemical groups represented through a quantum mechanical potential (right). Here, the oxyanion thiohemiacetal intermediate is stabilized by hydrogen bonding from the Asn169 side chain and the Cys302 main chain amide while the hydride is poised to transfer to the C4 of NAD+. Umbrella sampling along a hydride transfer reaction coordinate from this structure led to formation of the adduct.
Modeling ALDH Reactions with a QM/MM hybrid potential energy function
We have, during the course of our investigations into the ALDH mechanism, explored a variety of possible scenarios and simulation parameters. We will discuss here in-depth only the ones that shed the most light on the mechanism. The QM region was modeled with the semiempirical molecular orbital method, PM3, in all simulations and has demonstrated to return reasonable results for the reactions investigated as part of this study. The MM atoms are modeled with the OPLS force field (35) for the protein chains and a modification of the CHARMM force field for NAD+ (36) to work with the OPLS formalism. The QM region for modeling Cys302 nucleophilic attack on propanal with NAD in the HY position consisted of the Asn169 side chain, a region spanning the Cα of Cys301 to the Cα of Cys303 including the side chains, the nicotinamide moiety of NAD, the propanal substrate and two water molecules in close proximity to the substrate. This required the addition of three QM hydrogen link-atoms to satisfy the valency of the QM region were bonded to 1) Cα of Cys301 along the main chain nitrogen-Cys301 bond axis, 2) Cα of Cys302 along the main chain carbonyl carbon Cys302 bond axis and 3) N1 of nicotinamide along the carbon-ribose bond axis. The Dynamo library does this automatically once the QM atoms have been designated (29). The QM region spanning Cys301-Cys303 was necessary since Cys302 not only provides the nucleophile but also provides critical hydrogen bonding interactions to the substrate. By moving the link atom region across one residue on each side of Cys302, artifacts are less likely. The QM region is electrically neutral.
The QM region is modeled the same way when NAD is in the HT position except that the two water molecules in close proximity to the substrate are now further displaced and modeled as MM atoms. The QM region for the addition of Cys302 to NAD+ is also the same except only one water molecule is modeled as QM. During minimizations and MD simulations using the PM3/OPLS potential, residues with all atoms greater than 20.0 Å from the Sγ of Cys302 were fixed, all others were free to move according to the potential energy function. The non-bonded interactions were calculated with a force-switch with an inner cutoff of 9.5 Å and outer cutoff of 13.5 Å with a non-bonded list cutoff of 15.5 Å updated heuristically.
The reaction coordinate was defined as the distance between the Sγ of Cys302 and carbonyl carbon of propanal for the reaction forming the oxyanion intermediate. Umbrella sampling molecular dynamics were performed with 64 windows harmonically constrained to reaction coordinate values from 3.5 Å to 1.61 with spacing of 0.03 Å. A force constant of 2000 kJ mol−1 Å−2 was used for the umbrella potentials. Each window was minimized for 2000 steps using a conjugate gradient. Then 5 ps of equilibration was performed with the velocity Verlet algorithm at a temperature of 300.0 K followed by 10 ps of production sampling. A 1 fs time-step is used in all QM/MM-MD simulations. Simulations were initiated both from the Michaelis complex and the oxyanion intermediate to determine any sampling errors. Each free energy profile consisted of 960 ps of simulation. The sampled values of the reaction coordinate were used with the weighted histogram analysis method (WHAM) to derive the potential of mean force (free energies) along the reaction coordinate (37). The distance between Sγ of Cys302 and C4 of NAD was used as the reaction coordinate to describe the formation of the adduct. The simulation procedure was exactly the same as for formation of the oxyanion intermediate except that the reaction coordinate ran from 3.7 Å to 1.8 Å, the production part was 20 ps instead of 10 ps and the umbrella potential was 4000 kJ mol−1 Å−2 which was required in order to sample the separated reactants properly. The total simulation time was 1.6 nanoseconds (ns). Due to the increased simulation time and because there were no entities in the simulation that underwent any significant relative changes in position, simulations were not performed from other starting points.
Two-dimensional umbrella sampling was performed for the protonation of the oxyanion intermediate where one coordinate was the distance between the Sγ of Cys243 and the carbonyl carbon of propanal (q2). An appropriate reaction coordinate for the proton transfer from the main chain amide to the oxyanion intermediate (atom type of donor and acceptor are different) is the asymmetric stretch:
where rNH and rOH are the distances of the proton from the donor nitrogen and the acceptor oxygen respectively with mN and mO as their masses. The reaction coordinate, q2, went from 2.0 Å to 1.85 Å in 0.05 Å increments while q1 went from −0.41 (the proton located on the intermediate) to 0.85 (the proton located on the main chain) in 0.02 Å increments. Construction of this 2D free energy surface thus required 3.84 ns of simulation. Snapshots were taken from all trajectories every 100 steps. Visualization of structures and construction of Figures 3 and 5 were done with VMD (38). PSI-Plot (39) was used to construct the free energy surface in Figure 8.
Figure 5.

Free energy profile for the nucleophilic attack of Cys302 on the carbonyl carbon of acetaldehyde with NAD in the HY or HT position.
Figure 8.

The 2-dimensional free energy profile for proton transfer (q1) from the Cys302 main chain amide to the oxyanion intermediate as a function of the S-C bond distance (q2).
Results
Ab initio and semiempirical Quantum Mechanical Calculations on systems relevant to ALDH mechanism
In order to calculate such molecular features as geometry, proton affinity, and heats of formation, the molecule must have a stationary point on the electronic energy surface. For the oxyanion thiohemiacetal species that would result from a methylthiolate attack on acetaldehyde or propanal, there is no stationary point for the product state in the gas phase. We were able to create a stationary point for the thiohemiacetal molecule by substituting a fluorine for a hydrogen in acetaldehyde. Bond lengths are given for the oxyanion and protonated form of the fluorinated thiohemiacetal molecule for MP2 and for the semiempirical method PM3 in Figure 3. The thiohemiacetal bond lengths are each 2.12 Å while the carbon-oxygen bond length is 1.28 and 1.27 Å at the MP2 and PM3 level respectively. The carbon-oxygen bond is lengthened by 0.07 Å upon addition of methylthiolate in both geometry optimizations. The Oxygen-carbon-carbon-fluorine dihedral angle is different in MP2 versus PM3. At the MP2 level the fluorine is anti (178°) while in PM3 it is staggered (78°). Upon protonation of the thiohemiacetal molecule, the thiohemiacetal bond length is reduced by 0.31 Å at MP2 and 0.26 Å at PM3. The carbon-oxygen bond length is lengthened by 0.13 Å at MP2 and PM3. The reduction in the sulfur-carbon bond length is indicative of a much stronger bond. Furthermore, the vibrational frequency that is most associated with the stretching of this bond has shifted from 479 cm−1 to 686 cm−1 at the MP2 level. The enthalpy of reaction and proton affinity are given in Table 1. The enthalpy of reaction is more favorable at the MP2 level than at the PM3 level by 7.3 kcal/mol (this difference will affect interpretation of QM/MM results as described in the Discussion). The proton affinity of the thiohemiacetal molecule is larger at the MP2 level than at the PM3 level by 5.1 kcal/mol. The geometrical features of the addition of methylthiolate to a nicotinamide analogue are shown in Figure 3 and the enthalpy of reaction in Table 1. The most critical feature, the sulfur-carbon(NAD) bond length, is essentially the same at both levels of theory while the enthalpy of reaction is less favorable at MP2 versus PM3 by 12.7 kcal/mol. Again, this difference will affect the interpretations of the QM/MM results.
Table 1.
Heats of reaction (in kcal/mol) for small model reactions important to ALDH.
| MP2a | PM3 | |
|---|---|---|
| CH3S- addition to | ||
| CH2FCHO | −16.8 | −9.5 |
| Nicotinamide | −125.7 | −138.4 |
| Proton Affinity | ||
| CH2FC-(SCH3)OH | 347.8 | 342.7 |
| Dimethylamide (−1) | 360.4 | 353.2 |
| Proton Transfer | ||
| From D to E (Fig. 2) | 1.2 | −1.6 |
| Hydride Transfer | ||
| From A to NAD+ | −112.6 | −130.0 |
MP2/6−311++G(2df,2p) optimized structure and energies with conversion to enthalpies taken from corrections performed at MP2/6−31+G(d,p). BSSE at the MP2/6−311++G(2df,2p) level was still significant at 5.1 kcal/mol for the thiohemiacetal molecule and 7.1 kcal/mol for the adduct.
Since the proton affinity for the oxyanion thiohemiacetal is large and comparable to the proton affinity of dimethylamide we decided to examine the energetic difference between a proton located on a thiohemiacetal moiety versus on the main chain moiety in a structure similar to the ALDH active site. The structures at both levels of theory are very similar except with respect to the hydrogen bonding distances. The PM3 distances are longer than the MP2 distances by 0.2 Å in both structures. The MP2 results favor the presence of a proton on the main chain amide moiety by only 1.2 kcal/mol while PM3 favors location of the proton on the thiohemiacetal oxygen atom by 1.6 kcal/mol. Thus both methods suggest an isoenergetic proton transfer with small preferences on opposite sides. The use of PM3 method in QM/MM simulations of our ALDH enzymatic reactions is therefore reasonable.
The transfer of a hydride from the fluorinated thiohemiacetal (Figure 3A) to the NAD+ moiety to form NADH and the thioester in these model reactions was highly exothermic in the gas phase though not as exothermic as formation of the adduct (see Table 1). The MP2 results favor formation of the adduct over hydride transfer by 13.1 kcal/mol ; the PM3 results give 8.4 kcal/mol for the same difference. Because the adduct formation is more favorable, calculating a transition state structure/energy for this reaction is difficult if not impossible without adding groups to stabilize the thiohemiacetal.
Cys302 nucleophilic attack on propanal, NAD in HY position
Figure 5 shows the free energy profile for formation of the oxyanion intermediate with NAD+ in the HY position versus NAD+ in the HT position. Simulations initiated from the Michaelis complex with NAD+ in the HY position show a minima at a Cys302 Sγ—propanal carbonyl carbon (referred to as the S-C bond throughout the rest of the text) separation of 3.0 Å that decreases to 2.4 Å at the transition state and finally to 2.0 Å at the oxyanion intermediate. The calculated free energy change is 5.5 kcal/mol with a barrier of 8.6 kcal/mol. Simulations that were initiated from the oxyanion structure resulted in a transition state with a S-C bond length of 2.3 Å, a free energy change of 6.9 kcal/mol, and a barrier of 8.7 kcal/mol. Initiating simulations from the product structure results in a decrease of 0.1 Å in the transition state S-C bond length and an increase of 0.1 kcal/mol in the free energy barrier with 1.4 kcal/mol in free energy change. Thus, only minor hysteresis is present in these values. The Michaelis complex structure was analyzed by observing snapshots from the trajectory in which the reaction coordinate was restrained to 3.0 Å. The hydrogen bonding donors that make up the oxyanion hole (one proton from Nδ of Asn169 and one proton from the main chain amide of Cys302) interact with the carbonyl oxygen of propanal, but only weakly. In the simulation that was initiated from the Michaelis complex and from the oxyanion intermediate, the distance from one Asn169 Nδ proton to the propanal carbonyl oxygen averages 2.8 ± 0.3 Å and 2.5 ± 0.5 Å respectively (we analyzed the one that was closest). The distance from the main chain amide proton of Cys302 to the carbonyl oxygen of propanal averages 3.4 ± 0.4 Å and 3.0 ± 0.3 Å respectively. The Cys302 thiolate interacts primarily with the aldehydic proton of propanal in both simulations and lies roughly in the same plane as the aldehyde group. In the simulation initiated from the intermediate, a water molecule that was defined to be part of the QM region also interacts with the thiolate. This interaction is what most likely creates the slight differences in the free energy profiles that were initiated from different starting structures. From both QM/MM simulation and crystallography, the aldehyde is not tightly bound in the oxyanion hole region while in the Michaelis complex.
The oxyanion intermediate structure was characterized by examining snapshots from the trajectory when the S-C bond was restrained to 2.0 Å. These snapshots showed three proton donors surrounding the thiohemiacetal oxygen atom in a ring; these were the main chain amide of Cys302, one from Nδ of Asn169 and one from a water molecule. The first two are what are commonly regarded as making up the oxyanion hole while the latter has only been seen by simulation. All proton-to-thiohemiacetal oxygen distances average 1.8 Å with fluctuations of 0.1 Å. The carbon-oxygen bond in the oxyanion intermediate structure is 1.31 Å which is 0.1 Å longer than in the Michaelis complex and 0.1 Å shorter than a typical C-O single bond. The distance from the transferring hydride to C4 of NAD+ is 6.9 ± 0.4 Å and thus has an insignificant effect on the free energy profile. Figure 6 shows a representative snapshot from the dynamics for the oxyanion intermediate. The transition state structures were examined by analyzing structures from the trajectory where the reaction coordinate was 2.4 Å. These show essentially the same features as the intermediate except that the hydrogen bond distance from the main chain amide nitrogen of Cys302 to the carbonyl oxygen is longer, averaging 2.9 ± 0.3 Å.
Figure 6.

Representation of the oxyanion thiohemiacetal intermediate showing the three hydrogen bond donors that contribute to stabilizing this structure.
Cys302 nucleophilic attack on propanal, NAD in HT position
In contrast, the free energy profile for formation of the oxyanion intermediate with NAD in the HT position is dramatically different. There are no free energy wells for the Michaelis complex or the oxyanion intermediate. At a S-C distance of 2.0 Å, the free energy is 34.0 kcal/mol higher than at a separation of 3.4 Å. The structures along the reaction coordinate are similar in many respects to those with NAD+ in the HY position. We expected to obtain a structure for the thiohemiacetal intermediate similar to the one depicted in Figure 4. The major differences are that Cys302 is always bound to NAD+ by a bond formed between Sγ of Cys302 and C4 of NAD+. Also, the water molecule is absent that hydrogen bonded first to the thiolate of Cys302 in the Michaelis complex and was then primarily hydrogen bonded to the carbonyl oxygen atom as the oxyanion intermediate is formed. Finally, several snapshots showed the propanal molecule outside of the oxyanion hole region.
Cys302 addition to NAD+
The free energy profile for the addition of Cys302 to C4 of NAD+ in the presence or absence of propanal is shown in Figure 7. In the holo form (without propanal), the free energy well for the reactant state with a Cys302 Sγ –NAD+ C4 separation of 3.3 Å decreases to 2.95 Å at the transition state and ends at 1.85 Å at the Cys302-NAD adduct. There are no critical intermolecular interactions that stabilize the adduct, the structure of the adduct is as shown in Figure 3C. The free energy barrier for this reaction is 5.0 kcal/mol and the free energy change is −28.0 kcal/mol. In the presence of propanal, the free energy barrier is neglible and the free energy change is −31.7 kcal/mol. Both these reactions are exergonic in contrast to the nucleophilic attack on propanal which is endergonic. The thiolate anion in the reactant state (in holo form) is stabilized by direct interactions with adjacent main chain amides and from one water molecule. The adjacent cysteines, poorly conserved in other ALDHs, might also help stabilize the thiolate but they are observed to be hydrogen bonded to Asp457 in our simulations. These direct interactions and the polar environment of the active site accounts for the different energetics calculated in the gas phase (40). In the presence of propanal, the direct interactions direct that help stabilize the thiolate anion in the holo form have been replaced by interactions with the substrate which are weaker. These weaker interactions result in a more favorable reaction with the cofactor.
Figure 7.

Free energy profile for the addition of Cys302 to the C4 of NAD+ with propanal located in the oxyanion hole and in holo form. The zero of energy has been set to the reactant minimum.
Stabilization of thiohemiacetal intermediate through proton transfer from main chain amide
In order to avoid the breakdown of the oxyanion intermediate with formation of the Cys302-NAD adduct, it appears reasonable to search for possible proton donors to the oxygen atom of the oxyanion intermediate. The effect would be to strengthen the S-C bond. We have already demonstrated through small molecule ab initio calculations that an anionic amide has a proton affinity similar to that of the oxyanion intermediate. Therefore, we performed 2D free energy simulations of proton transfer from the main chain amide of Cys302 to the oxyanion intermediate. The free energy landscape is shown in Figure 8. The simulation results show essentially an isoenergetic reaction (the proton is favored to be on the oxygen atom by 0.1 kcal/mol) with a free energy barrier of 14.1 kcal/mol. Separating the anionic oxygen from the Cys302 amide proton costs about 3.0 kcal/mol. The S-C bond distance changes from 2.0 Å in the fully ionized oxyanion form to 1.93 at the transition state and 1.89 at the protonated, α-hydroxy sulfide form. Moving to larger values of the S-C bond length (q2 moving from 1.86 Å to 2.0 Å) when the intermediate is protonated (q1 = −0.30) shows an increase in the free energy. In contrast, moving the same distance along q2 when the intermediate is in oxyanion form (q1 = 0.46) does not result in a free energy change. This feature of the free energy landscape indicates that the S-C bond is stronger when the intermediate is protonated. The carbon-oxygen bond increases from 1.3 Å in the oxyanion form to 1.4 Å in the protonated form. Two water molecules are hydrogen bonded to the carbonyl oxygen of Cys301. The Asn169 Nδ protons are each 3 Å away from the oxygen atom of the substrate while the water molecule that was hydrogen bonded to the oxyanion intermediate has been displaced from this region.
Discussion
Our results using the PM3/OPLS hybrid potential (and taking into account the error associated with the PM3 method) suggest that formation of the oxyanion intermediate when NAD+ is in the HY position is nearly isoenergetic with a small barrier. The intermediate is stabilized through three hydrogen bonds to the anionic oxygen atom. Two of these hydrogen bond donors are those seen in crystal structures with inhibitors/substrates located in ALDH active sites. The third comes from a water molecule located between the substrate and NAD+. The oxyanion intermediate exists in a very shallow free energy well (3.1 kcal/mol) made possible by the hydrogen bonding interactions. A shallow free energy well would appear to be problematic considering that it must exist long enough for NAD+ to undergo transition from the HY to the HT position. Furthermore, the S-C bond length of the oxyanion intermediate, 2.0 Å, is much longer than typical carbon-sulfur single bonds of about 1.85 Å. This bond could be classified as a coordinate covalent bond in which the sulfur electrons are not fully shared with the carbon.
In contrast, the oxyanion intermediate with NAD+ in the HT position does not exist using the PM3/OPLS potential since there are no free energy wells along this reaction coordinate (Figure 4). A straightforward explanation of this result is found by examining the free energy change from separated reactants to an S-C bond distance of 2.0 Å. This is equal to breaking the Cysteine-C4(NAD) bond and forming the S-C bond. One can conclude from these results that formation of the oxyanion intermediate only occurs with NAD+ removed from the HT position. This is the first study to present results indicating the preferred location of NAD+ in ALDH during the cysteine nucleophilic attack and is contrary to suggestions that NAD+ plays a direct role in this reaction (6,7). The binding of the cofactor could still play an indirect role in catalyzing the nucleophilic attack. Binding of the cofactor excludes some solvent molecules from the active site and must surely organize the active site to make activation of the nucleophilic Cys302 more favorable by a general base.
Using the PM3/OPLS hybrid potential, formation of the cysteine-cofactor adduct is thermodynamically favorable in the presence of propanal or in holo form, with NAD in HT position. Given the error associated with the PM3 method, we predict that the reaction is exergonic by approximately 15 kcal/mol. Thus, from this particular arrangement of atoms taken from the crystal structure, formation of the adduct is much more favorable than formation of the oxyanion intermediate even given the errors associated with the PM3 method. Experiments (9) show degradation of the adduct by soaking crystals in aldehydes but provide no information as to the thermodynamics of each individual reaction. Although formation of the adduct must surely hinder the efficiency of catalysis in ALDH, the equilibrium between Cys302 with NAD+ in the HY position (reactants separated) and the adduct is heavily weighted toward the separated reactants (9).
Formation of the adduct may also serve to protect Cys302 from reactions with oxidizing agents other than aldehydes at times when the enzyme is not functioning catalytically (41). At least, the adduct in ALDH2 clearly serves to finally provide a plausible explanation for the refractiveness of that residue to complete modification with iodoacetamide (42). Iodoacetamide was long ago shown to behave as an active-site-directed inhibitor, selectively modifying Cys-302 to the exclusion of the other eight cysteine residues in the enzyme (43,44). However, despite satisfying other criteria for active site-directed inhibitors, the inescapable facts were that iodoacetamide could neither achieve more than ∼50% inactivation of ALDH2 catalytic activity nor be incorporated quantitatively, with typical incorporation of one equivalent per homotetramer (42). This cast a pall over the conclusion that Cys-302 was the catalytic cysteine for over a decade, a pall that was only removed on determination of the first crystal structures (45, 46).
A further in vitro aspect of ALDH2 known to most who have worked with it but which received scant attention is that in spectrophotometric assays, the enzyme typically displays a lag phase. This was already noticed in some of the earliest purifications of the enzyme, and seemed dependent on the order of addition of reagents (47). Both the non-quantitative incorporation of iodoacetamide and the kinetic lag phase are readily explained by adduction of some fraction of the catalytic thiols with NAD.
Additionally, it may be recalled that ALDH3 constitutes 10−40% of the soluble protein of the corneal epithelia of most mammals. Should the adduct also exist in ALDH3, covalent binding of NAD to the enzyme could provide a basis for enhancing the ability of ALDH3 to provide direct UV protection to the eye, as has been considered (48,49), or to maintain an added defense against products UV-induced lipid peroxidation (in addition to 4-hydroxynonenal) which could be directly deleterious to the thiol. At the moment, this remains conjecture, since no evidence has been found to suggest adduct formation in ALDH3. In addition, at least one enzyme that we are aware of employs a unique mechanism to protect the reactive cysteine from irreversible oxidation. On reacting with H2O2, Protein Tyrosine Phosphatase 1B forms a sulphenic acid that can then undergo a nucleophilic attack by the adjacent main chain nitrogen to form a sulphenyl-amide species (50). This interesting regulation mechanism, also involving the main chain, protects the enzyme from forming an irreversible sulphonic acid.
The most interesting problem that the adduct presents is how to avoid it during the ALDH catalytic cycle. As NAD+ moves from the HY position to HT position, the S-C bond must move from a shallow energy well to the slope of a free energy surface as shown in Figure 5 In other words, it is clear that positioning the coenzyme for hydride transfer destabilizes the oxyanion intermediate. The most obvious way to stabilize the oxyanion is through proton transfer. We chose to examine proton transfer from the main chain for several reasons. First, simulations carried out with ALDH3 revealed this to be the most thermodynamically favorable donor, more favorable than a water molecule or from the Asn114 side chain (23). Second, there appears to be a mechanism for labilizing the main chain proton. In our past MM-MD simulations of ALDH3A1, we observed a 5 Å conformational transition of Lys235 (the “proximal lysine” (23,51)) from a surface location to one that was hydrogen bonded to the main chain carbonyl oxygen of Thr242, adjacent to the catalytic Cys243 (23). This interaction would decrease the pKa of the main chain amide of Cys243. While this lysine is strictly conserved only in class 3 ALDHs and in succinic semialdehyde dehydrogenases (25), we note that in the crystal structure of a ternary complex of non-phosphorylating aldehyde dehydrogenase (GAPN) another lysine residue (Lys288, the “distal lysine”) in a geometrically analogous position hydrogen bonds instead to the equivalent main chain amide oxygen (51, 52). The distal lysine residue is conserved in many classes of ALDH. Furthermore, certain mutations of residues in the proximity of these lysines in two different but related ALDHs lead to two different metabolic diseases, Sjögren-Larsson syndrome (53) and Hyperprolinemia Type II (54). These mutations (Asp245Asn and Ser352Leu respectively) are removed from the active site of ALDH and are the focus of some of our current investigations.
In the simulations reported here, the distal Lys (Lys272) essentially retained its crystallographic coordinates though it was not fixed in the simulation. Longer molecular dynamics simulations using a MM force field will be performed in the near future to determine if this conformational transition is reasonable for ALDH2 since the QM/MM simulations are too short to observe a conformational fluctuation such as this. Positioning Lys272 closer to the main chain carbonyl oxygen of Cys301 would likely make the proton transfer from the main chain to the oxyanion intermediate more thermodynamically favorable thus altering the equilibrium between protonated and unprotonated form in favor of the former. Finally, any donor to the oxyanion intermediate should be positioned to have its proton donated back once the hydride transfer reaction is initiated. Binding of Cys302 to the substrate further restricts the motion of this residue, which should assist in satisfying this requirement. (Footnote: We considered a mechanism in which the substrate was first protonated and then attacked by the cysteine but the calculated thermodynamics are such that formation of the adduct is unavoidable.)
Thus, we propose the following mechanism to account for the experimental and theoretical results: With NAD in the HY position, formation of the oxyanion thiohemiacetal intermediate is predicted to be close to isoenergetic with a small free energy barrier. Proton transfer from the main chain amide of Cys302 to the oxyanion intermediate occurs after nucleophilic attack to form an α-hydroxy sulfide. The proton transfer reaction is also predicted to be isoenergetic which would give a 50:50 mixture of the two forms at equilibrium. The calculated barrier for this proton transfer of 14.1 kcal/mol would likely be lower with more accurate methods although this does not affect our mechanistic interpretations. Furthermore, the short length of the hydrogen bond for both structures obtained from high level QM calculations suggests that quantum dynamical effects (proton tunneling) most likely play a role which would further reduce the barrier to proton transfer (55). This type of hydrogen bond is sometimes classified as a low-barrier hydrogen bond (56). Subsequently, the cofactor undergoes a conformational transition to be positioned for hydride transfer. The effect of this conformational transition on the equilibrium between protonated and unprotonated forms of the intermediate has not as yet been investigated with computational techniques since the path from the HY to the HT position involves changes in several dihedral angles of NAD. To avoid reverting to the cysteine-NAD adduct from the protonated form and insure efficient catalysis, the free energy barrier for 1) the hydride transfer step from the α-hydroxy sulfide to form NADH and the thioester (coupled with proton transfer back to the main chain) must be sufficiently lower than 2) proton transfer from the α-hydroxy sulfide back to the main chain amide alone (see Figure 9). The quantitative simulation of a hydride transfer reaction in probable concert with a proton transfer is extremely challenging and will require the development of new methods similar to those reported by Ferrer et al (15). The position of the distal lysine may also have to be placed in a much closer position analogous to that observed in GAPN (51,52) to assess its possible role in facilitating formation of the α-hydroxy sulfide.
Figure 9.
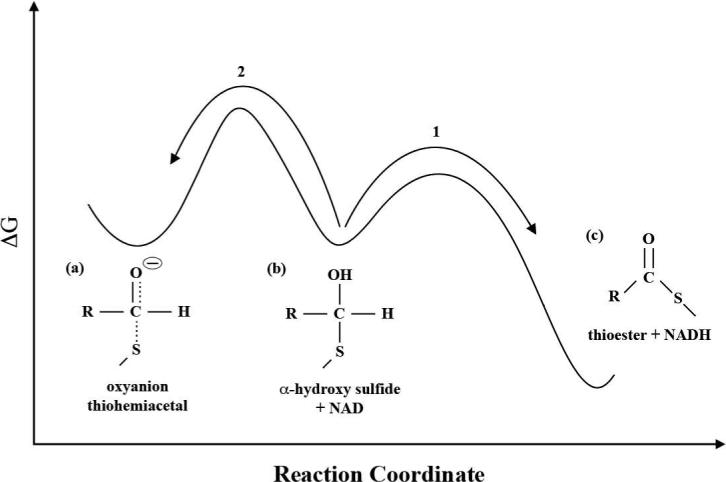
Conceptualized energetics: protonation of the oxyanion thiohemiacetal (a) to form the α-hydroxy sulfide (b) prevents the scission of the S-C bond and formation of the adduct. The hydride transfer reaction (1) must be both kinetically (lower barrier) and thermodynamically more favorable than proton transfer back to the main chain amide (2) with NAD in HT position.
Finally, the calculated energetics of forming the adduct differed significantly in the gas phase versus in the enzyme because of favorable interactions that stabilized the thiolate anion in the enzyme. In contrast, energetics for the hydride transfer reaction in the enzyme will be similar to the gas phase (ΔH = −112.6 kcal/mol at MP2/6−311++G(2df,2p)) due to absence of interactions that provide preferential stabilization of reactant species vs. product species. Thus, the hydride transfer reaction is expected to be thermodynamically favored over adduct formation.
Conclusion
We have employed both high level ab initio QM methods and hybrid QM/MM potentials in order to understand ALDH mechanisms at an atomic level. Using crystal structures of ALDH2 as a basis for modeling these reactions, we determined that formation of a Cys302-NAD adduct was highly favorable with NAD+ in the HT position and Cys302 in thiolate form. Prior to our presentation on the adduct in July 2006 (22), this reaction had only been reported by adding the respective reactants (thiols plus nicotinamides) together in solution (8). We surmised that another configuration of atoms was probably responsible for describing the chemistry of ALDH until recent crystallographic data (9) confirmed that this simulation result was correct in that this adduct is not only observed in the crystal structure of formyltetrahydrofolate dehydrogenase but also in the ALDH2 structures from re-examination of the earlier data of Perez-Miller and Hurley (1). The mechanistic implications of this adduct are many-fold. According to our calculations, NAD+ must be located in the HY position (or at least not in the HT position) to form the oxyanion intermediate, since formation of the adduct is thermodynamically favored over formation of the oxyanion intermediate with NAD in the HT position. Our data show that a mechanism for avoiding the breakdown of the oxyanion intermediate as NAD+ moves into the HT position is proton donation to the oxyanion to form an α-hydroxy sulfide. This is indicated, through observation of shorter bond lengths, to create a stronger S-C bond and an energy barrier that may withstand the attraction to form the adduct. We have presented theoretical calculations that show the main chain amide of Cys302 to be an attractive candidate to perform this function. This proton transfer is likely a low-barrier (fast) isoenergetic reaction that occurs after formation of the oxyanion intermediate. We argue that this is a necessary chemical reaction in the ALDH catalytic cycle; important for a more complete understanding of structure-function relationships in this enzyme family. The proton transfer back to the original donor would then be coupled to the hydride transfer reaction in a fashion similar to other enzymes that catalyze hydride transfer.
Supplementary Material
Acknowledgements
The computations were performed on the National Institutes of Health HP GS1280 system at the Pittsburgh Supercomputing Center.
This research was supported by funding from NIH-NCRR (RR06009).
Abbreviations
- ALDH
aldehyde dehydrogenase
- NAD
nicotinamide adenine dinucleotide
- QM/MM
Quantum Mechanical/Molecular Mechanical
- PM3
Parameterized Method 3
- HY
hydrolysis
- HT
hydride transfer
- WHAM
weighted histogram analysis method
- rmsd
root-mean-square-deviation
Footnotes
Supporting Information Available
Coordinates of optimized product structures presented in Figure 3 are given in XYZ format. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Perez-Miller SJ, Hurley TD. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry. 2003;42:7100–7109. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 2.Farrés J, Wang TTY, Cunningham SJ, Weiner H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry. 1995;34:2592–2598. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 3.Hammen PK, Allali-Hassani A, Hallenga K, Hurley TD, Weiner H. Multiple conformations of NAD and NADH when bound to human cytosolic and mitochondrial aldehyde dehydrogenase. Biochemistry. 2002;41:7156–7168. doi: 10.1021/bi012197t. [DOI] [PubMed] [Google Scholar]
- 4.Jones HK, Lindahl R, Baker DC, Timkovich R. Hydride transfer stereo specificity of rat liver aldehyde dehydrogenase. J. Biol. Chem. 1987;262:10911–10913. [PubMed] [Google Scholar]
- 5.Abriola DP, Fields R, Stein S, MacKerell AD, Jr., Pietruszko R. Active site of human liver aldehyde dehydrogenase. Biochemistry. 1987;26:5679–5684. doi: 10.1021/bi00392a015. [DOI] [PubMed] [Google Scholar]
- 6.Marchal S, Branlant G. Evidence for the chemical activation of essential Cys-302 upon cofactor binding to nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 1999;38:12950–12958. doi: 10.1021/bi990453k. [DOI] [PubMed] [Google Scholar]
- 7.Mann CJ, Weiner H. Differences in the roles of conserved glutamic acid residues in the active site of human class 3 and class 2 aldehyde dehydrogenases. Protein Sci. 1999;8:1922–1929. doi: 10.1110/ps.8.10.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dittmer DC, Kolyer JM. Addition compounds of thiols and 1-substituted nicotinamides. J. Org. Chem. 1963;28:1720–1722. [Google Scholar]
- 9.Tsybovsky Y, Donato H, Krupenko NI, Davies C, Krupenko SA. Crystal structures of the carboxyl terminal domain of rat 10-formyltetrahydrofolate dehydrogenase: implications for the catalytic mechanism of aldehyde dehydrogenases. Biochemistry. 2007;46:2917–2929. doi: 10.1021/bi0619573. [DOI] [PubMed] [Google Scholar]
- 10.Burgner JW, II, Ray WJ., Jr. Acceleration of the NAD-cyanide adduct reaction by lactate dehydrogenase: the equilibrium binding effect as a measure of the activation of bound NAD. Biochemistry. 1984;23:3620–3626. doi: 10.1021/bi00311a008. [DOI] [PubMed] [Google Scholar]
- 11.Benach J, Atrian S, Gonzalez-Duarte R, Ladenstein R. The catalytic reaction and inhibition mechanism of Drosophila alcohol dehydrogenase: observation of an enzyme-bound NAD-ketone adduct at 1.4 Å resolution by x-ray crystallography. J. Mol. Biol. 1999;289:335–355. doi: 10.1006/jmbi.1999.2765. [DOI] [PubMed] [Google Scholar]
- 12.Hewitt CO, Eszes CM, Sessions RB, Moreton KM, Dafforn TR, Takei J, Dempsey CE, Clarke AR, Holbrook JJ. A general method for relieving substrate inhibition in lactate dehydrogenases. Prot. Engineer. 1999;12:491–496. doi: 10.1093/protein/12.6.491. [DOI] [PubMed] [Google Scholar]
- 13.Jacques SL, Mirza A, Ejim L, Koteva K, Hughes DW, Green K, Kinach R, Honek JF, Lai HK, Berghuis AM, Wright GD. Enzyme-assisted suicide: molecular basis for the antifungal activity of 5-hydroxy-4-oxonorvaline by potent inhibition of homoserine dehydrogenase. Chem. Biol. 2003;10:989–995. doi: 10.1016/j.chembiol.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 14.Hempel J, Perozich J, Chapman T, Rose J, Boesch JS, Liu Z, Lindhal R, Wang B. Aldehyde dehydrogenase catalytic mechanism. Enzym. Mol. Biol. Carbonyl Met. 1999;7:53–59. doi: 10.1007/978-1-4615-4735-8_7. [DOI] [PubMed] [Google Scholar]
- 15.Ferrer S, Ruiz-Pernía JJ, Tunón I, Moliner V, Garcia-Viloca M, González-Lafont A, Lluch JM. A QM/MM exploration of the potential energy surface of pyruvate to lactate transformation catalyzed by LDH. Improving the accuracy of semiempirical descriptions. J. Chem. Theory Comput. 2005;1:750–761. doi: 10.1021/ct050016l. [DOI] [PubMed] [Google Scholar]
- 16.Messiha HL, Munro AW, Bruce NC, Barsukov I, Scrutton NS. Reaction of morphinone reductase with 2-cyclohexen-1-one and 1-nitrocyclohexene: Proton donation, ligand binding, and the role of residues Histidine 186 and Asparagine 189. J. Biol. Chem. 2005;280:10695–10709. doi: 10.1074/jbc.M410595200. [DOI] [PubMed] [Google Scholar]
- 17.Jencks WP, Gilbert HF. General acid-base catalysis of barbonyl and acyl group reactions, Pure & Appl. Chem. 1977;49:1021–1027. [Google Scholar]
- 18.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annu. Rev. Biochem. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 19.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 20.Friesner RA, Guallar V. Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Ann. Rev. Phys. Chem. 2005;56:389–427. doi: 10.1146/annurev.physchem.55.091602.094410. [DOI] [PubMed] [Google Scholar]
- 21.Field MJ. Simulating enzyme reactions: challenges and perspectives. J. Comput. Chem. 2002;23:48–58. doi: 10.1002/jcc.1156. [DOI] [PubMed] [Google Scholar]
- 22.Hempel J, Nicholas HB, Jr., Brown ST, Wymore T. Unexpected encounters in simulations of the ALDH mechanism. In: Weiner H, Plapp B, Lindahl R, Maser E, editors. Enzymology and Molecular Biology of Carbonyl Metabolism. Vol. 13. Purdue University Press; Lafayette, IN: 2007. pp. 9–13. [Google Scholar]
- 23.Wymore T, Deerfield DW, II., Field MJ, Hempel J, Nicholas HB., Jr. Initial catalytic events in class 3 aldehyde dehydrogenase: MM and QM/MM simulations. Chemico-Biological Interactions. 2003;143−144:75–84. doi: 10.1016/s0009-2797(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 24.Wymore T, Hempel J, Cho SS, MacKerell AD, Jr., Deerfield DW., II Molecular recognition of aldehydes by aldehyde dehydrogenase and mechanism of nucleophilic activation. Proteins Struct. Func. Bioinf. 2004;57:758–771. doi: 10.1002/prot.20256. [DOI] [PubMed] [Google Scholar]
- 25.Hempel J, Perozich J, Wymore T, Nicholas HB., Jr. An algorithm for identification and ranking of family-specific residues, applied to the ALDH3 family. Chemico-Biological Interactions. 2002;143−144:23–28. doi: 10.1016/s0009-2797(02)00165-5. [DOI] [PubMed] [Google Scholar]
- 26.Stewart JJP. Optimization of parameters for semiempirical methods. J. Comp. Chem. 1989;10:209–220. [Google Scholar]
- 27.Simon S, Duran M, Dannenberg JJ. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers. J. Chem. Phys. 1996;105:11024–11031. [Google Scholar]
- 28.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 29.Field MJ, Albe M, Bret C, Proust-De Martin F, Thomas A. The Dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J. Comp. Chem. 2000;21:1088–1100. [Google Scholar]
- 30.Thomas A, Jourand D, Bret C, Amara P, Field MJ. Is there a covalent intermediate in the viral neuraminidase reaction? A hybrid potential free-energy study. J. Am. Chem. Soc. 1999;121:9693–9702. [Google Scholar]
- 31.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983;4:187–217. [Google Scholar]
- 32.Brunger AT, Karplus M. Polar hydrogen positions in proteins: empirical energy placement and neutron diffraction comparison, Proteins. 1988;4:148–156. doi: 10.1002/prot.340040208. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 34.Brooks CL, III., Karplus M. Solvent effects on protein motion and protein effects on solvent motion. Dynamics of the active site region of lysozyme. J Mol Biol. 1989;208:159–181. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 36.Pavelites JJ, Bash PA, Gao J, MacKerell AD., Jr. Molecular mechanics force field for NAD+, NADH, and the pyrophosphate groups of nucleotides. J. Comp. Chem. 1997;18:221–239. [Google Scholar]
- 37.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free energy calculations on biomolecules. I. The method. J. Comp. Chem. 1992;13:1011–1021. [Google Scholar]
- 38.Humphrey W, Dalke A, Schulten K. VMD - Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.PSI-Plot, Version 7.0. Poly Software International; New York: [Google Scholar]
- 40.Pliego JR, Jr., Riveros JM. Gibbs energy of solvation of organic ions in aqueous and dimethyl suloxide solutions. Phys. Chem. Chem. Phys. 2002;4:1622–1627. [Google Scholar]
- 41.Lindahl R. Aldehyde dehydrogenases and their role in carcinogenesis. Critical Rev. Biochem. Mol. Biol. 1992;27:283–335. doi: 10.3109/10409239209082565. [DOI] [PubMed] [Google Scholar]
- 42.Hempel J, Pietruszko R. Selective Chemical Modification of Human Liver Aldehyde Dehydrogenases E1 and E2 by Iodoacetamide. J. Biol. Chem. 1981;256:10889–10896. [PubMed] [Google Scholar]
- 43.Hempel J, Pietruszko R, Fietzek P, Jörnvall H. Identification of a Segment Containing a Reactive Cysteine Residue in Human Liver Cytoplasmic Aldehyde Dehydrogenase. Biochemistry. 1982;21:6834–6838. doi: 10.1021/bi00269a032. [DOI] [PubMed] [Google Scholar]
- 44.Hempel J, Kaiser R, Jörnvall H. Mitochondrial Aldehyde Dehydrogenase from Human Liver: Primary Structure. Eur. J. Biochem. 1985;256:13–28. doi: 10.1111/j.1432-1033.1985.tb09260.x. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z, Sun Y, Rose J, Chung Y, Hsiao C, Chang W, Kuo I, Perozich J, Lindahl R, Hempel J, Wang B-C. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossman fold. Nat Struct Biol. 1997;4:317–326. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 46.Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. Structure. 1997;5:701–711. doi: 10.1016/s0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 47.Weiner H. (personal communication)
- 48.Pappa A, Chen C, Koutalos Y, Townsend AJ, Vasiliou V. ALDH3A1 Protects Human Corneal Epithelial Cells from Ultraviolet- and 4-hydroxy-2-nonenal-Induced Oxidative Damage. Free Radical Biol Med. 2003;34:1178–1189. doi: 10.1016/s0891-5849(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 49.Piatigorsky J. Enigma of the Abundant Water-Soluble Cytoplasmic Proteins of the Cornea. Cornea. 2002;21(S1):S7–S12. doi: 10.1097/00003226-200111000-00015. [DOI] [PubMed] [Google Scholar]
- 50.Salmeen A, Andersen JN, Myers MP, Meng T, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 51.Hempel J, Stanley S, Perozich J, Wymore T, Nicholas HB., Jr. Residue conservations in aldehyde dehydrogenase gene fusion products reemphasize functional interpretations. In: Weiner H, Plapp B, Lindahl R, Maser E, editors. Enzymology and Molecular Biology of Carbonyl Metabolism. Vol. 12. Purdue University Press; Lafayette, IN: 2006. pp. 8–14. [Google Scholar]
- 52.Cobessi D, Tete-Favier F, Marchal S, Branlant G, Aubry A. Structural and biochemical investigations of the catalytic mechanism of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol. 2000;300:141–152. doi: 10.1006/jmbi.2000.3824. [DOI] [PubMed] [Google Scholar]
- 53.Rizzo WB, Carney G, Lin Z. Molecular basis of Sjögren-Larsson syndrome: mutation analysis of the fatty ALDH gene. Am. J. Hum. Genet. 1999;65:1547–1560. doi: 10.1086/302681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geraghty MT, Vaughn D, Nicholson AJ, Lin W-W, Jimenez-Sanchez G, Obie C, Flynn MP, Valle D, Hu CA. Mutations in the Δ1-pyrroline 5-carboxylate dehydrogenase gene cause type II hyperprolinemia. Hum. Mol. Genet. 1998;7:1411–1415. doi: 10.1093/hmg/7.9.1411. [DOI] [PubMed] [Google Scholar]
- 55.Liang Z, Klinman JP. Structural bases of hydrogen tunneling in enzymes: process and puzzles. Curr. Opin. Struct. Biol. 2004;14:648–655. doi: 10.1016/j.sbi.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 56.Cleland W, Northop DB. Energetics of substrate binding, catalysis, and product release. Methods in Enzymology. 1999;308:3–27. doi: 10.1016/s0076-6879(99)08003-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.