

Abstract
Alterations in activities of one family of proteases, the matrix metalloproteinases (MMPs), have been implicated in primary and metastatic tumor growth, angiogenesis, and pathological degradation of extracellular matrix (ECM) components, such as collagen and laminin. Since hydrolysis of the collagen triple-helix is one of the committed steps in ECM turnover, we envisioned modulation of collagenolytic activity as a strategy for creating selective MMP inhibitors. In the present study, a phosphinate transition state analog has been incorporated within a triple-helical peptide template. The template sequence was based on the α1(V)436-450 collagen region, which is hydrolyzed at the Gly439~Val440 bond selectively by MMP-2 and MMP-9. The phosphinate acts as a tetrahedral transition state analog, which mimics the water-bound peptide bond of a protein substrate during hydrolysis. The phosphinate replaced the amide bond between Gly-Val in the P1-P1′ subsites of the triple-helical peptide. Inhibition studies revealed Ki values in the low nanomolar range for MMP-2 and MMP-9 and low to middle micromolar range for MMP-8 and MMP-13. MMP-1, MMP-3, and MT1-MMP/MMP-14 were not inhibited effectively. Melting of the triple-helix resulted in a decrease in inhibitor affinity for MMP-2. The phosphinate triple-helical transition state analog has high affinity and selectivity for the gelatinases (MMP-2 and MMP-9), and represents a new class of protease inhibitors that maximizes potential selectivity via interactions with both prime and non-prime active site subsites as well as with secondary binding sites (exosites).
Introduction
Proteolysis has often been cited as an important contributor to cancer initiation and progression.1 The 565 proteases identified in humans constitute 1.7% of coding regions in the human genome.2 The identification and validation of specific proteases as anti-cancer targets and the development of appropriate inhibitors is thus a daunting task. Alterations in activities of one family of proteases, the matrix metalloproteinases (MMPs),a have been implicated in primary and metastatic tumor growth, angiogenesis, and pathological degradation of extracellular matrix (ECM) components, such as collagen and laminin.3 In fact, the destruction of collagen by tumor cell extracts was observed 30 years ago.4 MMP inhibitor programs began in earnest in the 1980s, using the destruction of ECM components as a model for inhibitor design.5 Most of these programs evaluated MMP inhibitors for treatment cancer or other inflammatory diseases such as arthritis.6,7
The first generation of MMP inhibitors were peptidic, broad spectrum compounds, whereas the second generation were non-peptidic compounds designed based on MMP active site structural features.3,7 However, in general, neither generation of MMP inhibitors were successful in clinic trials. Compounds either showed no significant therapeutic advantage or had considerable side effects, such as musculoskeletal syndrome (MSS).7 One problem was the design of the clinic trials themselves. MMP inhibitors had been successful in animal models of early stage disease, but were only tested in late-stage disease in clinic trials.3 There were also concerns over whether adequate doses of inhibitors were given.2 The lack of selectivity of the first generation of MMP inhibitors may well have contributed to MSS.3 In addition, some MMPs have host-beneficial functions, making them anti-targets.2 To date, the vast majority of MMP inhibitors contain a hydroxamic or carboxylic acid group which chelates the active site Zn2+.5,7–10 However, the hydroxamic or carboxylic acid usually represents a terminal point in the chain, and thus residues that interact with only one side of the enzyme active site can be incorporated into the inhibitor. Hydroxamates may also chelate Zn2+ too strongly, overwhelming contributions (and hence specificity elements) from the rest of the compound.11,12 This may be why some small molecule MMP inhibitors bind to other, unrelated metalloproteases, such as neprilysin, leucine aminopeptidase, and dipeptidylpeptidase.13 An additional concern is that hydroxamates are known to have unfavorable pharmacokinetics and poor solubilities, and may be metabolically activated.7,11,14 Attempts to produce selective MMP inhibitors have been somewhat thwarted by flexibility in MMP active site subsites, particularly S1′.7,11
One way to circumvent the selectivity problem is to add sequence diversity, using an inhibitory molecule that, rather than terminate a chain, can be incorporated within a chain. This allows for inhibitor interaction with both the primed and non-primed sides of the active site (Figure 1).3,7,11 Additionally, use of a zinc binding group (ZBG) with lesser affinity than a hydroxamate may be advantageous.
Figure 1.

Nomenclature used for enzyme and substrate subsites.87 The arrow marks the site of protease hydrolysis.
Two classes of proteases, the aspartyl proteases and the metallo(zinc)-proteases, use the nucleophilic attack of a water molecule as one of the steps of amide bond hydrolysis.14 The tetrahedral intermediate that results from water addition to the amide carbonyl has been the focus of many protease inhibitor designs and has given rise to two robust classes of inhibitors, namely the statines and phosphorus-based amide bond replacements (Figure 2). An effective enzyme substrate almost invariably produces an inhibitor of the enzyme by incorporation of a statine (mainly for aspartyl proteases) or phosphorus (aspartyl and Zn2+ proteases) tetrahedral intermediate mimic.
Figure 2.

Tetrahedral intermediate (boxed) and statine and phosphorus-based transition state analog inhibitors.
There are three general types of phosphorus-based inhibitors: phosphonamidates [Ψ{PO2H-NH}; Y = NH in Figure 2]; phosphonate esters [Ψ{PO2H-O}; Y = O in Figure 2]; and phosphinic peptides/phosphinates [Ψ{PO2H-CH2}; Y = CH2 in Figure 2]. Phosphonamidates have been shown previously to inhibit MMP-1,5,15,16 MMP-3,17 and MMP-818,19 by behaving as transition state analogs. Unfortunately, phosphonamidates tend to be the least stable of the three phosphorus-based inhibitors. A phosphonate peptide inhibitor has been described for MMP-8,20,21 but the selectivity was not investigated.
Phosphinic peptides have also been shown to behave as transition state analog inhibitors of MMPs.22 Phosphinic peptide combinatorial libraries have been used to design selective MMP-12 inhibitors with Ki values in the low or sub-nM range.23–25 Phosphinic peptide inhibitors have also been described for MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, MMP-11, MMP-13, and MT1-MMP.5,16,22,26–32 The most effective of these inhibitors had Ki values in the low nM range but offered varying degrees of selectivity between the nine aforementioned MMPs.
Phosphinates exhibit weaker binding to Zn2+ than hydroxamates, and thus may limit off target inhibitor binding.12 Density functional theory calculations have indicated that hypophosphite will bind to the Zn2+ in MMPs in a half-bidentate fashion, i.e. one oxygen coordinates directly with the Zn2+ while the other does so partially.33 This prediction is borne out by X-ray crystallographic analyses of phosphinic peptides bound to MMPs. For phosphinic acid peptide inhibitors of MMP-1, MMP-7, and MMP-11, the solvent accessible phosphinate oxygen has been found to bind to the active site Zn2+ while the buried phosphinate oxygen is hydrogen bonded to the active site Glu.14,22,26 The buried phosphinate oxygen also interacts with the MMP-11 active site Zn2+.22 Overall, phosphinic acid peptides have been shown to be good analogs of the H2O-bound tetrahedral transition state upon binding to MMPs.22
The present study has utilized the phosphinate tetrahedral intermediate mimic to create a potentially selective MMP inhibitor. The phosphinate moiety has been incorporated within a triple-helical peptide template. We had previously described a triple-helical substrate that was selective for MMP-2 and MMP-9.34 The substrate sequence was based on residues 436-450 from α1(V) collagen, where the Gly439~Val440 bond is cleaved. To create an MMP-2/MMP-9 inhibitor, a 9-fluorenylmethoxycarbonyl (Fmoc)-GlyΨ{PO2H-CH2}Val building block was synthesized (Figure 3) and used in the assembly of a triple-helical collagen mimic sequence. Selectivity of the inhibitor may originate from both the sequence and triple-helical structure, as these elements were found to play a role in the selectivity of the substrate.34 To evaluate the role of triple-helicity in MMP inhibition, activity has been examined as a function of triple-helical content. This has required the use of “peptide-amphiphiles,” in which the thermal stability of the triple-helix is enhanced by pseudo-lipids attached to the N-terminus of the peptide.35,36
Figure 3.

Synthesis of (R,S)-2-isopropyl-3-((1-(N-(9-fluorenylmethoxycarbonyl)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid.
Materials and Methods
All standard chemicals were peptide synthesis or molecular biology grade and purchased from Fisher Scientific. O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP), 1-hydroxybenzotriazole (HOBt), Fmoc-amino acid derivatives, and MMP inhibitor III (a hydroxamic acid-Leu-homoPhe dipeptide) were obtained from Novabiochem (San Diego, CA). Amino acids are of L-configuration (except for Gly). Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2 was synthesized by methods described previously.37,38
For the synthesis of the protected Fmoc-GlyΨ{PO2H-CH2}Val building block, intermediates and products were characterized by 1H, 13C, and 31P NMR spectroscopy, mass spectrometry, and elemental analysis. Assignment of the NMR signals was accomplished with the aid of DEPT and HSQC experiments. 1H NMR spectra were recorded at 250 or 300 MHz. 13C NMR spectra were recorded at 63 or 75 MHz. 31P NMR spectra were recorded at 101 or 202 MHz. 13C and 31P NMR spectra were fully proton decoupled. Chemical shifts were expressed in parts per million (δ) relative to tetramethylsilane for 1H NMR and d6-DMSO or CDCl3 for 13C NMR. 31P-chemical shifts were reported downfield from 85% H3PO4. Electrospray mass spectrometry (ES-MS) was performed on a Hitachi MS-8000 3DQ LC-ion trap mass spectrometer by the LSU Department of Chemistry Mass Spectrometry Facility. Elemental analyses were obtained from M-H-W Laboratories (Phoenix, AZ).
Synthesis of (R,S)-2-Isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid (1)
The synthesis of the protected Fmoc-GlyΨ{PO2H-CH2}Val building block (1) is outlined in Figure 3. The procedure follows the strategy devised by Yiotakis and coworkers,39 which requires a Michael addition of the Arbuzov-like nucleophile, 1-(N-(9-Fmoc)amino)-methyl phosphinic acid (5), to the acceptor, 2-isopropylacrylic acid allyl ester (6).
1-(N-(9-Fmoc)amino)-methyl phosphinic acid (5) was prepared as recently described.40 Briefly, the phosphinate analog of Gly, 1-aminomethylphosphinic acid (4), was prepared from the slow addition of N-tritylmethanimine (3) to the in situ formed bistrimethylsilylphosphinic acid, followed by hydrolysis to cleave the trityl group.41 Next, following a modification of the Bolin-procedure for amino acid protection,40 amino acid (4) was silylated with trimethylsilyl chloride (TMS-Cl), N,N-diisopropylethylamine (DIEA) in CH2Cl2 and then treated with Fmoc-Cl to yield 1-(N-(9-Fmoc)amino)-methyl phosphinic acid (5).28
2-Isopropylacrylic acid allyl ester (6) was prepared as follows.42 To a suspension of t-BuOK (8.5 g, 72.0 mmol) in THF (200 mL) was added allyl acetoacetate (9.9 mL, 70.4 mmol) at 0 °C. The resulting clear solution was stirred for 30 min, and then iodopropane (7.9 mL, 77.7 mmol) was added to the solution. The solution was stirred at 70 °C for 12 h. The reaction was quenched with water, and then a saturated aqueous sodium bicarbonate solution was added. The aqueous layer was extracted with diethyl ether (3 × 100 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated to give a yellow oil. The crude product was purified by silica gel column chromatography (1:5 EtOAc/hexanes) to give 2-isopropyl-3-oxobutyric acid allyl ester (2) (9.4 g) in 72% yield. 1H (250 MHz, CDCl3): δ 5.9 (m, 1H), 5.3 (m, 2H), 4.6 (d, J = 5.7 Hz, 2H), 3.2 (d, J = 9.5 Hz, 1H), 2.4 (m, 1H), 2.2 (s, 3H), 1.0 (dd, J = 6.7 Hz, 11.0 Hz, 3H each); 13C (63 MHz, CDCl3): δ 202.4, 168.4, 131.3, 118.4, 67.0, 65.2, 28.8, 28.3, 20.1, 20.0 (Supporting Information, Figures S4 and S5); [M+H]+ 184.1 Da.
To a stirred solution of 2-isopropyl-3-oxobutyric acid allyl ester (2) (4.61 g, 25.0 mmol) in THF (170 mL) was added lithium hexamethyldisilazide (LiHMDS) (27.5 mL, 27.5 mmol, and 1.0 M solution in THF) at −78 °C. The solution was stirred for 30 min, and then paraformaldehyde (3.5 g, excess) was added as a solid in one portion. The resulting suspension was stirred at room temperature for 12 h and then filtered through Celite to remove the excess paraformaldehyde. The filtrate was concentrated, and the residue was purified by column chromatography (1:9 EtOAc/hexanes) to give 2-isopropylacrylic acid allyl ester (6) (2.7 g) in 69% yield. 1H (300 MHz, CDCl3): δ 6.2 (s, 1H), 6.0 (m, 1H), 5.5 (s, 1H), 5.3 (m, 2H), 4.7 (m, 2H), 2.8 (m, 1H), 1.1 (d, J = 6.8 Hz, 6H); 13C (75 MHz, CDCl3): δ 166.7, 146.9, 132.2, 121.7, 117.7, 65.0, 29.2, 21.6 (Supporting Information, Figures S6 and S7); [M+H]+ 154.1 Da.
Condensation of 1-(N-(9-Fmoc)amino)-methyl phosphinic acid (5) and 2-isopropylacrylic acid allyl ester (6) proceeded as follows.39 To an ice cold suspension of 5 (532 mg, 1.68 mmol) in CH2Cl2 (8 mL), DIEA (0.94 mL, 5.38 mmol) and TMS-Cl (0.68 mL, 5.38 mmol) were added under argon atmosphere. This solution was stirred for 3 h at room temperature. Then, the mixture was cooled to 0 °C and compound 6 (310 mg, 2.02 mmol) was added dropwise for 30 min. When the addition was over, the solution was stirred for 36 h at 40 °C. Then, absolute ethanol (2 mL) was added dropwise and the mixture was stirred for 15 min. The solvent was evaporated. To the residue, H2O was added and the resulting suspension was acidified with 1 M HCl to pH 1 and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated to dryness. The oily residue was purified by column chromatography (7:0.4:0.4 chloroform/methanol/acetic acid) to give (R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-phosphinic acid) propanoic acid allyl ester (7) (423 mg) crude in 54% yield. 1H (300 MHz, CD3SOCD3): δ 7.9 (m, 11H), 7.7 (m, 6H), 7.4 (m, 15H), 5.9 (m, 1H), 5.3 (m, 2H), 4.5 (d, J = 5.6 Hz, 2H), 4.3 (m, 8H), 2.0 (m, 6H), 0.8 (d, J = 5.1 Hz, 6H); 13C (75 MHz, CD3SOCD3): δ 173.1, 170.2, 156.3, 147.4, 143.7, 140.6, 139.9, 132.7, 127.5, 127.0, 125.4, 125.2, 120.0, 119.7, 117.7, 65.8, 64.4, 59.7, 46.6, 44.7, 41.5, 32.3, 31.2, 27.4, 26.2, 20.7, 19.6, 19.1, 14.0; 31P (101 MHz, CD3SOCD3): δ 44.0, 43.8 (Supporting Information, Figures S8-S10); [M+H]+ 472.2 Da (calculated 472.2 Da), [M+Na]+ 494.5 (calculated 494.2).
Phosphinate dipeptide (7) (360 mg, 0.77 mmol) and 1-adamantyl bromide (199 mg, 0.92 mmol) were dissolved in chloroform (10 mL), and the reaction mixture was refluxed. To this refluxing mixture, silver oxide (214 mg, 0.92 mmol) was added in 5 equal portions over 50 min. The reaction mixture was refluxed for two additional hours. Then the solvents were removed in vacuo and the residue was treated with diethylether. The silver bromide and the excess of silver oxide were removed by filtration through celite. The filtrate was concentrated to dryness and the residue was purified by column chromatography (98:2 chloroform/2-propanol) to give (R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid, allyl ester (8) (451 mg) crude in 97% yield. 1H (250 MHz, CDCl3): δ 7.8 (m, 3H), 7.6 (m, 2H), 7.4 (m, 6H), 5.9 (m, 1H), 5.3 (m, 2H), 4.6 (m, 2H), 4.4 (m, 2H), 4.2 (m, 1H), 3.6 (m, 2H), 2.1 (m, 15H), 1.7 (m, 17H), 1.2 (m, 1H), 0.9 (m, 6H); 13C (63 MHz, CDCl3): δ 173.9, 173.7, 156.2, 147.1, 146.8, 141.2, 140.6, 140.5, 132.0, 127.7, 127.4, 127.0, 125.3, 125.1, 124.8, 119.9, 119.8, 119.7, 118.6, 83.5, 83.3, 68.1, 67.3, 67.1, 65.4, 64.3, 47.1, 45.3, 44.3, 41.5, 40.9, 36.0, 35.6, 31.1, 30.7, 25.3, 19.7, 19.4, 19.3; 31P (101 MHz, CDCl3): δ 47.9, 47.4, 46.5, 46.3 (Supporting Information, Figures S11-S13); [M+H]+ 606.0 Da (calculated 606.3 Da), [M+Na]+ 628.3 (calculated 628.3).
Removal of the allyl ester proceeded as follows.43 [CpRu(CH3CN)3]PF6 (8.7 mg, 0.02 mmol), quinaldic acid (3.5 mg, 0.02 mmol), and ethanol (3 mL) were placed in a 10 mL two-neck round bottom flask under argon stream. After standing for 30 min at 25 °C, the reddish brown solution was transferred into a 25 mL two-neck round bottom flask containing (R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid, allyl ester (8) (1.0 g, 1.65 mmol) and ethanol (15 mL). The brown solution was stirred for 12 h at 70 °C. The reaction mixture was concentrated under reduced pressure to give a crude product. This was purified by column chromatography (95:5 chloroform/2-propanol) to give (R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid (1) (332 mg) crude in 35% yield. 1H (250 MHz, CD3SOCD3): δ 7.9 (m, 2H), 7.7 (m, 2H), 7.4 (m, 4H), 4.3 (m, 3H), 2.0 (m, 16H), 1.5 (m, 11H), 1.2 (m, 1H), 0.8 (m, 7H); 13C (75 MHz, CD3SOCD3): δ 174.8, 174.7, 156.2, 143.7, 140.7, 133.3, 131.9, 131.5, 131.4, 128.7, 128.6, 127.6, 126.9, 125.1, 120.1, 115.3, 111.5, 80.9, 80.8, 79.1, 65.9, 46.6, 45.2, 43.4, 35.8, 35.3, 30.4, 29.9, 19.3; 31P (101 MHz, CD3SOCD3): δ 47.7, 47.4 (Supporting Information, Figures S1-S3); [M+H]+ 567.0 Da (calculated 566.3 Da), [M+Na]+ 588.2 (calculated 588.3); elemental analysis for C32H40NO6P: C, 66.75% (calculated 67.95%); H, 7.05% (calculated 7.13%); N, 2.34% (calculated 2.48%).
Peptide Synthesis
Peptide-resin assembly of the triple-helical peptide (THP) [(Gly-Pro-Hyp)4-Gly-Pro-Pro-GlyΨ{PO2H-CH2}Val-Val-Gly-Glu-Gln-Gly-Glu-Gln-Gly-Pro-Pro-(Gly-Pro-Hyp)4-NH2] was performed by Fmoc solid-phase methodology44 in continuous-flow mode on an Applied Biosystems Pioneer Peptide Synthesizer. The peptide was synthesized as a C-terminal amide using Fmoc-PAL-PEG-PS resin (0.16 mmol/g initial loading) to prevent diketopiperazine formation.45 Standard Fmoc-amino acid coupling utilized 4 equiv each of amino acid, TBTU, and HOBt (0.25 M final concentration of each) dissolved in 0.5 M DIEA in DMF for 1 h. For difficult couplings (Pro14, Pro26, and Hyp39), 4 equiv of each amino acid and PyAOP (0.25 M final concentration of each) were dissolved in 0.5 M DIEA in DMF and double-coupled for 1 h each time. For addition of (R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid (1), 2 equiv of 1 and PyAOP (0.25 M final concentration of each) were dissolved in 0.5 M DIEA in DMF and coupled in a shaker for 12 h. Capping was performed following the coupling of 1 by addition of Ac2O–DIEA–DMF (0.5:0.6:8.9). Washings between reactions were carried out with DMF. Fmoc group removal was achieved with piperidine–1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)–DMF (1:1:48) for 5 min. A portion of the peptide-resin was lipidated with 20 equiv hexanoic acid [CH3(CH2)4CO2H, designated C6] and PyAOP (0.25 M final concentration each) dissolved in 0.5 M DIEA in DMF for 12 h to create a peptide-amphiphile.35,46 Cleavage and side-chain deprotection of the peptide-resin and peptide-amphiphile-resin proceeded for 2 h using 10 mL of triisopropylsilane–phenol–water–trifluoroacetic acid (TFA) (2:5:5:88),47 followed by filtration of the resin. The resin was rinsed with 2 mL of TFA, and the combined filtrates were reduced under pressure at 35 °C to half the original volume. The remaining filtrate was precipitated by dropwise addition into a 10-fold excess of diethylether. The peptide and peptide-amphiphile were allowed to precipitate at −27 °C for 24 h. The peptide/ether solution was centrifuged and the resulting pellet washed 3 times with diethylether and dried for 4 h under vacuum.
Peptide Analyses
Analytical RP-HPLC was performed on a Waters 600E multisolvent delivery system with a Model 486 tunable dectector controlled by Empower Software, equipped with a Vydac C18 RP column (15 mm particle size, 300 Å pore size, 100 × 8 mm). Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). The elution gradient was 15–35% B in 60 min with a flow rate of 1.0 mL/min. Detection was at λ = 220 nm. Fractions from analytical RP-HPLC were analyzed by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF-MS) on a Bruker Proflex III instrument with XMASS software. Mass values were as follows: f1, [M+H]+ 3579.2 Da (theoretical 3577.9 Da); f2, [M+H]+ 3579.2 Da (theoretical 3577.9 Da); f3, [M+H]+ 3673.4 Da (theoretical 3676.1 Da); and f4, [M+H]+ 3673.4 Da (theoretical 3676.1 Da). For size-exclusion chromatography (SEC), peptides were analyzed in TSB (50 mM Tris•HCl, pH 7.5 containing 100 mM NaCl, 10 mM CaCl2, 0.05% Brij-35, pH 7.5) on an AKTA Purifier (GE Biosciences) equipped with a Superdex 75 (GE Biosciences) column at a flow rate of 1 ml/min. Both solvent and column were maintained at the appropriate temperature, either 10 or 37 °C, for at least 2 h prior to injection. Retention times of standard proteins did not vary with changes in temperature.
Peptide Purification
Semi-preparative RP-HPLC was performed on a Waters Deltaprep System using a Vydac C18 RP column (15 μm particle size, 300 Å pore size, 200 × 25 mm) at a flow rate of 10.0 mL/min. Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). The elution gradient was 10–30% B in 80 min, with detection at λ = 220 nm. Isolated fractions were analyzed by PDA RP-HPLC. PDA RP-HPLC was performed on a Waters 625 pump with a Model 996 diode array detector controlled by Millennium software, using a Vydac C18 RP column (5 mm particle size, 120 Å pore size, 250 × 4.6 mm) at a flow rate of 1.0 mL/min. Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). The elution gradient was 10–30% B in 20 min, with detection at λ = 200–400 nm.
Circular Dichroism (CD) Spectroscopy
CD spectra were recorded over the range λ = 190–250 nm on a JASCO J-810 spectropolarimeter using a 0.1 cm path-length quartz cell. Thermal transition curves were obtained by recording the molar ellipticity ([Θ]) at λ = 222 nm while the temperature was continuously increased in the range of 5–95 °C at a rate of 0.2 °C/min. Temperature was controlled using a JASCO PFD-425S temperature control unit. For samples exhibiting sigmoidal melting curves, the inflection point in the transition region (first derivative) is defined as the melting temperature (Tm). To compare transitions for different triple-helical species, the highest [Θ]222 nm value was designated as 100% folded and the lowest [Θ]222 nm value was designated as 0% folded.48–51
Matrix Metalloproteinases
ProMMP-1 and proMMP-3 were expressed in E. coli and folded from the inclusion bodies as described previously.52,53 ProMMP-1 was activated by reacting with 1 mM 4-aminophenyl mercuric acetate (APMA) and 0.1 equiv of MMP-3(Δ248-460) at 37 °C for 6 h. After activation, MMP-3(Δ248-460) was completely removed from MMP-1 by affinity chromatography using an anti-MMP-3 IgG Affi-Gel 10 column. ProMMP-3 was activated by reacting with 5 μg/mL chymotrypsin at 37 °C for 2 h. Chymotrypsin was inactivated with 2 mM diisopropylfluorophosphate. ProMMP-2 was purified from the culture medium of human uterine cervical fibroblasts54 and activated by incubating with 1 mM APMA for 2 h at 37 °C. ProMMP-8 was expressed in CHO-K1 cells as described previously.55 ProMMP-8 was activated by incubating with 1 mM APMA for 2 h at 37 °C. Recombinant proMMP-9 was purchased from Chemicon International (Temecula, CA) and activated with 1 mM APMA for 1 h at 37 °C. Recombinant proMMP-13 was purchased from R&D Systems (Minneapolis, MN) and activated by incubating with 1 mM APMA for 2 h at 37 °C. The concentrations of active MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, and MMP-13 were determined by titration with recombinant TIMP-1 or N-TIMP-1 over a concentration range of 0.1–3 μg/mL.56 Recombinant MT1-MMP with the linker and C-terminal hemopexin-like domains deleted [residues 279-523; designated MT1-MMP(Δ279-523)] was purchased from Chemicon. MT1-MMP(Δ279-523) was expressed and activated, resulting in Tyr112 at the N-terminus. MT1-MMP(Δ279-523), which, in contrast to MT1-MMP, does not undergo rapid autoproteolysis, was used in the present studies due to the relatively small differences in MT1-MMP(Δ279-523) and MT1-MMP triple-helical peptidase activities noted previously.57 The concentration of active MT1-MMP(Δ279-523) was determined by titration with recombinant N-TIMP-3.57 ProMMP-3(Δ248-460) were expressed in E. coli using the expression vector pET3a (Novagen), folded from inclusion bodies and purified as described previously.58 The zymogen was activated as described above for the full-length proMMP-3. Active site titrations utilized either Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2 or NFF-3 as substrate.37,38,59
Inhibition Kinetic Studies
Peptide substrates and inhibitors were dissolved in TSB buffer. 1–2 nM enzyme was incubated with varying concentrations of inhibitors for 2 h at room temperature. A 2 h incubation was utilized based on the generally observed behavior of slow on and off rates for tight-binding inhibitors60 and studies demonstrating that high affinity phosphinate inhibitors of Zn2+ metalloproteinases are slow binding.61 Residual enzyme activity was monitored by adding 0.1 volume of Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2 to produce a final concentration of < 0.1 KM. Initial velocity rates were determined from the first 20 min of hydrolysis when product release is linear with time. Fluorescence was measured on a Molecular Devices SPECTRAmax Gemini EM Dual-Scanning Microplate Spectrofluorimeter using λexcitation = 324 nm and λemission = 393 nm. Apparent Ki values were calculated from the following formulas:
| equation 1 |
| equation 2 |
where It is the total inhibitor concentration, Et is the total enzyme concentration, At is the total substrate concentration, vo is the activity in the absence of inhibitor, and KM is the Michaelis constant. In our assays the value of Et/Ki(app) does not exceed 100 so that the inhibitor is distributed in both free and bound forms, and Ki(app) can be calculated by fitting inhibition data to equation 1. Because the substrate concentration is less than KM/10, Ki(app) values are insignificantly different from true Ki values. In cases where weak inhibition occurred, Ki(app) values were calculated using vi = vo/(1 + It/ Ki(app)).
Results
Synthesis and Characterization of Triple-Helical Transition State Analogs
The type V collagen-derived sequence Gly-Pro-Pro-Gly439~Val440-Val-Gly-Glu-Gln, when incorporated into a triple-helical structure, is hydrolyzed efficiently by MMP-2 and MMP-9 but not by MMP-1, MMP-3, MMP-13, or MT1-MMP.34 The sequence was thus used as a template for the design of a potentially selective MMP inhibitor. In order to create the desired phosphinate transition state analog, a GlyΨ{PO2H-CH2}Val building block needed to be synthesized. A conjugate addition of 1-(N-(9-Fmoc)amino)-methyl phosphinic acid (5) to 2-isopropylacrylic acid allyl ester (6) was required to prepare the building block (Figure 3). N-Protected α-aminophosphinic acids are necessary intermediates in the synthesis of a variety of transition state analogs such as phosphinate, phosphonate, and phosphonamide dipeptides.23 The Fmoc protecting group is desirable for peptide synthesis, but, until recently, there was no facile process reported for its incorporation onto α-aminophosphinic acids. The previous methods using aqueous/organic solvent mixtures and classic Schotten-Baumann conditions are successful,62 but are not generally high yielding due to hydrolysis side reactions and solubility problems. An in situ silylation procedure popularized for Fmoc derivatization of L-amino acids63 was utilized for protection of α-aminophosphinic acid with an approximate yield of 80%.40
Mild silylation of Fmoc-aminophosphinic acid with TMS-Cl and DIEA gave the trivalent phosphinate, which reacted readily in an Arbusov-like reaction with allyl acrylate 664 to give the phosphinic acid 7. Finally, the phosphinate dipeptide analog was protected as the adamantyl ester by reaction with silver oxide and 1-adamantyl bromide, and the terminal allyl ester removed by CpRu(CH3CN)3PF6 catalysis to give the protected phosphinate dipeptide mimic 1 [(R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid]. The use of allyl protection for 2-isopropylacrylic acid, and removal of the allyl group in the presence of the Fmoc group, allowed for a more convenient preparation of the Fmoc-phosphinate dipeptide building block then reported previously.27,29,39,65 The adamantyl ester in compound 1 does not prematurely hydrolyze but it is readily removed under TFA cleavage conditions used in Fmoc solid-phase strategies.66,67
(R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl) propanoic acid was incorporated by solid-phase methods to create (Gly-Pro-Hyp)4-Gly-Pro-Pro-GlyΨ{PO2H-CH2}(R,S)Val-Val-Gly-Glu-Gln-Gly-Glu-Gln-Gly-Pro-Pro-(Gly-Pro-Hyp)4-NH2. A portion of the peptidyl-resin was lipidated on the N-terminus with hexanoic acid to create a peptide-amphiphile construct.
RP-HPLC analysis of the triple-helical peptide and peptide-amphiphile revealed two major peaks in each chromatogram (Figure 4). The material in the major peaks (f1-f4) was isolated and analyzed by RP-HPLC (Figure 5) and MALDI-TOF-MS. Peaks f1 and f2 had masses that corresponded to the desired triple-helical peptide, while f3 and f4 had masses that corresponded to the desired triple-helical peptide-amphiphile. Thus, it appeared that in each case that the diastereomers, where the Val analog in the P1′ subsite was either the S or R configuration, could be separated. Based on this data and relative inhibitory activities (see later discussion), we hypothesize that f1 and f3 contain the S configuration in the P1′ position (which is equivalent to an L-amino acid) and that f2 and f4 contain the R configuration in the P1′ position (which is equivalent to a D-amino acid). Prior studies on phosphinate peptide inhibitors of MMPs indicated that the earlier eluting component by RP-HPLC contains the S configuration in the P1′ position and is the more potent inhibitor.22,25,27,31
Figure 4.
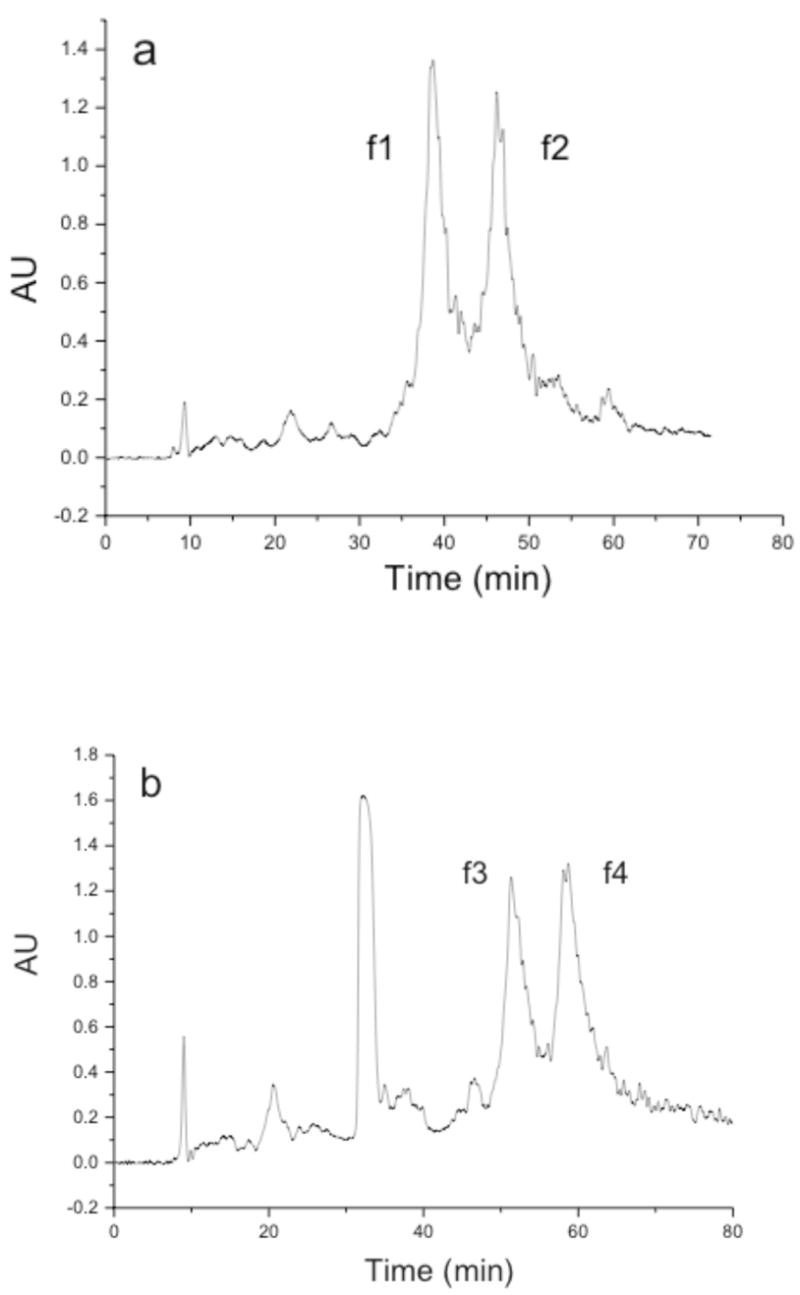
Semi-preparative RP-HPLC analysis of (a) phosphinate triple-helical peptide and (b) phosphinate triple-helical peptide-amphiphile. Semi-preparative RP-HPLC was performed using a C18 RP column, an elution gradient of 10–30% B in 80 min, and a flow rate of 10.0 mL/min. Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). Detection was at λ = 220 nm. Other conditions are given in Material and Methods.
Figure 5.
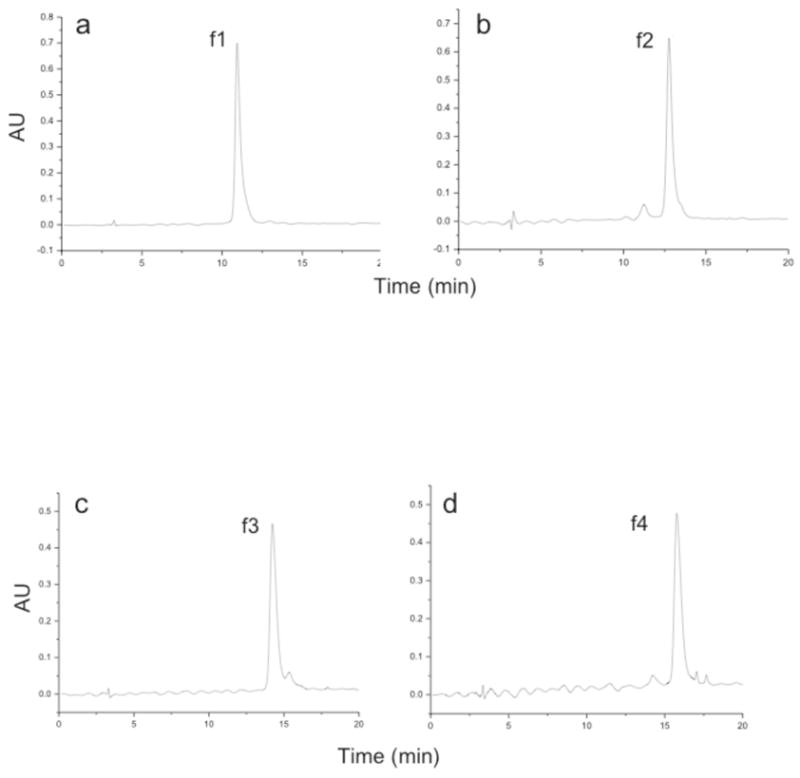
PDA RP-HPLC analysis of isolated (a) f1, (b) f2, (c) f3, and (d) f4 products (see designations in Figure 4). PDA RP-HPLC was performed using a C18 RP column, an elution gradient of 10–30% B in 20 min, and a flow rate of 1.0 mL/min. Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). Other conditions are given in Material and Methods.
CD spectra indicated weak triple-helices for f1 and f2 and more pronounced triple-helical structure for f3 and f4 (Figure 6). To examine the thermal stability of each construct, the molar ellipticity ([Θ]) at λ = 225 nm was monitored as a function of increasing temperature. All structures exhibited cooperative transitions, indicative of the melting of a triple-helix to a single-stranded structure (Figure 7). Melting temperatures (Tm values) were then determined for f1-f4. The f1 and f2 peptides had similar melting temperatures (Tm ~ 17.5 °C), as did the f3 and f4 peptide-amphiphiles (Tm ~ 25 °C). The Tm values for the phosphinate peptide-amphiphiles were considerably lower than that for the analogous peptide-amphiphile substrate (Tm = 49.5 °C).34 SEC analysis of f4 showed that, at 37 °C, f4 appears as a single species of near identical molecular weight as f2, but upon cooling to 10 °C both f4 and f2 shift to a trimeric species (data not shown). The trimeric species of f4 had virtually the identical retention time as the analogous, trimeric peptide-amphiphile substrate α1(V)436-450 THP.34 There were no aggregates of trimeric species observed.
Figure 6.
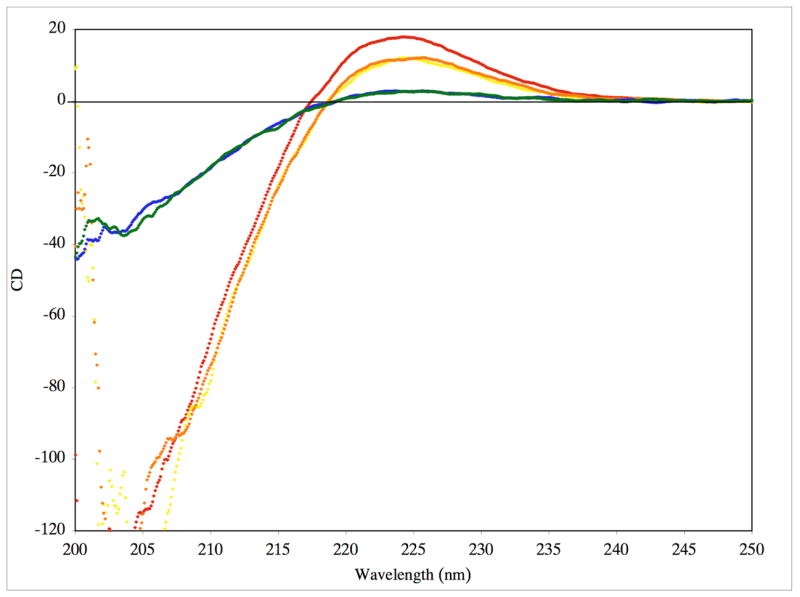
CD spectra of purified f1 (blue), f2 (green), f3 (yellow), f4 (orange), and the α1(V)436-450 THP substrate (red) in 0.25% (v/v) TSB buffer at a substrate concentration of 25 μM. Molar ellipticity ([θ]) values are in units of deg•cm2•dmol−1 × 10−3.
Figure 7.

Thermal transition curve for purified f1 (blue diamonds), f2 (green diamonds), f3 (yellow diamonds), f4 (orange diamonds), and the α1(V)436-450 THP substrate (red diamonds) in 0.25% (v/v) TSB buffer at a substrate concentration of 25 μM. Molar ellipticities ([θ]) were recorded at λ = 225 nm while the temperature was increased from 5 °C to 85 °C.
Inhibition of MMPs
The phosphinate peptide-amphiphiles (f3 and f4) were initially tested against MMP-2 (Figures 8 and 9; Table 1) and MMP-9 (Table 1). Due to the low melting temperatures of the potential inhibitors, Ki values were first determined at 10 °C. Both f3 and f4 were found to be very effective inhibitors of MMP-2 and MMP-9, with Ki values in the 4–6 and 2 nM ranges, respectively. When inhibition assays were repeated at 37 °C, Ki values increased for MMP-2 but not for MMP-9 (Table 1). Thus, triple-helical structure modulated inhibition of MMP-2 but not MMP-9. Inhibition of MMP-2 was then examined with peptides f1 and f2 at 25 °C, which corresponds to ~15–20% folded conformation (see Figure 7). The Ki values for f1 and f2 (Table 1) were worse than for the analogous peptide-amphiphiles f3 and f4 at 37 °C (which corresponds to ~15–20% of their folded conformation). Although the low melting temperatures of f1 and f2 precluded their study as inhibitors when primarily in triple-helical conformation, partially unfolded conditions demonstrated that the more stable triple-helices (f3 and f4) were better MMP-2 inhibitors than their less stable counterparts (f1 and f2).
Figure 8.

Inhibition of MMP-2 by f3 at varying temperatures. MMP-2 was incubated with varying concentrations of f3 at 10 (blue diamonds), 25 (pink squares), 30 (green triangles), or 37 °C (orange circles). Residual activity was monitored as indicated in Materials and Methods. Standard deviations are indicated with error bars.
Figure 9.

Inhibition of MMP-2 by f4 at varying temperatures. MMP-2 was incubated with varying concentrations of f3 at 10 (blue diamonds), 25 (pink squares), 30 (green triangles), or 37 °C (orange circles). Residual activity was monitored as indicated in Materials and Methods. Standard deviations are indicated with error bars.
Table 1.
Inhibition of MMP-2 and MMP-9.
| Enzyme | Inhibitor | Temperature
(°C) |
Ki(app) (nM) |
|---|---|---|---|
| MMP-2 | f3 | 10 | 4.14 ± 0.47 |
| “ | “ | 37 | 19.23 ± 0.64 |
| “ | f4 | 10 | 5.48 ± 0.00 |
| “ | “ | 37 | 138.32 ± 27.85 |
| “ | MMP inhibitor III | 10 | 3.17 ± 0.23 |
| “ | “ | 37 | 0.83 ± 0.03 |
| “ | f1 | 25 | 50.05 ± 9.56 |
| “ | f2 | 25 | 451.18 ± 89.48 |
| MMP-9 | f3 | 10 | 1.76 ± 0.05 |
| “ | “ | 37 | 1.29 ± 0.00 |
| “ | f4 | 10 | 2.20 ± 0.34 |
| “ | “ | 37 | 2.34 ± 0.23 |
At 10 and 37 °C, f3 was a better inhibitor than f4 for both MMP-2 and MMP-9. Interestingly, the increase in Ki value as a function of temperature was not the same for f3 and f4. For f3, the increase in temperature resulted in a 5-fold increase in Ki for MMP-2. For f4, the increase in temperature resulted in a 25 times increase in Ki for MMP-2. Removing the R configuration of the isopropyl side chain from a triple-helical context made f4 a considerably worse inhibitor than removing the S configuration from a triple-helical context (f3).
To determine if an increase in Ki as a function of temperature was general trend for inhibition of MMP-2, inhibition of MMP-2 by MMP inhibitor III was examined. At 10 °C, the Ki value for MMP-2 inhibition was 3 nM (Table 1). Increasing the temperature to 37 °C decreased the Ki to 0.8 nM (Table 1). Thus, for a small molecule inhibitor, an increase in temperature slightly increased the affinity towards MMP-2, most likely due to an enhancement of hydrophobic interactions. This further suggests that the decreased inhibition of MMP-2 by f3 and f4 as a function of increasing temperature is due to unfolding of the inhibitor triple-helical structure.
MMP-1, MMP-3, MMP-8, MMP-13, and MT1-MMP were tested for inhibition by f3 and f4. No inhibition of MMP-1, MMP-3, or MT1-MMP was observed up to a concentration of 25 μM for either f3 or f4 (data not shown). MMP-8 and MMP-13 were inhibited weakly, with Ki values in the range of 50 and 10 μM, respectively (data not shown).
Discussion
The present study has utilized a GlyΨ{PO2H-CH2}Val transition state analog to bind selectively at the S1-S1′ site of MMP-2 and MMP-9. MMP-2 has been validated as an anti-cancer drug target, while MMP-9 inhibition may be useful in treating early-stage cancers.2 An MMP-2 and MMP-9 selective substrate was used as a framework for a phosphinic acid-based inhibitor. Two variants of the phosphinate triple-helical peptide were produced, one in which the P1′ isopropyl side-chain is in the S configuration and one in which the side-chain is in the R configuration. The difference in triple-helical thermal stability between the S and R forms of the inhibitor is negligible. As shown previously, the use of stabilizing regions [such as (Gly-Pro-Hyp)n] on both the N- and C-termini can fold and order a central non-Gly-Xxx-Yyy region, minimizing relative thermal destabilization.68,69 Also, the presumed S diastereomer (natural “L-amino acid like” stereochemistry) is a slightly better inhibitor than the R diastereomer. This is consistent with prior studies on inhibition of MMPs by P1′ subsite S versus R phosphinic peptides.22,25,27,31
The triple-helical transition state analog had a Tm value 25–27°C lower than the triple-helical substrate it was modeled after (Figure 7). It is likely that the GlyΨ{PO2H-CH2}Val transition state analog destabilizes the triple-helix via charge repulsion and/or steric clashes. Alternatively, the H2O-bound tetrahedral intermediate may destabilize native collagen, which would explain why the MMP active site Zn2+ is required for triple-helix unwinding to proceed.70 It is possible that MMPs utilize H2O to destabilize the substrate, thus allowing for unwinding without an external source of energy (such as ATP). Future experiments will address the role of the tetrahedral intermediate in substrate unwinding.
The present THP inhibitors exhibit low nM Ki values for MMP-2 and MMP-9 while having little or no activity against collagenolytic MMPs (MMP-1, MMP-8, MMP-13, and MT1-MMP) nor MMP-3. This selectivity is important, as MMP-3 and MMP-8 have been identified as anti-targets for cancer therapeutics.2 It was previously noted that an MMP inhibitor should be selective over anti-target MMPs by ~3 log orders of difference in Ki values,12 which is satisfied in the present case. Prior work created a series of phosphonic acid pseudotripeptide inhibitors of MMP-2 and MMP-9 with Ki values in low nM range, but offered little selectivity towards MMP-8 and MT1-MMP.28 Similarly, peptidyl biphenylalkylphosphinates inhibited both MMP-2 and MMP-8 with IC50 values in the low μM range.32
Phosphinic acid inhibitors of MMPs have often utilized GlyΨ{PO2H-CH2}Leu transition state analogs. Ala-Gly-Pro-Leu-GlyΨ{PO2H-CH2}Leu-Tyr-Ala-Arg-Gly was found to inhibit MMP-9 with Ki values of 0.6 and 3.4 nM for the S and R diastereomers, respectively.27 β-Napthoyl-GlyΨ{PO2H-CH2}Leu-Trp-NHBzl had a Ki value of 10 nM for MMP-1.16 Selectivity amongst MMPs for these inhibitors was not reported. Reiter et al. developed a pseudotripeptide phosphinic acid MMP-1 inhibitor with an IC50 value of 60 nM; replacement of the isobutyl side-chain in the P1′ subsite with phenethyl or phenoxybutyl resulted in inhibitors more selective for MMP-13 than MMP-1 and with IC50 values of 14 and 30 nM, respectively, for MMP-13.26 These studies did not examine inhibitor selectivity for a variety of MMPs; an 85-fold selectivity was observed between MMP-13 and MMP-3 for one inhibitor. A latter study from the same group identified a phosphinic acid MMP-13 inhibitor that had an IC50 of 4.5 nM for MMP-13 and offered 270–360-fold selectivity over MMP-1 and MMP-3.71 However, low nM IC50 values were also observed for MMP-2, MMP-8, and MMP-12, and thus the inhibitor was only partially selective.71
A solid-phase combinatorial chemistry approach, which incorporated the GlyΨ{PO2H-CH2}Leu mimic with amino acid substitutions in the P4, P3, P2, P2′, P3′, P4′, and P5′ subsites, facilitated the development of inhibitors selective for MMP-12.23,24 The best MMP-12 inhibitors had Ki values in the 6–25 nM range, and exhibited up to 2000-fold selectivity for MMP-12 over MMP-9 and 250–300-fold selectivity for MMP-12 over MMP-13 and MT1-MMP.23,24 A selective MMP-12 inhibitor was also obtained by screening peptide libraries of the general structure p-Br-Ph-(PO2-CH2)-Xaa′-Yaa′-Zaa′.25 p-Br-Ph-(PO2-CH2)-(3-[3-chlorobiphenyl-4-yl]isoxazol-5-yl)methyl-Glu-Glu was found to inhibit MMP-12 with a Ki value of 0.19 nM and offer a 200–100,000-fold selectivity over other MMPs.25 Combinatorial approaches were also utilized to create isoxazole-containing phosphinic peptides with low nM Ki values towards MMP-13 and MT1-MMP.31 Finally, a series of phosphinic peptides of general structure Z-Ala/PheΨ{PO2H-CH2}Xaa′-Yaa′-NH2 or R-PheΨ{PO2H-CH2}Xaa′-Trp-NH2 were utilized to develop high affinity MMP-11 inhibitors (low and sub-nM Ki values).28
There are no prior examples of phosphinic peptides offering the MMP-2/MMP-9 selectivity observed here, nor are there documented triple-helical phosphinic peptides. Triple-helical peptide inhibitors of MMPs have been constructed previously using a hydroxamate as the ZBG. In one study, (Pro-Pro-Gly)6-NHOH and (Pro-Pro-Gly)12-NHOH were found to inhibit MMP-2 with IC50 values of 160 and 90 μM, respectively.72 These constructs showed similarly weak inhibition of MMP-1 and MMP-3.72 The weak inhibition may have been the result of interaction with only the S subsites of the enzyme and/or poor alignment of the ZBG into the active site. It should be noted that the melting temperatures of these inhibitors were not evaluated. Inhibition assays were performed at 37 °C; since (Pro-Pro-Gly)10 has a Tm value of 25 °C,73 the inhibitors were most likely not triple-helical unless the hydroxamate group conferred considerable stability to the system. A second study utilized a solid-phase C-terminal branching protocol74 to incorporate a hydroxamate-containing peptidomimetic onto the N-terminus of (Gly-Pro-Hyp)4-Gly-Pro-Pro-Gly-Ser-Ser.75 Inhibition of MMP-1 was achieved with an IC50 value of ~9 nM. Constructs in which fewer Gly-Pro-Hyp repeats were present, and thus presumably had less or no triple-helicity, exhibited IC50 values of ~100–500 nM. Thus, MMP-1 inhibition was dependent upon triple-helical structure. While interesting, this particular inhibitor would offer little selectivity amongst collagenolytic MMPs.
Phosphinate triple-helical MMP inhibitors have several advantages over other inhibitor constructs. These analogs allow incorporation of specificity elements for both the S and S′ subsites of the enzyme. Although binding to the non-primed region of the active site is generally weaker than the primed site to prevent product inhibition,14 it does add sequence diversity and potential selectivity. The triple-helical structure allows for interaction with both the active site and secondary substrate binding sites (exosites).2,3,12 Inhibitors that recognize exosites can enhance selectivity.76–78 The triple-helical conformation is also less susceptible to proteolysis than peptides and other folded proteins.79 On its own, a phosphinate seems to reduce proteolytic processing of peptides in vivo,26,80 and thus its inclusion in a triple-helical context may create an inhibitor that is reasonably stable to metabolic processes. The phosphinate triple-helical inhibitor is also faster binding then the natural MMP inhibitors, TIMPs.
The present inhibitors are too thermally unstable for in vivo applications, but can be modified by inclusion of additional Gly-Pro-Hyp repeats and/or longer N-terminal alkyl chains or replacement of Hyp residues by 4-fluoroproline to improve triple-helical stability.35,73,81–83 For other collagenolytic enzymes, diversity of the phosphinate pseudodipeptide may be efficiently achieved by a recently described three-component condensation reaction.65 It has been proposed that the negative charge of the phosphoryl group may prevent peptide membrane permeability and uptake by liver cells and promote peptide interaction with non-specific proteases in the plasma.12,80 However, a phosphinic MMP inhibitor has been to shown to decrease necrosis and apoptosis in ischemic rat livers.30 In addition, monopril (fosinopril sodium tablets; L-proline, 4-cyclohexyl-1-[[[2-methyl-1-(1-oxopropoxy) propoxyl](4-phenylbutyl) phosphinyl]acetyl]-, sodium salt, trans-) is an orally bioavailable phosphinate tripeptide analog inhibitor of angiotensin converting enzyme.84,85 However, because of the solid-phase methodology employed here, one can potentially utilize new, higher affinity ZBGs7,86 to replace the phosphinate if needed.
The present MMP-2/MMP-9 selective transition state analog inhibitor has several potential applications. First, it can be utilized for metalloproteinase structural studies and to evaluate exosite interactions. Second, it can allow for the mechanistic study of collagenolysis. Third, it can be evaluated for inhibition of experimental metastasis. Finally, it could serve as a molecular probe and a lead compound for drug development.
Supplementary Material
1H and 13C NMR spectra for compounds 1, 2, 6, 7, and 8, 31P NMR spectra for compounds 1, 7, and 8, and the complete reference 26. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge Dr. Hideaki Nagase for supplying MMP-1, MMP-2, and MMP-3 and Dr. Christopher Overall for supplying MMP-8. This work was supported by the National Institutes of Health (CA 98799 to GBF/RPH), a Glenn/American Federation for Aging Research (AFAR) Scholarship (to JLL-F), an ASBMB Travel Award (to JLL-F), and an NSF IGERT (CHE-9987603) Fellowship (to JKW).
Footnotes
Abbreviations used: MMP, matrix metalloproteinase; Ac2O, acetic anhydride; APMA, 4-aminophenyl mercuric acetate; CD, circular dichroism; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; DIEA, N,N-diisopropylethylamine; Dnp, 2,4-dinitrophenyl; ECM, extracellular matrix; Fmoc, 9-fluorenylmethoxycarbonyl; HOBt, 1-hydroxybenzotriazole; Hyp, 4-hydroxy-L-proline; LiHMDS, lithium hexamethyldisilazide; MALDI-TOF-MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; Mca, (7-methoxycoumarin-4-yl)acetyl; MSS, musculoskeletal syndrome; PyAOP, (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate; SEC, size-exclusion chromatography; TBTU, O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate; TFA, trifluoroacetic acid; THP, triple-helical peptide; TIMP, tissue inhibitor of metalloproteinase; TMS-Cl, trimethylsilyl chloride; ZBG, zinc binding group.
References
- 1.Turk B. Nat Rev Drug Discovery. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 2.Overall CM, Kleifeld O. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 3.Overall CM, Lopez-Otin C. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 4.Labrosse KR, Liener IE, Hargrave PA. Anal Biochem. 1976;70:218–223. doi: 10.1016/s0378-5173(83)90103-5. [DOI] [PubMed] [Google Scholar]
- 5.Whittaker M, Floyd CD, Brown P, Gearing AJH. Chem Rev. 1999;99:2735–2776. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 6.Whittaker M, Ayscough A. Celltransmissions. 2001;17:3–14. [Google Scholar]
- 7.Rao BG. Curr Pharm Design. 2005;11:295–322. doi: 10.2174/1381612053382115. [DOI] [PubMed] [Google Scholar]
- 8.Beckett RP. Exp Opin Ther Patents. 1996;6:1305–1315. [Google Scholar]
- 9.Beckett RP, Whittaker M. Exp Opin Ther Patents. 1998;8:259–282. [Google Scholar]
- 10.Lauer-Fields JL, Fields GB. Exp Opin Ther Patents. 2000;10:1873–1884. [Google Scholar]
- 11.Cuniasse P, Devel L, Makaritis A, Beau F, Georgiadis D, Matziari M, Yiotakis A, Dive V. Biochimie. 2005;87:393–402. doi: 10.1016/j.biochi.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 12.Overall CM, Kleifeld O. Br J Cancer. 2006;94:941–946. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Proc Natl Acad Sci USA. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babine RE, Bender SL. Chem Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 15.Kortylewicz ZP, Galardy RE. J Med Chem. 1990;33:263–273. doi: 10.1021/jm00163a044. [DOI] [PubMed] [Google Scholar]
- 16.Galardy RE, Grobelny D, Kortylewicz ZP, Poncz L. Matrix Suppl. 1992;1:259–262. [PubMed] [Google Scholar]
- 17.Izquierdo-Martin M, Stein RL. Bioorg Med Chem. 1993;1:19–26. doi: 10.1016/s0968-0896(00)82099-4. [DOI] [PubMed] [Google Scholar]
- 18.Bartlett PA, Marlowe CK, Giannousis PP, Hanson JE. Cold Spring Harbor Symp Quant Biol. 1987;52:83–90. doi: 10.1101/sqb.1987.052.01.012. [DOI] [PubMed] [Google Scholar]
- 19.Mookhtiar KA, Marlowe CK, Bartlett PA, Van Wart HE. Biochemistry. 1987;26:1962–1965. doi: 10.1021/bi00381a026. [DOI] [PubMed] [Google Scholar]
- 20.D’Alessio S, Gallina C, Gavuzzo E, Giordano C, Gorini B, Mazza F, Paglialunga Paradisi M, Panini G, Pochetti G, Sella A. Bioorg Med Chem Lett. 1999;7:389–394. doi: 10.1016/s0968-0896(98)00243-0. [DOI] [PubMed] [Google Scholar]
- 21.Gavuzzo E, Pochetti G, Mazza F, Gallina C, Gorini B, D’Alessio S, Pieper M, Tschesche H, Tucker PA. J Med Chem. 2000;43:3377–3385. doi: 10.1021/jm9909589. [DOI] [PubMed] [Google Scholar]
- 22.Gall AL, Ruff M, Kannan R, Cuniasse P, Yiotakis A, Dive V, Rio MC, Basset P, Moras D. J Mol Biol. 2001;307:577–586. doi: 10.1006/jmbi.2001.4493. [DOI] [PubMed] [Google Scholar]
- 23.Buchardt J, Schiodt CB, Krog-Jensen C, Delaissé JM, Foged NT, Meldal M. J Comb Chem. 2000;2:624–638. doi: 10.1021/cc000031q. [DOI] [PubMed] [Google Scholar]
- 24.Schiodt CB, Buchardt J, Terp GE, Christensen U, Brink M, Larsen YB, Meldal M, Foged NT. Current Med Chem. 2001;8:967–976. doi: 10.2174/0929867013372670. [DOI] [PubMed] [Google Scholar]
- 25.Devel L, Rogakos V, David A, Makaritis A, Beau F, Cuniasse P, Yiotakis A, Dive V. J Biol Chem. 2006;281:11152–11160. doi: 10.1074/jbc.M600222200. [DOI] [PubMed] [Google Scholar]
- 26.Reiter LA, et al. Bioorg Med Chem Lett. 1999;9:127–132. doi: 10.1016/s0960-894x(98)00729-x. [DOI] [PubMed] [Google Scholar]
- 27.Buchardt J, Ferreras M, Krog-Jensen C, Delaisse JM, Foged NT, Meldal M. Chem Eur J. 1999;5:2877–2884. [Google Scholar]
- 28.Vassiliou S, Mucha A, Cuniasse P, Georgiadis D, Lucet-Levannier K, Beau F, Kannan R, Murphy G, Knauper V, Rio MC, Basset P, Yiotakis A, Dive V. J Med Chem. 1999;42:2610–2620. doi: 10.1021/jm9900164. [DOI] [PubMed] [Google Scholar]
- 29.Matziari M, Georgiadis D, Dive V, Yiotakis A. Org Lett. 2001;3:659–662. doi: 10.1021/ol0069103. [DOI] [PubMed] [Google Scholar]
- 30.Cursio R, Mari B, Louis K, Rostagno P, Saint-Paul MC, Giudicelli J, Bottero V, Anglard P, Yiotakis A, Dive V, Gugenheim J, Auberger P. FASEB J. 2002;16:93–95. doi: 10.1096/fj.01-0279fje. [DOI] [PubMed] [Google Scholar]
- 31.Makaritis A, Georgiadis D, Dive V, Yiotakis A. Chem Eur J. 2003;9:2079–2094. doi: 10.1002/chem.200204456. [DOI] [PubMed] [Google Scholar]
- 32.Bianchini G, Aschi M, Cavicchio G, Crucianelli M, Preziuso S, Gallina C, Nastari A, Gavuzzo E, Mazza F. Bioorg Med Chem. 2005;13:4740–4749. doi: 10.1016/j.bmc.2005.04.079. [DOI] [PubMed] [Google Scholar]
- 33.Cheng F, Zhang R, Luo X, Shen J, Li X, Gu J, Zhu W, Shen J, Sagi I, Ji R, Chen K, Jiang H. J Phys Chem B. 2002;106:4552–4559. [Google Scholar]
- 34.Lauer-Fields JL, Sritharan T, Stack MS, Nagase H, Fields GB. J Biol Chem. 2003;278:18140–18145. doi: 10.1074/jbc.M211330200. [DOI] [PubMed] [Google Scholar]
- 35.Yu YC, Tirrell M, Fields GB. J Am Chem Soc. 1998;120:9979–9987. [Google Scholar]
- 36.Minond D, Lauer-Fields JL, Nagase H, Fields GB. Biochemistry. 2004;43:11474–11481. doi: 10.1021/bi048938i. [DOI] [PubMed] [Google Scholar]
- 37.Nagase H, Fields CG, Fields GB. J Biol Chem. 1994;269:20952–20957. [PubMed] [Google Scholar]
- 38.Neumann U, Kubota H, Frei K, Ganu V, Leppert D. Anal Biochem. 2004;328:166–173. doi: 10.1016/j.ab.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 39.Georgiadis D, Matziari M, Yiotakis A. Tetrahedron. 2001;57:3471–3478. [Google Scholar]
- 40.Li S, Whitehead JK, Hammer RP. J Org Chem. 2007;72:3116–3118. doi: 10.1021/jo070266p. [DOI] [PubMed] [Google Scholar]
- 41.Jiao XY, Verbruggen C, Borloo M, Bollaert W, Groot AD, Dommisse R, Haemers A. Synthesis. 1994:23–24. [Google Scholar]
- 42.Lee HS, Park JS, Kim BM, Gellman SH. J Org Chem. 2003;68:1575–1578. doi: 10.1021/jo026738b. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka S, Saburi H, Ishibashi Y, Kitamura M. Org Lett. 2004;6:1873–1875. doi: 10.1021/ol0493397. [DOI] [PubMed] [Google Scholar]
- 44.Lauer-Fields JL, Broder T, Sritharan T, Nagase H, Fields GB. Biochemistry. 2001;40:5795–5803. doi: 10.1021/bi0101190. [DOI] [PubMed] [Google Scholar]
- 45.Fields GB, Lauer-Fields JL, Liu R-q, Barany G. In: Synthetic Peptides: A User’s Guide. 2. Grant GA, editor. W.H. Freeman & Co.; New York: 2001. pp. 93–219. [Google Scholar]
- 46.Yu YC, Berndt P, Tirrell M, Fields GB. J Am Chem Soc. 1996;118:12515–12520. [Google Scholar]
- 47.Solé NA, Barany G. J Org Chem. 1992;57:5399–5403. [Google Scholar]
- 48.Beck K, Chan VC, Shenoy N, Kirkpatrick A, Ramshaw JAM, Brodsky B. Proc Natl Acad Sci USA. 2000;97:4273–4278. doi: 10.1073/pnas.070050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Persikov AV, Ramshaw JAM, Brodsky B. Biopolymers (Peptide Sci) 2000;55:436–450. doi: 10.1002/1097-0282(2000)55:6<436::AID-BIP1019>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 50.Persikov AV, Ramshaw JAM, Kirkpatrick A, Brodsky B. Biochemistry. 2000;39:14960–14967. doi: 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- 51.Persikov AV, Xu Y, Brodsky B. Protein Sci. 2004;13:893–902. doi: 10.1110/ps.03501704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung L, Shimokawa K, Dinakarpandian D, Grams F, Fields GB, Nagase H. J Biol Chem. 2000;275:29610–29617. doi: 10.1074/jbc.M004039200. [DOI] [PubMed] [Google Scholar]
- 53.Lauer-Fields JL, Nagase H, Fields GB. J Biomolecular Techniques. 2004;15:305–316. [PMC free article] [PubMed] [Google Scholar]
- 54.Itoh Y, Binner S, Nagase H. Biochem J. 1995;308:645–651. doi: 10.1042/bj3080645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pelman GR, Morrison CJ, Overall CM. J Biol Chem. 2005;280:2370–2377. doi: 10.1074/jbc.M409603200. [DOI] [PubMed] [Google Scholar]
- 56.Huang W, Suzuki K, Nagase H, Arumugam S, Van Doren S, Brew K. FEBS Lett. 1996;384:155–161. doi: 10.1016/0014-5793(96)00304-3. [DOI] [PubMed] [Google Scholar]
- 57.Hurst DR, Schwartz MA, Ghaffari MA, Jin Y, Tschesche H, Fields GB, Sang QXA. Biochem J. 2004;377:775–779. doi: 10.1042/BJ20031067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki K, Kan CC, Huang W, Gehring MR, Brew K, Nagase H. Biol Chem. 1998;379:185–191. doi: 10.1515/bchm.1998.379.2.185. [DOI] [PubMed] [Google Scholar]
- 59.Knight CG, Willenbrock F, Murphy G. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 60.Copeland RA. In: Evaluation of Enzyme Inhibitors in Drug Discovery. Copeland RA, editor. John Wiley & Sons, Inc.; Hoboken, NJ: 2005. pp. 178–213. [Google Scholar]
- 61.Yiallouros I, Vassiliou S, Yiotakis A, Zwilling R, Stöcker W. Biochem J. 1998;331:375–379. doi: 10.1042/bj3310375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dumy P, Escale R, Girard JP, Parello J, Vidal JP. Synthesis. 1992:1226–1228. [Google Scholar]
- 63.Bolin DR. Int J Peptide Protein Res. 1989;33:353–359. [Google Scholar]
- 64.Huntington KM, Yi T, Wei Y, Pei D. Biochemistry. 2000;39:4543–4551. doi: 10.1021/bi992452y. [DOI] [PubMed] [Google Scholar]
- 65.Matziari M, Yiotakis A. Org Lett. 2005;7:4049–4052. doi: 10.1021/ol051622y. [DOI] [PubMed] [Google Scholar]
- 66.Yiotakis A, Vassiliou S, Jiracek J, Dive V. J Org Chem. 1996;61:6601–6605. doi: 10.1021/jo9603439. [DOI] [PubMed] [Google Scholar]
- 67.Georgiadis D, Dive V, Yiotakis A. J Org Chem. 2001;66:6604–6610. doi: 10.1021/jo0156363. [DOI] [PubMed] [Google Scholar]
- 68.Mohs A, Popiel M, Li Y, Baum J, Brodsky B. J Biol Chem. 2006;281:17197–17202. doi: 10.1074/jbc.M601763200. [DOI] [PubMed] [Google Scholar]
- 69.Hyde TJ, Bryan M, Brodsky B, Baum J. J Biol Chem. 2006;281:36937–36943. doi: 10.1074/jbc.M605135200. [DOI] [PubMed] [Google Scholar]
- 70.Chung L, Dinakarpandian D, Yoshida N, Lauer-Fields JL, Fields GB, Visse R, Nagase H. EMBO J. 2004;23:3020–3030. doi: 10.1038/sj.emboj.7600318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reiter LA, Mitchell PG, Martinelli GJ, Lopresti-Morrow LL, Yocum SA, Eskra JD. Bioorg Med Chem Lett. 2003;13:2331–2336. doi: 10.1016/s0960-894x(03)00413-x. [DOI] [PubMed] [Google Scholar]
- 72.Jani M, Tordai H, Trexler M, Banyai L, Patthy L. Biochimie. 2005;87:385–392. doi: 10.1016/j.biochi.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 73.Fields GB, Prockop DJ. Biopolymers (Peptide Sci) 1996;40:345–357. doi: 10.1002/(SICI)1097-0282(1996)40:4%3C345::AID-BIP1%3E3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 74.Fields CG, Lovdahl CM, Miles AJ, Matthias-Hagen VL, Fields GB. Biopolymers. 1993;33:1695–1707. doi: 10.1002/bip.360331107. [DOI] [PubMed] [Google Scholar]
- 75.Chen JM, Yeh LA. Frontiers Biosci. 2004;9:2788–2795. doi: 10.2741/1436. [DOI] [PubMed] [Google Scholar]
- 76.Dennis MS, Eigenbrot C, Skelton NJ, Ultsch MH, Santell L, Dwyer MA, O’Connell MP, Lazarus RA. Nature. 2000;404:465–470. doi: 10.1038/35006574. [DOI] [PubMed] [Google Scholar]
- 77.Overall CM. In: Methods in Molecular Biology 151: Matrix Metalloproteinase Protocols. Clark IM, editor. Humana Press; Totowa, NJ: 2001. pp. 79–120. [PubMed] [Google Scholar]
- 78.Roberge M, Santell L, Dennis MS, Eigenbrot C, Dwyer MA, Lazarus RA. Biochemistry. 2001;40:9522–9531. doi: 10.1021/bi010592d. [DOI] [PubMed] [Google Scholar]
- 79.Lauer-Fields JL, Juska D, Fields GB. Biopolymers (Peptide Sci) 2002;66:19–32. doi: 10.1002/bip.10201. [DOI] [PubMed] [Google Scholar]
- 80.Dive V, Lucet-Levannier K, Georgiadis D, Cotton J, Vassiliou S, Cuniasse P, Yiotakis A. Biochem Soc Trans. 2000;28:455–460. [PubMed] [Google Scholar]
- 81.Holmgren SK, Taylor KM, Bretscher LE, Raines RT. Nature. 1998;392:666–667. doi: 10.1038/33573. [DOI] [PubMed] [Google Scholar]
- 82.Holmgren SK, Bretscher LE, Taylor KM, Raines RT. Chem Biol. 1999;6:63–70. doi: 10.1016/S1074-5521(99)80003-9. [DOI] [PubMed] [Google Scholar]
- 83.Malkar NB, Lauer-Fields JL, Borgia JA, Fields GB. Biochemistry. 2002;41:6054–6064. doi: 10.1021/bi012071w. [DOI] [PubMed] [Google Scholar]
- 84.Murdoch D, McTavish D. Drugs. 1992;43:123–140. doi: 10.2165/00003495-199243010-00009. [DOI] [PubMed] [Google Scholar]
- 85.Weber MA. J Cardiovasc Pharmacol. 1992;20 Suppl 10:S7–S12. doi: 10.1097/00005344-199200101-00003. [DOI] [PubMed] [Google Scholar]
- 86.Puerta DT, Lewis JA, Cohen SM. J Am Chem Soc. 2004;126:8388–8389. doi: 10.1021/ja0485513. [DOI] [PubMed] [Google Scholar]
- 87.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:159–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H and 13C NMR spectra for compounds 1, 2, 6, 7, and 8, 31P NMR spectra for compounds 1, 7, and 8, and the complete reference 26. This material is available free of charge via the Internet at http://pubs.acs.org.