

Abstract
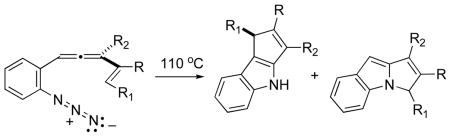
Thermolysis of 2-(allenyl)phenylazides leads to a cascade cyclization sequence furnishing both C(2)-C(3) and N-C(2) cyclopentennelated indoles.
Organic synthesis via diyl cyclization has had an episodic history, with surges in activity quickly following discovery of novel diradical generating reactions.1 One of the more notable advances to emerge from this area of chemistry stemmed from the observation that N2 extrusion from 4-methylene-1,2-diazenes led efficiently to triplet trimethylenemethane (TMM) diyls, species that have served a central role in numerous cyclization- and cycloaddition-based approaches to cyclopentanoid natural products.2 One little studied modification of the TMM diyl, azatrimethylenemethane (ATMM), can be constructed, at least in principle, by simply exchanging one methylene fragment for a nitrogen (cf. 4, Scheme 1). By analogy with TMM chemistry, the ATMM variant might provide ready access to polycyclic cyclopentanoid alkaloid frameworks. The development of ATMM chemistry has laged far behind its all-carbon cousin, but early studies that hinted at its existence3 and more recent suggestions of its intermediacy in azide-allene cyclization cascades4 (Scheme 1) may yet elevate this species to a position of prominence in lkaloid synthesis.
Scheme 1.

Intramolecular azide-allene cycloaddition cascades for nitrogen heterocycle synthesis.
C(2)-C(3) annelated indoles represent one potential target class for this chemistry, and given the success of the 1→5 conversion, wherein only cyclization through the imine species 4b was observed, it seemed plausible to expect that incorporation of an aryl residue in the allene-azide tether (cf. 6) would lead by analogy to the C(2)-C(3) cyclopentennelated indole products 7. As described in this report, this goal was achieved in practice. However, the unanticipated intervention of subtle electronic (?) effects, presumably as a consequence of the electronic connectivity supplied by the intervening aryl ring, served to divert some of the reactive intermediate(s) down alternative channels, leading to N-C(2) annelated products as well. A description of the scope of this process for cyclopentennelated indole synthesis with both alkenyl- and aryl-substituted 2-(allenyl)phenylazides is detailed below.
The syntheses of suitable 2-(allenyl)phenylazides of the type 6 were accomplished by straightforward chemistry using Konno’s procedure for palladium-mediated aryl(alkenyl)zinc addition to propargylic acetates 9,5 or the cuprate-based alternative procedure of Palenzuela,6 Table 1. The azide function survived exposure to these organometallic reagents with no detectable decomposition, but attempts to add (Bu3Sn)CuSPh to 9a met with concomitant azide reduction to furnish an amide product following O-to-N acetyl transfer. The (alkenyl)allenyl azides 6a–6i were stable, isolable, and characterizable compounds, but the aryl-substituted analogues 6j–6m were sensitive to chromatography and could not be isolated in pure form. Therefore, these species were routinely carried on to the thermolysis procedure as crude materials.
Table 1.
2-Allenylphenylazide cyclization substrate synthesis
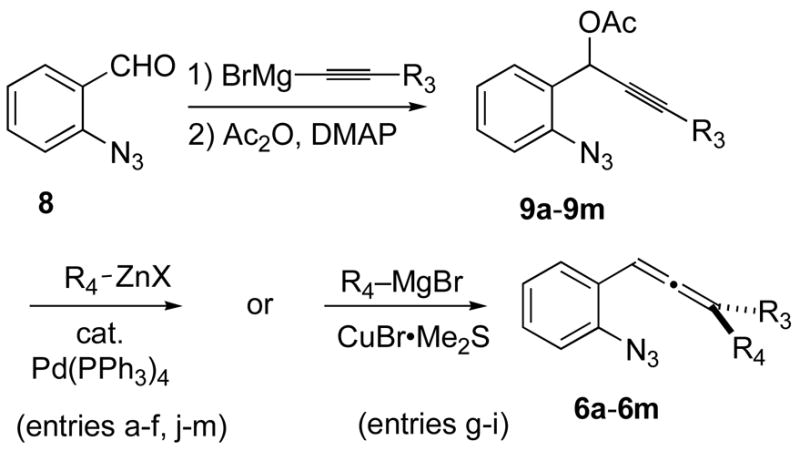 | ||||
|---|---|---|---|---|
| entry | R3 | R4 | yield 9 (%)a | yield 6 (%)a |
| a | CH3 | H2C=CH– | 48 | 58 |
| b | CH2OTBS | H2C=CH– | 91 | 55 |
| c | CH2CH2OTBS | H2C=CH– | 73 | 57 |
| d | t-Bu | H2C=CH– | 78 | 63 |
| e | CH3 | H2C=C(Ph)– | 48 | 67 |
| f | CH3 | H2C=C(CH3)– | 48 | 59 |
| g | PhCH=CH– | CH3 | 47 | 34 |
| h | 1-cyclohexenyl | CH3 | 31 | 37 |
| i | 1-cyclopentenyl | CH3 | 49 | 30 |
| j | CH3 | Ph | 48 | b |
| k | CH3 | p-(CH3O)C6 H4 | 48 | b |
| l | CH3 | m-(CH3O)C6 H4 | 48 | b |
| m | CH3 | m-FC6H4 | 48 | b |
Yield for chromatographically pure, characterized material.
These allenes were too reactive to isolate and characterize, and so they were carried into the thermolyses as crude materials.
The thermolysis/cyclization studies commenced with the prototype substrate 6a featuring unelaborated methyl and vinyl units at the allene terminus, Table 2. Heating a 0.1 M solution of 6a in toluene led cleanly to the formation of two new species whose gross spectral data were suggestive of the presence of tricyclic ring systems in both cases. Chromatographic purification of the crude thermosylate furnished samples of both compounds, and their spectroscopic data were entirely consistent with those expected for the desired C(2)-C(3) annelated indole 7a as well as the unanticipated pyrrole 11a. The more slowly eluting compound 7a exhibited the characteristic N–H resonance of an indole (δ 8.03 (s)) as well as signals for an alkene-bound CH3 (δ 2.22, d, J = 1.6 Hz) and for a single alkenyl proton (δ 6.22, m). The faster eluting species displayed spectral data that were strikingly similar to those reported for the characterized reference compound des methyl 11a,7 allowing ready assignment of 11a as a pyrrole. X-ray crystallographic analysis of 11a later confirmed the structural assignment (see Supporting Information). Examination of the crude reaction product by 1H NMR prior to purification indictated the presence of only 7 and 10; pyrrole 11 was not formed until exposure of the crude thermolysate to SiO2. The ratio of isolated 7 to isolated 11 occasionally was at variance with the 7:10 ratio as it appeared in the 1H NMR spectrum of the crude thermolysate, presumably as a consequence of differential chromatographic stability, and so the latter value is reported as well. In addition, 1H NMR-based examination of the crude thermolysate immediately after reaction allowed identification of signals consistent with the indole species 10, but these signals decayed over the course of a few hours as the spectroscopic signature for the pyrrole 11 grew in. In only a few cases (6g, 6i) was this N-C(2) cyclopentannelated indole isolable, but even in those instances, isomerization into the pyrrole followed after a few more hours at room temperature. The next three examples (6b–6d) demonstrated that both silyl ethers (6b–6c) and steric bulk (6d) at the R2 position are tolerated in the transformation with little impact on yield. Interestingly, comparing entries a-d reveals that the ratio of C(3)-to-N cyclization is responsive to the size of the R2 substituent, with the bulkiest entry (6d, R2 = t-Bu) leading to the greatest selectivity for the desired C(2)-C(3) cyclization regioisomer 7.
Table 2.
2-(Allenyl)phenylazide cyclization results
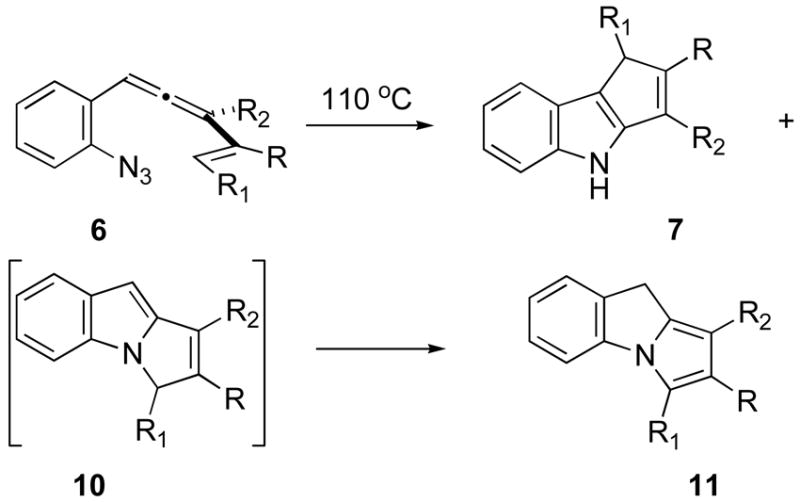 | ||||||
|---|---|---|---|---|---|---|
| entry | R | R1 | R2 | yield 7(%)a | yield11(%)a | Ratio7:10b |
| a | H | H | CH3 | 40 | 56 | 1:1.2 |
| b | H | H | CH2OTBS | 22 | 29 | 1.2:1 |
| c | H | H | CH2CH2OTBS | 52 | 43 | 1.5:1 |
| d | H | H | t-Bu | 57 | 20 | 2.7:1 |
| e | Ph | H | CH3 | 40 | 30 | 1:1.2 |
| f | CH3 | H | CH3 | 36 | 36 | 1:1.3 |
| g | H | Ph | CH3 | - - | 35 (10g) | 1:1.1 |
| h | –(CH2)4– | CH3 | 36 | 51 | 1:1.4 | |
| i | –(CH2)3– | CH3 | - - | 40 (10i) | 1:1.5 | |
Yield for chromatographically pure, characterized material.
Ratio by 1H NMR analysis of the crude thermoslyate.
The next two entries (6e, 6f) test the effect of a substituent at the internal (R) position of the alkene. For both substrates, the reaction proceeds similarly to the simpler R = H cases to afford nearly equal mixtures of the indole 7e/7f and pyrrole 11c/11f products. The R substituent resides at a position that apparently exerts little steric or electronic influence on the course of the reaction, as both the R=CH3 and the R=Ph cases proceed to product(s) in very similar yields/selectivities. The Ph-bearing substrate 6g introduces at the alkene terminus a group that might confer both electronically favorable (i.e., radical stabilizing, cf. 4) and sterically unfavorable characteristics, and the tradeoff between these possibly opposing effects appears to favor the latter. For the first time, the C(2)-C(3) cyclized indole product did not survive attempted chromatographic purification, and the regioisomeric N-C(2) indole 10g was the only identifiable species isolated. However, the indole 7g was detected in the crude thermolysate admixed with 10g (1:1.1 ratio, Table 2). Hydrogenation of 10g (Scheme 2) furnished the cis disposed indole product 12g whose structure was secured by single crystal X-ray analysis (see Supporting Information). The final two entries, 6h and 6i, probed the capability of this cascade cyclization sequence to deliver tetracyclic material. The cyclohexenyl case 6h proceeded uneventfully to deliver a slightly biased mixture of the pyrrole 11h and the indole product 7h in excellent overall yield. The cyclopentyl lower homologue 6i, in contrast, yielded only a moderate amount of the N-C(2) cyclized indole product 10i as the only tetracyclic material to survive chromatography. The corresponding C(3)-cyclized material 7i was observed in the crude thermolysate’s 1H NMR spectrum, at the ratio reported in Table 2, but it decomposed upon attempted purification.
Scheme 2.
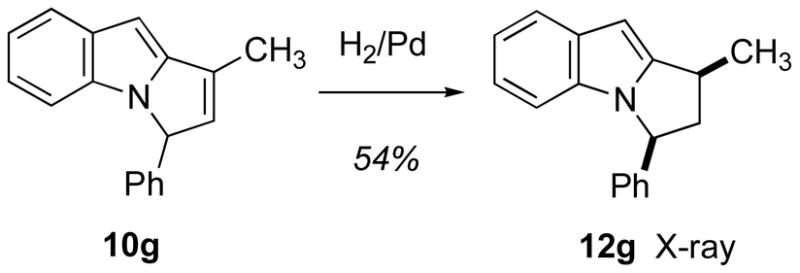
Formation of a derivative characterizable by X-ray crystallography.
Thermolyses of the aryl-substituted 2-(allenyl)phenylazides 6j–6m did not provide tetracyclic material, Scheme 3. In each instance, the methylidene-containing products 14j–14m were isolated in moderate yields. Presumably, a diyl intermediate represented by 13 underwent H-atom transfer, possibly via an intramolecular pathway as shown, to yield the product alkene. These results stand as another point of departure from the saturated tether series 1, where pendant aryl rings (R⏜R1 = aryl ring) did participate in diyl cyclizations to afford benzannelated products 5 (R⏜R1 = aryl ring via alkene isomerization). The failure of the analogous species 13 to cyclize remains a mystery, but a rationale might be tied to the different nature of the diyl intermediate in the unsaturated series vis á vis the saturated series 1, as discussed below.
Scheme 3.
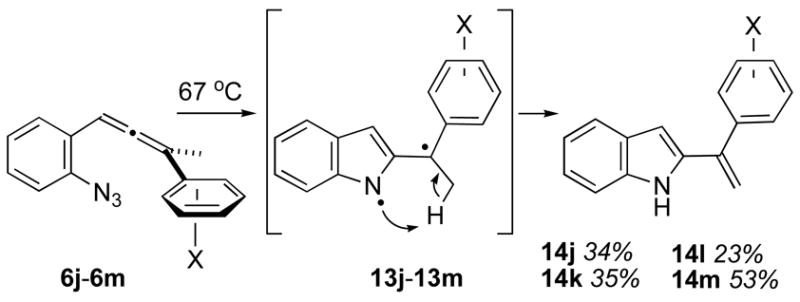
Thermolysis of phenyl-substituted 2-(allenyl)phenylazides.
Given the similarities and the differences between the reactions of the diyls derived from 1 and from 6, it is appropriate to consider the conceivable roles that the different candidate diradicals might play in product formation. Figure 1 details the plausible diyl (and related) options. Singlet σ/π orthogonal diyl 15a is the likely first-formed species immediately following N2 extrusion. This presumably short-lived diyl could cyclize via initial C–C bond rotation and then C–N closure to give products of the type 10, or it could undergo singlet-to-triplet interconversion to give a new diyl that could not cyclize directly and therefore might have a long enough lifetime to realize other chemistry. As a alternative to direct cyclization, singlet or triplet 15a could undergo an electronic reorganization that is tantamount to placing one electron from the nitrogen’s π orbital into its half-occupied sp2 orbital, giving the orthogonal π/π diyl 15b (singlet or triplet).
Figure 1.

Possible intermediates along the cyclization pathway.
This species provides the means to access the C–C bonded product 7 via (singlet) diyl closure following bond rotation. In fact, this rotation (indicated by the blue arrow in 15b) may hold the key to understanding the regiochemical divergence of diyl closure.8 Rotation of singlet 15b in the clockwise direction (observing down the C(2)→C(·) bond) would lead to the C–C bond formation product 7, whereas the opposite direction of rotation would bring N(·) and C(·)R1 together to form 10. To the extent that the C(3)–H/R2 steric interaction shown in 15b becomes energetically penalizing upon counterclockwise rotation, larger R2 groups arguably should differentially favor the alternative clockwise motion, leading to a preference for C–C bond formation (→7). This expectation is borne out by the limited results with 6a–6d.
Triplet 15b, on the other hand, could participate in similar divergent rotations to furnish the planar (E or Z) ATMM intermediate 15c. The E/Z preference could be tied to the same steric features (avoiding C(3)–H/R2 steric interactions) discussed for singlet 15b. Upon intersystem crossing, the now singlet 15c could cyclize (electrocyclize?) with either C–C bond formation from the E species or C–N bond formation from the Z isomer. Finally, the singlet version of 15c might best be represented as the closed-shell (E/Z) alkylidene indolinene 15d, a species whose (electro)cyclization manifold is identical to singlet ATMM 15c. At this point, it is not possible to sort between these options and provide a comprehensive description of this 2-(allenyl)phenylazide cyclization cascade. Work in that direction will continue.
Supplementary Material
Experimental procedures and characterization data for 6a–6m, 7a–7f, 7h, 10g, 10i, 11a–11f, 11h, and 14j–14m. In addition, details for the X-ray crystal structure determination of 11a and 12g are included. This material is is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The National Institutes of Health, General Medical Sciences division, is gratefully acknowledged (GM72572), as is support from NSF CHE 0131112 for the X-ray crystallographic facility.
References
- 1.(a) Little RD. Chem Rev. 1986;86:875–884. [Google Scholar]; (b) Moore H. Advances in Strain in Organic Chemistry. 1995;4:81–162. [Google Scholar]; (c) Little RD. Chem Rev. 1996;96:93–114. doi: 10.1021/cr950017d. [DOI] [PubMed] [Google Scholar]; (d) Grissom JW, Gunawardena GU, Klingberg D, Huang D. Tetrahedron. 1996;52:6453–6518. [Google Scholar]; (e) Feldman KS, Mareska DA. J Org Chem. 1999;64:5650–5660. doi: 10.1021/jo9907538. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dowd PA. Acc Chem Res. 1972;5:242–248. [Google Scholar]; (b) Berson JA. In: Diradicals. Borden WT, editor. Wiley-VCH; New York: 1982. pp. 151–194. [Google Scholar]; (c) Allan AK, Carroll GL, Little RD. Eur J Chem. 1998:1–12. [Google Scholar]
- 3.(a) Bleiholder RF, Shechter H. J Am Chem Soc. 1968;90:2131–2137. [Google Scholar]; (b) Bingham EM, Gilbert JC. J Org Chem. 1975;40:224–228. [Google Scholar]; (c) Quast H, Weise Vélez CA. Angew Chem Int Ed Engl. 1978;17:213–214. [Google Scholar]; (d) Quast H, Fub A, Heublein A. Angew Chem Int Ed Engl. 1980;19:49–50. [Google Scholar]; (e) Barraclough D, Moorhouse NP, Onwuyali EI, Scheinmann F, Hursthouse MB, Galas AMR. J Chem Res (S) 1984:102–103. [Google Scholar]; (f) Quast H, Meichsner G. Chem Ber. 1987;120:1049–1058. [Google Scholar]; (g) Quast H, Fub A, Heublein A, Jakobi H, Seiferling B. Chem Ber. 1991;124:2545–2554. [Google Scholar]; (h) Barker SJ, Storr RC. J Chem Soc, Perkin Trans 1. 1990:485–488. [Google Scholar]
- 4.Feldman KS, Iyer MR. J Am Chem Soc. 2005;127:4590–4591. doi: 10.1021/ja050757w. [DOI] [PubMed] [Google Scholar]
- 5.Konno T, Tanikawa M, Ishihara T, Yamanaka H. Collect Czech Chem Commun. 2002;67:1421–1435. [Google Scholar]
- 6.Regás D, Afonso MM, Rodríguez ML, Palenzuela JA. J Org Chem. 2003;68:7845–7852. doi: 10.1021/jo034480z. [DOI] [PubMed] [Google Scholar]
- 7.Kashulin IA, Nifant’ev IE. J Org Chem. 2004;69:5476–5479. doi: 10.1021/jo049504w. [DOI] [PubMed] [Google Scholar]
- 8.Shields TC, Billups WE, Lepley AR. J Am Chem Soc. 1968;90:4749–4751.Roth WR, Schmidt T. Tetrahedron Lett. 1971:3639–3642.Kende AS, Riecki EE. J Am Chem Soc. 1972;94:1397–1399.Billups WE, Leavell KH, Lewis ES, Vanderpool S. J Am Chem Soc. 1973;95:8096–8102.Gilbert JC, Higley DP. Tetrahedron Lett. 1973:2075–2078.Pikulin S, Berson JA. J Am Chem Soc. 1985;107:8274–8276.Shook CA, Romberger ML, Jung SH, Xiao M, Sherbine JP, Zhang B, Lin FT, Cohen T. J Am Chem Soc. 1993;115:10754–10773.Davidson ER, Gajewski JJ, Shook CA, Cohen T. J Am Chem Soc. 1995;33:8495–8501.Direct (IR) detection of triplet-4-methylene-2-pentene-1,5-diyl: Maier G, Senger S. J Am Chem Soc. 1997;119:5852–5861.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for 6a–6m, 7a–7f, 7h, 10g, 10i, 11a–11f, 11h, and 14j–14m. In addition, details for the X-ray crystal structure determination of 11a and 12g are included. This material is is available free of charge via the Internet at http://pubs.acs.org.