

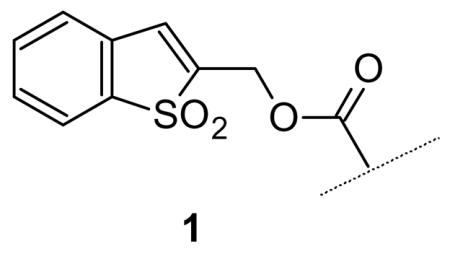
Recently1 we described an amino-protecting group, the Bsmoc residue 1, which is deblocked under conditions of Michael addition by means of a primary or secondary amine, most often piperidine. This process of addition to an α, β-unsaturated system contrasts with the standard β-elimination process involved in the deblocking of the Fmoc2 and related base-sensitive amino protecting groups. Because of its unique deblocking chemistry the Bsmoc group provides a number of advantages over Fmoc chemistry in terms of speed and completeness of deblocking3, selectivity, etc. In addition a remarkable reactivity difference has emerged between Bsmoc and Fmoc amino acids with the Bsmoc derivatives being more reactive in acylation reactions than their Fmoc counterparts to an extent which becomes greater and greater as the steric requirements between the coupling components increase.4 These various advantages of Bsmoc chemistry justify its continued evaluation and the amelioration of some limited but exact deficiencies of the Bsmoc system itself.
 |
One limitation is that four Bsmoc amino acids (and eight Bsmoc amino acid fluorides) could be obtained only as difficultly handled oils or foamy materials. Substitutes were sought among the higher molecular weight benzo derivatives. This led to consideration of the three monobenzo derivatives 2–4. With regard to the key deblocking step systems 2 and 3 are expected to be directly comparable to the Bsmoc residue whereas system 4 was predicted to be somewhat less reactive due to possible steric retardation5 of attack by piperidine at the β-carbon atom of the α, β-unsaturated sulfone unit due to the peri hydrogen atom shown.
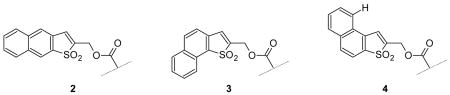 |
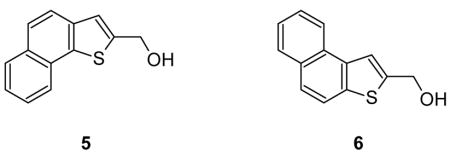
Linear system 2 was not examined in view of the sparseness of the chemical literature on potential precursors for this system which contrasts with the rich chemistry recorded for the two bent systems related to 3 and 4. The two sulfide alcohols 5 and 6, promising intermediates for the synthesis of protective systems 3 and 4 respectively, have been reported6 and require only oxidation to the corresponding sulfone alcohols for conversion to the two Bsmoc-related protectants.
 |
The two sulfide alcohols 5 and 6 were prepared by adaptations of the methods previously described. The α-derivative 5 was obtained readily from inexpensive α-tetralone 7 according to Scheme 1.7 Oxidation via the commercially available magnesium salt of perphthalic acid or use of sodium perborate8 gave the sulfone alcohol 11.
Scheme 1.

Although the isomeric alcohol 17 could be obtained similarly from β-tetralone, this compound is currently rather expensive and we examined alternative sources for the β-analog of key ester 10. The method chosen involved a standard rhodanine route to mercapto carboxylic acid 14 which underwent iodine-catalysed cyclization to acid 15 according to the method of Campaigne and Cline9 (Scheme 2). An attempt to reduce acid 15 directly to alcohol 6 by means of lithium aluminum hydride in ether failed, possibly due to its very low solubility. The acid was therefore esterified to give 16 which was reduced readily and the remainder of the synthesis proceeded normally.
Scheme 2.

With the two sulfone alcohols now readily available, each was converted to the appropriate chloroformates 18 and 19 by reaction with phosgene or triphosgene. Protected amino acid derivatives were made from the chloroformates by the Bolin technique10 which involves prior treatment of the amino acid with trimethylsilyl chloride. Table 1 collects the various amino acids made. For full characterization data see the Supporting Information section. As noted, for both series, all were obtained in solid form including those for proline, tryptophan, serine t-butyl ether and aspartic acid t-butyl ester for which in the Bsmoc series this was not possible. In some cases the acids exhibited wide melting point ranges although even then no difficulty was encountered in obtaining analytically pure material by recrystallization from DCM/hexane. Only in the cases of asparagine and glutamine was it necessary to use column chromatography to obtain pure samples. The acids were all converted to their acid fluoride derivatives via standard cyanuric fluoride methodology11 and again all proved to be obtainable in solid form. Using either the acid fluoride as coupling species or coupling to H-Ala-OMe via N-HATU/DIEA it was shown that no significant loss of configuration accompanied the synthesis and use of α-Nsmoc α-phenylglycine (see Supporting Information Section). A similar result had previously been reported for Bsmoc-Phg-OH (see ref. 1, p. 4333).
Table 1.
| α–Derivative
|
β-Derivative
|
|||
|---|---|---|---|---|
| Amino Acid | mp (°C) | Yield (%) | mp(°C)c | Yield (%) |
| Gly | 191–193 | 90 | 76–151 | 89.2 |
| Ala | 120–125 | 86.2 | 89–109 | 82.7 |
| Val | 138–139 | 75.4 | 89–115 | 73.3 |
| Leu | 75–96 | 84.6 | 78–105 | 83.8 |
| Ile | 148–150 | 88.5 | 76–123 | 84.0 |
| Pro | 192–195 | 77.6 | 91–146 | 80.0 |
| Phe | 81–135 | 89.2 | 81–158 | 91.8 |
| Met | 55–117 | 80.8 | 74–143 | 73.5 |
| Trp | 107–160 | 85.6 | 119–204 | 85.7 |
| Ser(tBu) | 76–135 | 82.0 | 79–139 | 78.5 |
| Thr(tBu) | 85–107 | 82.8 | 88–119 | 86.3 |
| Tyr(tBu) | 134–136 | 81.8 | 96–162 | 87.2 |
| Asp(O-tBu) | 67–142 | 84.0 | 85–151 | 83.0 |
| Glu(O-tBu) | 69–128 | 46.1 | 77–164 | 52.0 |
| Lys(BOC) | 83–125 | 72.0 | 86–149 | 68.5 |
| Phg | 93–146 | 78.8 | 98–159 | 80.3 |
| Aib | 181–182 | 84.4 | 184–187 | 77.8 |
| Asn(Trt) | 170–200 | 49.7 | 77–164 | 47.8 |
| Gln(Trt) | 120–160 | 56.8 | 238–240 | 56.3 |
All compounds were characterized on the basis of elemental analysis (± 0.3%, C, H, N) and consistent IR and 1H NMR spectral data. Complete data are presented in the supporting information section.
All compounds were recrystallized from DCM/hexane.
In many cases wide melting point ranges were observed. In spite of the mp behavior analytically pure samples were obtained in all cases.
 |
In order to compare deblocking rates for the two new protecting systems with those for the Bsmoc residue we used the same 1H NMR technique used previously involving model carbamate substrate 20a. Treatment of these derivatives of p-chloroaniline (PCA) with 2 eqv. of piperidine in CDCl3-DMSO-d6 showed that under the conditions and at the concentrations used, which were the same in all three cases, rough halftimes for release of p-chloroaniline were 2.0, 2.4 and 7.4 min. in the cases of the α-Nsmoc, Bsmoc and β-Nsmoc systems respectively. Similar halftimes were measured based on the disappearance of the starting urethanes by following the decrease for the CH2O peaks near δ 5.2 in the three cases. The lowered reactivity of the β-Nsmoc system is in accord with the peri-hindrance mentioned above. The enhanced reactivity of the α-Nsmoc system relative to the Bsmoc case may be due to the inductive effect of the added benzo substituent since steric factors are much the same for these two systems. The course of the reaction for the α-Nsmoc case is outlined in Scheme 3.12 The decrease in the intensity of the NMR peak at δ 5.24 due to the CH2O group of 20b is paralleled by increase of the olefinic peaks and the methine proton position at δ 6.3, 6.45 and 5.08, respectively, signaling the formation of initial piperidine adduct 21. At the same time aromatic peaks at δ 6.59 and 6.98 appear due to liberated PCA. Adduct 21 then undergoes piperidine-catalysed rearrangement to give stable adduct 22 with its CH2N peak appearing at δ 3.35.
Scheme 3.

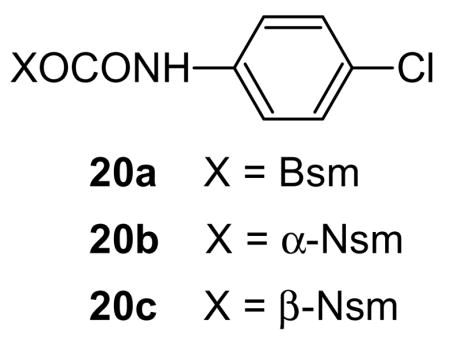 |
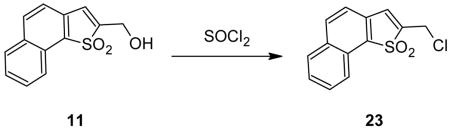
Comparable reactions occur with the β-Nsmoc analog 20c although complete liberation of PCA is slower as noted above. It proved to be difficult to isolate pure samples of either adduct 21 or 22 from the urethane-based reaction mixtures but when chloride 23, available from alcohol 11 by treatment with thionyl chloride, was substituted for the urethane in the reaction with piperidine it was possible to isolate stable adduct 22 in pure form. When piperidine was substituted by 2-methylpiperidine both intermediate and final adducts could be isolated in analytically pure form. With 20% 2- methylpiperidine in DMF HPLC analysis showed that after 1 min the initial adduct (2- methylpiperidine analog of 21) was present in a yield of 93.9%. After 3 h rearrangement had occurred to give the 2-Me analog of 22 in 97.3 % yield. With only 1 eqv. of 2- methylpiperidine only the initial adduct could be observed and it was still the only isomer visible even after 24 h.
 |
As a test for the use of the new α- and β-Nsmoc protectants in solid phase peptide synthesis two model peptides leucine enkephalin (H-Tyr-Gly-Gly-Phe-Leu-NH2)13 and ACP65–74 (H-Val-Gln-Ala-Ala-Ile-Asp-Tyr-Ile-Asn-Gly- NH2)14 were assembled. For the α-Nsmoc system in the case of leucine enkephalin, with a PAL-PEG-PS resin, optimum deblocking times varied from 20 sec to 5 min depending on the concentration of the piperidine reagent (Table 2). For the β-Nsmoc system, in line with its reduced sensitivity toward bases, best results were observed when 20% piperidine was used with a deblocking time of 5 min (96.9% purity of the crude pentapeptide). In contrast, with the 5%/5 min system which gave excellent results for the α-Nsmoc system (crude purity 97%) the crude pentapeptide purity was only 67.8%. For assembly of the ACP decapeptide using N-HATU in place of N-HBTU comparable results were obtained for both protectants provided that 20% piperidine was used with a deblocking time of 1 min for α-Nsmoc (crude purity 87.5 %) and 8 min for β-Nsmoc (crude purity 83.5%).
Table 2.
Synthesis of Leucine Enkephalin via α-Nsmoc Amino Acidsa
| Concentration (%) of piperidine in DMF | deblocking time (min) | crude purity by HPLC analysis (%) |
|---|---|---|
| 2 | 1 | 85.9 |
| 5 | 20 sec | 87.7 |
| 5 | 1 | 96.6 |
| 5 | 3 | 94.2 |
| 5 | 5 | 97.0 |
| 20 | 20 sec | 100 |
Three eqv. of the amino acid with 3 eqv. of N-HBTU and 6 eqv. of DIEA and a coupling time of 30 min.
In previous studies1,4a it was shown that one could selectively deblock the Fmoc group from dipeptide ester 24 using a hindered amine such as t-butyl amine with only minimal attack at the Bsm residue. Indeed treatment of 24 with 30% t-butyl amine in acetonitrile for 10 min followed by in situ coupling via Fmoc-Gly-OH in the presence of N-HATU (60 min) gave after work-up and column chromatography tripeptide 25 in a yield of 72.4%. Application of the same methodology to the β-Nsm analog of 24 gave the corresponding β-Nsm analog of 25 in a yield of only 31.8%. This suggests that factors other than steric hindrance toward attack at the α, β-unsaturated sulfone units of these systems are important in determining the relative sensitivity for attack at the Fmoc vs. the Bsm or β-Nsm ester functions.
| Fmoc-Phe-Leu-OBsm | Fmoc-Gly-Phy-OBsm |
| 24 | 25 |
Because of their greater availability and the closer correspondence of their deblocking sensitivity to the simple Bsmoc derivatives, the α-Nsmoc amino acid derivatives are recommended over the β-isomers as substitutes for the few Bsmoc amino acids which are not available in solid form. In addition the full complement of α-Nsmoc derivatives described here are potentially of special utility in view of their even greater sensitivity toward deblocking compared to the already highly sensitive Bsmoc analogs.
Experimental Section
Ethyl Naphtho[1,2-b]thiophene-2-carboxylate (10)
N-Bromosuccinimide (15 g, 84.2 mmol) was added in portions with stirring to a boiling solution of ethyl 4,5-dihydronaphtho[1,2-b]thiophene-2-carboxylate7(17.0 g, 65.8 mmol) and dibenzoyl peroxide (20 mg) in dry CCl4(100 ml) while being irradiated with a 200-W sun lamp. The hydrogen bromide which was formed was removed in a stream of nitrogen. When the addition was complete, the mixture was refluxed for a furthur 45 min. (NMR was used to check for reaction completion and if the reaction was not finished a small additional amount of N-bromosuccinimide was added, the refluxing continued and NMR checked again). The mixture was cooled, filtered and evaporation of the filtrate gave the crude ester which was recrystallized from EtOAc/hexane to give 14.5 g (87.5%) of the pure ester as off-white crystals, mp 86–88°C (lit.7 mp 86–87°C); IR (KBr): 1696 cm1(CO); 1 H NMR (CDCl3): δ 1.40–1.47 (t, 3), 4.38–4.49 (q, 2) 7.25–8.37 (m, 7).
Ethyl Naphtho[2,1-b]thiophene-2-carboxylate (16) from 2-tetralone
Ethyl mercaptoacetate (15.69 g, 14.31 ml, 130.8 mmol) was added to a cooled, stirred solution of sodium (3.77 g, 0.16 g-atom) in dry ethanol (58 mL). Crude 2-bromo-3,4-dihydronaphthalene-1-carboxaldehyde (30 g) prepared according to the method of Gilchrist and Summersell15 (see also the Supporting Information Section) was added portion wise during 30 min at 0–5°C. The mixture was stirred overnight at room temperature, refluxed for 30 min and poured into 300 mL of water. The ester was filtered, decolorized with activated charcoal and recrystallized from ethanol to give 21.5 g (64% based on 1H NMR analysis of the crude starting aldehyde showing that 25 g. of pure material was present) of the ester, mp 50–52°C; IR (KBr): 1708 cm−1 (C=O); 1H NMR (CDCl3): δ 1.35–1.42 (t, 3) 2.99–3.00 (s, 4), 4.30–4.41 (q, 2); 7.13–8.03 (m, 5). Without further purification the dihydro compound was treated with NBS as described above for ethyl naphtho[1,2-b]thiophene-2-carboxylate. The pure ester was obtained as an off-white solid in 85% yield, mp 83–84°C; IR (KBr): 1717 cm−1 (C=O); 1H NMR (CDCl3): δ 1.41–1.48 (t, 3), 4.39–4.50 (q, 2), 7.26–8.75 (m, 7). Anal. Calcd for C15H12O2S: C, 70.31; H, 4.68. Found: C, 70.36; H, 4.68.
2-(Hydroxymethyl)naphtho[1,2-b]thiophene (5)
A solution of 14.0 g (54.7 mmol) of ethyl naphtho[1,2-b]thiophene-2-carboxylate in 200 ml of dry THF/ether (1:1) was added at room temperature under N2 to a suspension of 3.11 g (82 mmol) of lithium aluminum hydride in 60 ml of dry THF/ether (1:1). After refluxing for 3.5 hours, the reaction mixture was cooled and carefully quenched by adding 50 ml of cold water and enough 20% HCl (~200 ml) to dissolve the inorganic salts. The aqueous layer was separated and extracted with ethyl acetate (3 × 300 ml). The organic layers were washed with water (3 × 200 ml) and saturated NaHCO3 (1 × 200 ml), dried over MgSO4, filtered and the solvent evaporated in vacuo. The residue was recrystallized from benzene/hexane to give 10.2 g (87.2%) of the alcohol; mp 85–86 (lit.6 mp 89–91°C); IR (KBr): 3252 cm−1(OH); 1H NMR (CDCl3): δ 2.10 (s, 1), 4.99 (s, 2) 7.32(s, 1), 7.30 (s, 1), 7.43 (m, 2), 7.54 (m, 2), 7.91 (d, 1), 8.1 (d, 1).
2-(Hydroxymethyl)naphtho[2,1-b]thiophene (6)
The reaction was carried out as described above for 2-(hydroxymethyl)naphtho[1,2-b]thiophene. There was obtained 9.5 g (87.5%) of the alcohol, mp 105–106°C (lit.6 mp 106°C); IR (KBr): 3220 cm−1(OH); 1 H NMR (CDCl3): δ 2.07 (s, 1), 4.99 (s, 2), 7.24–8.25 (m, 7).
1,1-Dioxo-2-(hydroxymethyl)naphtho[1,2-b]thiophene (11)
(a) via MMPP oxidation
To a stirred solution of 10 g (46.7 mmol) of the sulfide alcohol in 120 ml of methanol at 0°C, was added portionwise 34.7 g (70 mmol) of monoperoxyphthalic acid, magnesium salt hexahydrate (Aldrich Chemical Co.) with 30 ml of water also added slowly over the same period. The reaction mixture was stirred overnight at room temperature. The white precipitate was filtered and the solvent was removed in vacuo. The residue was dissolved in 400 ml of ethyl acetate and the solution washed with saturated NaHCO3, saturated NaCl, water (3 × 75 ml each), dried over MgSO4, filtered and the solvent removed in vacuo. The resulting off-white solid was recrystallized from hot ethyl acetate/hexane to give 9.2 g (80.1%) of the sulfone alcohol, mp 169–170°C; IR (KBr): 1152, 1292 (SO), 3522 (OH) cm−1; 1H NMR (CDCl3 + TFA) δ 4.91 (s, 2) 7.30 (s,1), 7.4 (d, 1), 7.63 (m, 1), 7.71 (m, 1), 7.91 (d, 1), 8.1 (d, 1), 8.23 (d, 1). Anal Calcd for C13H10O3S: C, 63.41; H, 4.06, S, 13.01. Found: C, 63.38; H, 4.11; S, 12.93.
(b) via sodium perborate oxidation
To a stirred solution of 26.5 (0.13 mol) of the sulfide alcohol in glacial acetic acid (500 ml) at 45–50°C was added portionwise during 20 min 95.4 g (0.62 mol) of sodium perborate tetrahydrate. Stirring was continued at this temperature until TLC analysis (EtOAc-hexane (3/2 v/v)) indicated completion of the reaction (5–6 h). The acetic acid was removed in vacuo and the residue stirred with 150 ml of water. The crude alcohol which separated was filtered, washed with water, dried in air and recrystallized from absolute ethanol to give 22.5 g (73.6%) of the pure sulfone alcohol, mp 169–170°C; 1H NMR (CDCl3): δ 2.35 (s, 1); 4.79 (s, 2); 7.17 (t, 1), 7.37–8.29 (m, 6). In the case of the sample of alcohol derived by MMPP oxidation the hydroxyl proton was not observed in the NMR spectrum, presumably because TFA had been added to the CDCl3 solvent.
3,3-Dioxo-2-(hydroxymethyl)naphtho[2,1-b]thiophene (17)
The alcohol was obtained in the same manner as described above for the α-isomer in 81.3% yield (8.4 g) mp 127–129°C; IR (KBr): 1130, 1279 (SO2) 3508 (OH) cm−1; 1H NMR (CDCl3 + 1 drop TFA): δ 4.96 (s, 2), δ 7.26–8.00 (m, 7). Anal. Calcd for C13H10O3S: C, 63.41; H, 4.06. Found 63.53; H, 4.05
Ethyl Naphtho[2,1-b]thiophene-2-carboxylate (16) from the corresponding carboxylic acid
To a stirred solution of 40 g (175.4 mmol) of naphtho[2,1-b]thiophene-2-carboxylic acid prepared as described previously 9 (full details in supporting information section) in 600 ml of absolute ethanol was added dropwise 45 ml of concentrated H2SO4 and the reaction mixture was refluxed overnight. Ethanol was evaporated at reduced pressure and the residue was dissolved in 1200 ml of EtOAc. The organic solution was washed with water, saturated NaHCO3, saturated NaCl and water (3 × 300 mL each). After drying over MgSO4 evaporation of ethyl acetate and recrystallization of the residue from EtOAc/hexane gave 35.2 g (78.4%) of the pure ester for which the mp, IR and 1H NMR characterization data were the same as described above for the sample prepared from β-tetralone. The ester obtained in this way was also reduced to its sulfide alcohol and the sulfide alcohol oxidized to the sulfone alcohol by the same methods described above. The physical and spectral data collected on these samples of the sulfide and sulfone alcohols also agreed with the data given above and in references 6 and 9.
1,1-Dioxonaphtho[1,2-b]thiophene-2-methyl Chloroformate (α-Nsmoc-Cl) (18)
(a) via phosgene: CAUTION!
Working with highly toxic gaseous phosgene requires a good hood, special precautions and an experienced operator. It is highly recommended that the safer reagent triphosgene be used as a substitute (see part (b) below). To a stirred solution of 10 g (40.6 mmol) of 1,1-dioxo-2-(hydroxymethyl) naphtho[1,2-b]thiophene in 150 ml of dry DCM and 50 ml of dry THF at −78°C was added in one portion 15 ml (21.0 g; 212.5 mmol) of phosgene which had been condensed into a graduated cylinder. The solution was allowed to come to room temperature and stirred overnight. Excess phosgene and solvent were removed under reduced pressure with the aid of a water aspirator, the process being repeated using an additional 200 ml of DCM and then 200 ml of ether. Recrystallization of the residue from DCM/hexane gave 11.3 g (90.5%) of the chloroformate as off-white crystals, mp 134–135°C; IR (KBr): 1151, 1295 (SO2), 1766 (CO) cm−1; 1H NMR (CDCl3): δ 7.30(s, 1), 7.41 (d, 1), 7.61 (m, 1), 7.69 (m, 1), 7.90 (d, 1), 8.0 (d, 1) 8.25 (d, 1). Anal. Calcd for C14H9ClO4S: C, 54.46; H, 2.93; S, 10.38; Cl, 11.48. Found C, 54.70; H. 2.85; S, 10.46; Cl, 11.53.
(b) via triphosgene
A mixture of the alcohol (10.0 g, 40.7 mmol), triphosgene (6.0 g, 20.2 mmol) and triethylamine hydrochloride (0.244 g, 1.77 mmol) in 70 ml of dry epoxide-free THF was heated to 40°C (outside oil bath temperature) for 3 h. A test by infrared examination showed that after 1 h some alcohol was still present, but after 3 h, alcohol was no longer visible. The solvent was removed in vacuo with a rotary evaporator to give 10.4 g (82.8%) of the α-Nsmoc-Cl after recrystallization from dry ether. Triphosgene was used similarly to prepare β-Nsmoc-Cl (85.6%) and Bsmoc-Cl (88.6%).
3,3-Dioxonaphtho[2,1-b]thiophene-2-methyl Chloroformate (β-Nsmoc-Cl) (19)
The reaction was carried out as described in (a) above for the α-analog to give 11.7 g (93.8%) of the chloroformate as cream-colored crystals, mp 135–136°C; IR (KBr): 1152, 1298 (SO2), 1785 (CO) cm−1; 1H NMR (CDCl3) δ 5.37 (d, 2), 7.26–8.09 (m, 7). Anal. Calcd for C14H9O4ClS: C, 54.46; H, 2.93; S, 10.38. Found: C, 54.47; H, 3.02; S, 10.38.
1,1-Dioxonaphtho[1,2-b]thiophene-2-methyl N-Succinimidyl Carbonate (α-Nsmoc-OSu)
N-Hydroxysuccinimidyl DCHA salt16 [3.39 g, 11.6 mmol] was added in small portions to a stirred solution of α-Nsmoc-Cl (3.57 g, 11.6 mmol) in 100 ml of dry DCM. The reaction mixture was stirred overnight at room temperature after which the separated precipitate was filtered and washed with DCM. The filtrate was washed with 10% aqueous citric acid, saturated NaHCO3, saturated NaCl and water (30 ml each), dried over MgSO4 and the solvent removed in vacuo. The resulting off-white solid was recrystallized from EtOAc/hexane to give 2.5 g (55.8%) of the ester as white crystals, mp 188–190°C; IR (KBr): 1152, 1303 (SO2), 1740 (C=O, imide), 1815 (OCOO) cm−1; 1H NMR (CDCl3 + TFA) δ 2.99 (s, 4), 5.4 (s, 2), 7.45 (d, 2), 7.71 (m, 1), 7.82 (m, 1), 7.96 (d, 1), 8.15 (d, 1), 8.22 (d, 1). Anal. Calcd for C18H13NO7S: C, 55.82; H, 3.35; N, 3.61. Found: C, 55.63; H, 3.29; N, 3.50.
1,1-Dioxonaphtho[1,2-b]thiophene-2-methyl N-p-Chlorophenyl Carbamate (α-Nsmoc-PCA) (20 b)
A solution of 1 g (4.06 mmol) of 1,1-dioxo-2-(hydroxymethyl) naptho[1,2-b]thiophene and 0.73 g (4.8 mmol) of p-chlorophenyl isocyanate in 20 ml of benzene was refluxed for 24 h. The precipitate was filtered, washed with benzene and recystallized from EtOAc/hexane to give 0.98 g (60.5%) of the urethane as off-white crystals, mp 212–214°C; IR (KBr): 1143, 1282 (SO2), 1738 (CO), (urethane) cm−1; 1H NMR (CDCl3 + DMSO-d6) δ 5.24 (s, 2), 7.22–8.20 (m, 11), 9.97 (bs, 1). Anal. Calcd for C20H14 ClO4S: C, 60.12; H, 3.50; N, 3.50. Found: 59.98; H, 3.43; N, 3.52
3,3-Dioxonaphtho[2,1-b]thiophene-2-methyl N-p-Chlorophenyl Carbamate (β-Nsmoc-PCA) (20c)
The preparation was carried out as described above for the isomeric alcohol. The urethane was obtained in a yield of 61.7% as off-white crystals, mp 181–183°C; IR (KBr): 1151, 1295 (SO2), 1701 (CO2, urethane) cm−1; 1H NMR (CDCl3 + DMSO-d6) δ 5.27 (s, 2), 7.21–8.33 (m, 11), 9.94 (bs, 1). Anal. Calcd for C20H14 ClO4S: C, 60.12; H, 3.50; N, 3.50. Found: 59.96; H, 3.60; N, 3.77
General Procedure for the Preparation of 1,1-Dioxonaphtho[1,2-b]thiophene-2-methoxycarbonyl Amino Acids (α-Nsmoc-AAs) and 3,3-Dioxonaphtho[2,1-b]thiophene-2-methoxycarbonyl Amino Acids (β-Nsmoc-AAs) using the Bolin Technique10
To a suspension of 3.24 mmol of an amino acid in 20 ml of dry DCM was added in one portion 0.82 ml (6.5 mmol) of dry chlorotrimethylsilane. The mixture was refluxed for 1.5 h and cooled in an ice bath. Diisopropylethylamine (1.1 ml, 6.2 mmol) was added slowly followed by 1 g (3.24 mmol) of α-Nsmoc-Cl (or β-Nsmoc-Cl). The reaction mixture was allowed to stand at 0°C for 30 min., and for 3 h at room temperature. The solvent was removed in vacuo and the resulting oil distributed between 50 ml of ether and 100 ml of 2.5% NaHCO3 solution. The combined aqueous layers were acidified to pH 2 with concentrated HCl and extracted with EtOAc (3 × 40 ml). The extracts were combined and washed with 40 ml of saturated NaCl, 40 ml of water, dried over MgSO4, filtered and solvent evaporated in vacuo. The solid or oily residue was recystallized from the appropriate solvent mixture to give the corresponding α-Nsmoc (or β-Nsmoc)- protected amino acids.
Note
In the cases of the Lys(Boc), Asn(Trt) and Gln(Trt) derivatives the regular Bolin technique was not suitable therefore we modified the procedure by adding 2.5 eq of TEA in the case Lys and Gln and 3.5 eq of TEA in the case of Asn to the amino acid suspension in 40 ml of dry DCM. Chlorotrimethylsilane (2 eq) was added and the mixture was refluxed for 2 h. The chloroformate was added in one portion at room temperature and the mixture was stirred overnight. The solution was washed once with 10 ml of water, dried over MgSO4, concentrated in vacuo and column chromatographed using ethyl acetate/hexane (3/2). The isolated material was then recrystallized from DCM/hexane.
For details see Table 1 and the Supporting Information Section.
General Procedure for the Preparation of α- and β-Nsmoc Amino Acid Fluorides11
To a stirred solution of the amino acid (1 mmol) in dry DCM (20 ml) and dry pyridine (1 mmol) kept under a N2 atmosphere was added cyanuric fluoride (5 mmol) at −20 to −10°C. A precipitate or emulsion was formed and gradually increased in amount. After stirring at this temperature for 1 h and at room temperature for 2 h, crushed ice was added followed by 20 ml of DCM. The organic layer was separated and the aqueous layer was extracted with DCM (2 × 20 ml). The combined DCM layers were washed with ice-cold water (3 × 15 ml), dried over MgSO4 and the solvent was removed in vacuo. The residue was recrystallized from an appropriate solvent. In some cases the reaction mixture had to be refluxed rather than being held at room temperature for 2 h. The acid fluorides were identified by their IR and 1H NMR spectra.
Solid-Phase Peptide Synthesis
Peptide segments were synthesized from both α-Nsmoc and β-Nsmoc amino acids and their corresponding fluorides via manual synthesis with a plastic syringe being used as a reaction vessel.17 The syringe was fitted with a sintered disc and connected to a water aspirator.
Synthesis of Leucine Enkephalin via α-Nsmoc and β-Nsmoc amino acids
Peptide segments were synthesized using either a coupling reagent or the isolated acid fluorides. The starting resin (Fmoc-PAL-Peg-PS, 100 mg, 0.24 mmol/g) was washed with DMF, DCM and DMF (3 × 4 mL each). The Fmoc group on the resin was deblocked using 20% pip/DMF for 7 min. followed by washing with DMF, DCM and DMF (3 × 4 mL each). Coupling was carried out using 3 eq of either N-HATU or N-HBTU as a coupling reagent along with 3 eq of α-Nsmoc and β-Nsmoc amino acids and 6 eq of DIEA as a base. In the case of the corresponding α-Nsmoc or β-Nsmoc amino acid fluorides, only 3 eq of DIEA was used. In all cases the coupling time was 30 min. The α-Nsmoc and β-Nsmoc groups were cleaved for various times using different pip/DMF ratios. A washing cycle involving DMF, DCM and DMF (3 × 4 mL each) followed each deprotection and coupling step. After assembling the complete sequence of the desired peptide, the α-Nsmoc and β-Nsmoc N-terminal groups were deblocked and the resin washed with DMF, DCM, ethanol and ether (3 × 4 mL each). The resin was dried under vacuum and then treated with 4 mL of a mixture of 95% TFA and 5% water for 2 h. The filtrate was collected and the resin was further washed with DCM (3 × 4 mL). The combined filtrates were evaporated in vacuo at room temperature and the peptide was precipitated by adding cold ether, the solvent was decanted and the crude peptide was dried under vacuum and examined by HPLC analysis using a solvent system consisting of 5/35 CH3CN 0.1% TFA over 25 min.
Synthesis of Acyl Carrier Peptide ACP65–74 via α- or β-Nsmoc Amino Acids
Sequence
H-Val-Gln-Ala-Ala-Ile-Asp-Tyr-Ile-Asn-Gly-NH2. The method followed that described for leucine enkephalin, except that the deblocking time was 1 min using 20% piperidine in DMF for α-Nsmoc protection and 8 min using 20% piperidine in DMF for β-Nsmoc protection.
Possible Loss of Configuration Involving the Synthesis and Coupling of α-Nsmoc-Phg-OH
(a) α-Nsmoc-Phg-Ala-OMe
To a solution of 140 mg (1 mmol) of H-Ala-OMe.HCl in 15 ml of DMF was added 0.52 ml (3.0 mmol) of DIEA, 421 mg (1.0 mmol) of α-Nsmoc-Phg-OH and 380 mg (1 mmol) of N-HATU. The reaction mixture was stirred for 1 h at 0°C and at room temperature for 2 h. The solution was diluted with 50 ml of EtOAc and washed with 5% citric acid, saturated NaHCO3 and saturated NaCl (3 × 15 mL each) and then dried over MgSO4. The solvent was removed with a rotary evaporator to give a residue which prior to purification was shown by 1H NMR analysis to contain 1.46% of the DL- diastereomer. A sample for elemental analysis was prepared by recrystallization from DCM/hexane to give 302 mg (59.4%) of the dipeptide as an off-white solid. m.p 182–184°C; IR (KBr) 1154, 1303 (SO2),1648, 1691, 1741 (CO), 3309 (NH) cm−1 ; 1H NMR (CDCl3) δ 1.38 (d, 3), 3.63 (s, 3), 4.52 (m, 1), δ 5.17 (d, 1), 5.15 (m, 2) 6.25 (dd, 2), 7.21 (d, 1), 7.32–7.41 (m 6), 7.6 (t, 1), 7.7 (m, 1), 7.91 (d, 1), 7.8 (d, 1), 8.27 (d, 1). Anal.Calcd for C26H24N2O7S: C, 61.41; H, 4.76; N, 5.51 Found: C, 61.51; H, 4.82; N, 5.44
(b) Establishment of the Authentic Positions for the OMe Singlets in the 1H NMR Spectra of LL- and LD-α-Nsmoc-Phg-Ala-OMe
In order to establish the position of the OMe singlet in the 1H NMR spectrum for the DL-diastereomer (which would be the same as that for the LD-diastereomer) the same method was used as described above except that DL-H-Ala-OMe.HCl was substituted for the L-isomer. This gave a roughly 50–50 mixture of the LL- and LD-diastereomers (m.p 80°C) for which the OMe singlets were found at δ 3.63 and 3.70, respectively.
(c) Establishment of Minimal Loss of Configuration via Acid Fluoride Coupling
A second test for the loss of configuration was made using the acid fluoride (See Supporting Information) of α-Nsmoc-Phg-OH prepared in the normal manner.11 The acid fluoride (346 mg, 0.82 mmol) was added in one portion to a stirred solution of 104 mg (0.75 mmol) of H-Ala-OMe.HCl in 10 mL of DCM in the presence of 0.261 mL (1.5 mmol) of DIEA. The mixture was stirred at room temperature for 10 min. (IR analysis showed complete disappearance of the acid fluoride band at 1846 cm−1). An additional 25 ml of DCM was added and the DCM layer washed with 5% citric acid, saturated NaHCO3 and saturated NaCl solution (2 × 10 mL each), and the solvent dried over MgSO4. The solvent was evaporated with a rotary evaporator and examined as described above in the OMe singlet region of the 1H NMR spectrum. This showed that no more than 0.6% of the DL- diastereomer could have been present. The acid fluoride was also coupled to H-Ala-OMe.HCl by a two-phase method in the presence of sodium bicarbonate. In this case somewhat more loss of configuration was observed (1.23% of the DL-diastereomer). For the 1H NMR spectra detailing these tests see the Supporting Information Section.
1,1-Dioxo-2-(Chloromethyl)naphtho[1,2-b]thiophene (23)
To a solution of 3 g (12.2 mmol) of 1,1 dioxo-2-(hydroxymethyl)naphtho[1,2-b]thiophene in 20 ml of DMF at 0°C was slowly added 10 ml of thionyl chloride. The reaction mixture was stirred for 30 min at 0°C and for 5 h at room temperature, quenched with 200 ml of ice-water and the off-white precipitate filtered and dried. Recrystallization from DCM/hexane gave 2.3 g (72.2%) of the 2-chloromethyl derivative as off-white crystals, mp 158–160°C; IR (KBr) 1148, 1291 (SO2) cm−1; 1H NMR (CDCl3) δ 4.61 (d, 2), 7.41 (d, 2), 7.63 (m, 1), 7.71 (m, 1), 7.93 (d, 1), 8.12 (d, 1), 8.27 (d, 1). Anal. Calcd for C13H9ClO2S: C, 58.99; H, 3.40. Found: C, 58.90; H, 3.42
1,1-Dioxo-2-(1-piperidinomethyl)-naphtho[1,2-b]thiophene (22)
To 0.66 g (2.5 mmol) of 1,1-dioxo-2-(chloromethyl)naphtho[1,2-b]thiophene was added 20 ml of DMF and 5 ml of piperidine. The reaction mixture was stirred for 17 h and 200 ml of EtOAc was added and the solution washed with water and saturated NaCl (3 × 70 ml each). After drying over MgSO4 the solvent was evaporated in vacuo and the resulting residue was flash chromatographed using EtOAc/hexane (3/2) to give two substances which were separately recrystallized from DCM/hexane: (1) 235 mg of a white solid, Rf., 0.33, mp 155–160°C which could not be obtained in a pure state but is assigned structure 21 on the basis of its 1H NMR spectrum and (2) 130 mg of the title substance as bright yellow crystals, Rf., 0.83, mp 237–240°C; IR (KBr) 1157, 1263 (SO2) cm−1; 1H NMR (CDCl3) δ 1.75 (m, 6), 2.29 (s, 4), 3.35 (d, 2), 7.27–8.39 (m, 7). Anal. Calcd for C18H19NO2S: C, 69.01; H, 6.06; N, 4.46. Found: C, 68.75; H, 5.97, N, 4.41
Isolation of initial and final stable adducts by reaction of 1,1-dioxo-2-(chloromethyl)naphtho[1,2-b]thiophene (23) with 2-methylpiperidine/DMF
To 0.330 g (1.25 mmol) of 1,1-dioxo-2-(chloromethyl)naphtho[1,2-b]thiophene (23) was added 20% 2-methylpiperidine in DMF. The reaction mixture was stirred for 1 min or 3 h and then 100 ml of EtOAc (for each reaction) was added. The reaction mixture was washed with water and saturated NaCl (3 × 30 ml in both cases), dried over MgSO4 and the solvent removed in vacuo. Analytical samples were obtained by flash column chromatography using EtOAc/hexane (3/2). From the 1-min reaction mixture the initial adduct was obtained as off-white crystals from DCM/hexane in 42.5% yield, mp 164–166°C; IR (KBr) 1162, 1290 (SO2) cm−1; 1H NMR (CDCl3) δ 1.28–1.69 (m, 8), 2.19 (m, 3), 2.88 (m, 1), 5.45 (t, 1), 5.95 (t, 1), 6.47 (t, 1) 7.26–8.58 (m, 6). Anal. Calcd for C19H21NO2S: C, 69.72; H, 6.41; N, 4.27. Found: C, 69.54; H, 6.43; N, 4.30.
From the 3-h reaction mixture the final stable adduct was obtained as yellow crystals from DCM/hexane in 40.9% yield, mp 127–130°C; IR (KBr) 1162, 1285 (SO2) cm−1; 1H NMR (CDCl3) δ 1.18–1.20 (d, 3), 1.59–2.04 (m, 6), 2.26 (s, 2), 3.03 (m, 1), 3.47 (m, 1), 3.8 (m, 1), 7.27–8.38 (m, 7). Anal. Calcd for C19H21NO2S: C, 69.72; H, 6.41; N, 4.27. Found: 69.44; H, 6.41; N, 4.17
Fmoc-Phe-Leu-O-β-Nsm
To a solution of 0.747 g (3 mmol) of Boc-Leu-OH.H2O and 0.738 g (3 mmol) of β-Nsm-OH and 100 mg of DMAP in 30 ml of dry DCM was added 0.618 g (3 mmol) of DCC at 0`°C. After stirring at 0°C for 1 h and at room temperature overnight, the solvent was removed in vacuo, 90 ml of EtOAc was added and the mixture filtered to remove DCU. The organic layer was washed with citric acid, saturated NaHCO3 and saturated NaCl (3 × 25 ml each) and dried (MgSO4). The solvent was removed in vacuo and the residue was stored under vacuum for 2 h and then 30 ml of TFA/DCM (50% v/v) was added and the solution stirred for 2 h at room temperature. After the Boc group had been completely removed, the solvent mixture was evaporated in vacuo and the residue was recrystallized from EtOH/ether (1:100) to give the TFA salt, which was obtained as cream-colored crystals in 80% yield, mp 161–163°C; IR (KBr) 1157, 1303 (SO2), 1675, 1754 (CO amide, ester) cm−1; 1H NMR (CDCl3 + TFA) δ 0.95 (m, 6), 1.75–1.99 (m, 3), 4.24 (m, 1), 5.37 (s, 2), 7.26–8.09 (m, 10). Without further purification to 1.42 g (3 mmol) of the TFA salt in 30 ml of DMF was added 1.7 ml (9.9 mmol) of DIEA, 1.3 g (3.3 mmol) of Fmoc-Phe-OH and 1.26 g (3.3 mmol) of N-HATU. The reaction mixture was stirred for 30 min. at 0°C and at room temperature for 2 h. The solution was diluted with 120 ml of EtOAc and washed with 5% citric acid, saturated NaHCO3 and saturated NaCl (3 x30 ml each) and then dried over MgSO4. The solvent was removed in vacuo and the resulting residue was flash chromatographed using EtOAc/hexane (3/2) to give the dipeptide as cream-colored crystals from DCM/hexane in 79.4% yield, mp 128–130°C; IR (KBr) 1157, 1301 (SO2), 1675 (CO, amide), 1723 (CO ester, urethane), 3359 (NH) cm−1; 1H NMR (CDCl3) δ 0.9 (m, 6), 1.48–1.73 (m, 3), 3.09 (d, 2), 4.13–4.69 (m, 5), 5.15 (d, 2), 6.26 (d, 1), 7.16–8.08 (m, 21). Anal. Calcd for C43H40N2O7S: C, 70.89; H, 5.84; N, 3.84. Found: C, 70.87; H, 5.56; N, 3.78
Fmoc-Gly-Phe-Leu-O-β-Nsm
A solution of 0.728 g (1 mmol) of Fmoc-Phe-Leu-Oβ-Nsm in 10 ml of DMF containing 20% t-BuNH2 was stirred at room temperature for 3 min. The reaction was quenched by adding quickly 100 ml of EtOAc and the solution was washed with H2O (3 × 25 ml), dried over MgSO4 and evaporated in vacuo. The resulting oil was dissolved in 7 ml of DCM and 3 ml of DMF and 0.3 g (1.0 mmol) of Fmoc-Gly-F was added in addition to 174 μL (10 mmol) of DIEA. The mixture was stirred for 45 min at room temperature and then DCM was evaporated and 100 ml of EtOAc was added and the solution was washed with water, saturated Na2CO3 and saturated NaCl (3 × 25 ml each) and dried over MgSO4. The solvent was evaporated in vacuo and the resulting oil was precipitated from DCM/hexane, washed with hexane and then chromatographed using EtOAc/hexane (3/2) as an eluent. The residue was separated and recrystallized from DCM/hexane to give Fmoc-Gly-Phe-Leu-Oβ-Nsm which was obtained as cream-colored crystals in 31.8% yield, mp 95–120°C; IR (KBr) 1151, 1301 (SO2), 1664 (CO, amide), 1728 (CO ester and urethane), 3348 (NH) cm−1; 1H NMR (CDCl3) δ 0.85 (d, 6), 1.65 (m, 3), 3.09 (d, 2), 3.83 (d, 2), 4.20 (t, 1), 4.34 (d, 2) 4.57 (m, 1), 4.70 (m, 1), 5.21 (d, 2), 5.48 (t, 1), 6.57 (d, 2), 7.17–8.12 (m, 20). Anal. Calcd for C45H43N3O8S: C, 68.80; H, 5.47; N, 5.34. Found: C, 68.50; H, 5.62; N, 5.30
Relative Reactivities of Bsmoc, α-Nsmoc and β-Nsmoc PCA Derivatives
In an NMR tube 10 mg of carbamate was dissolved in a mixture of 0.5 ml of CDCl3 and 0.4 ml of DMSO-d6. The initial 1H NMR spectrum was recorded at 500 MHz and 4.9 μL (2 eqv) of piperidine was added and the progress of the reaction followed by changes in the NMR spectra. Thus for the α-Nsmoc system the peak at δ 5.24 (CH2O) decreases in intensity as peaks at δ 6.13, 6.45 and 5.08 appear due to the formation of the initial adduct 21. At the same time peaks at δ 6.59 and 6.98 appear from the liberated PCA. The peak at δ 3.35 then builds up as the initial adduct is converted to the final stable adduct 22. A similar series of changes was noted for the Bsmoc and β-Nsmoc derivatives. By measuring at 30-sec. intervals the integrated areas for the buildup of PCA at δ 6.59 or the decrease in area for the CH2O peaks near δ 5.2 and plotting the figures against time shows the rough half lives of the α-Nsmoc, Bsmoc and β-Nsmoc systems to be 2.0, 2.4 and 7.4 min. respectively.
Supplementary Material
Supporting Information Available: Additional experimental details for the synthesis of sulfone alcohols 11 and 17, full characterization data for all α- and β-Nsmoc amino acids and selected spectral data for these and other new compounds.
Acknowledgments
We are indebted to the National Science Foundation (NSF CHE-9003192) and the National Institutes of Health (GM-09706) for support of this work. Millipore, Inc., Perseptive Biosystems, Inc., and Polycarbon, Inc. kindly supplied the N-HATU used in this study. The National Science Foundation is also thanked for grants used to purchase the high-field NMR spectrometers used in this research.
Footnotes
Common names: α- and β-Napthothiophenesulfone-2-methyloxycarbonyl. Other abbreviations used: ACP = acyl carrier protein decapeptide (64–75); Aib = α-aminoisobutyric acid; α-Nsm = 1,1-dioxonaphtho[1,2-b]thiophene-2-methyl; α-Nsmoc = 1,1-dioxonaphtho[1,2-b]thiophene-2-methyloxycarbonyl; β-Nsm = 3,3-dioxonaphtho[2,1-b]thiophene-2-methyl; β-Nsmoc = 3,3-dioxonaphtho[2,1-b]thiophene-2-methyloxycarbonyl; Boc = tert-butyloxycarbonyl; Bsm = 1,1-dioxobenzo[b]thiophene-2-methyl; Bsmoc =1,1-dioxobenzo[b]thiophene-2-methyloxycarbonyl; DCC = dicyclohexylcarbodiimide; DCM = dichloromethane; DIEA = diisopropylethylamine; DMAP = 4-(dimethylamino)pyridine; DMF = N,N-dimethylformamide; Fmoc = 9-fluorenemethyloxycarbonyl; N-HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium hexafluorophosphate 3-oxide; N-HBTU =1-[bis(dimethylamino)methylene]-1H-1-benzotriazolium hexafluorophosphate 3-oxide; MMPP = magnesium monoperoxyphthalate hexahydrate; NBS = N-bromosuccinimide; PAL-PEG-PS = peptide amide linker (5-(4-aminomethyl)-3,5-dimethoxyphenoxy)valeric acid) on poly(ethyleneglycol)/polystyrene support; PCA = p-chloroaniline; TFA = trifluoroacetic acid; THF = tetrahydrofuran.
References
- 1.Carpino LA, Ismail M, Truran GA, Mansour EME, Iguchi S, Ionescu D, El-Faham A, Riemer C, Warrass R. J Org Chem. 1999;64:4324. [Google Scholar]
- 2.Carpino LA. Accts Chem Res. 1987;20:401. [Google Scholar]
- 3.For the use of exactly one equivalent of a secondary amine (4-piperidinopiperidine) to deblock a sensitive Bsmoc paclitaxel derivative see: Greenwald RB, Zhao H, Reddy P. J Org Chem. 2003;68:4894. doi: 10.1021/jo034077s.
- 4.(a) Carpino LA, Philbin M, Ismail M, Truran GA, Mansour EME, Iguchi S, Ionescu D, El-Faham A, Riemer C, Warrass R, Weiss MSJ. Am Chem Soc. 1997;119:9915. [Google Scholar]; (b) Carpino LA, Krause E, Sferdean CD, Schümann M, Fabian H, Bienert M, Beyermann M. Tetrahedron Lett. 2004;45:7519. [Google Scholar]
- 5.(a) Carpino LA, Philbin M. J Org Chem. 1999;64:4315. [Google Scholar]; (b) McDowell ST, Stirling CJM. J Chem Soc (B) 1967:343. [Google Scholar]; (c) McDowell ST, Stirling CJM. J Chem Soc (C) 1967:351. [Google Scholar]
- 6.Tedjamulia ML, Tominaga Y, Castle RN, Lee ML. J Het Chem. 1983;20:1143. [Google Scholar]
- 7.Clarke K, Gregory DN, Scrowston RM. J Chem Soc Perkin 1. 1973:2956. [Google Scholar]
- 8.McKillip A, Kemp D. Tetrahedron. 1989;45:3299. [Google Scholar]
- 9.Campaigne E, Cline RE. J Org Chem. 1956;21:32–39. [Google Scholar]
- 10.Bolin DR, Sytwu II, Humiec F, Meienhofer J. Int J Pept Protein Res. 1989;33:353. [Google Scholar]
- 11.Carpino LA, Sadat-Aalaee D, Chao HG, DeSelms RH. J Am Chem Soc. 1990;112:9651. [Google Scholar]
- 12.A similar course for the Bsmoc deblocking process was previously recorded. See the supporting information section of ref. 4 (a), page S13.
- 13.(a) Carpino LA, Chao HG, Tien JH. J Org Chem. 1989;54:4302. [Google Scholar]; (b) Carpino LA, Gao HS, Ti GS, Segev D. J Org Chem. 1989;54:5887. [Google Scholar]; (c) Carpino LA, Cohen BJ, Lin Y-Z, Stephens KE, Jr, Triolo SA. J Org Chem. 1990;55:251. [Google Scholar]
- 14.(a) Arshady R, Atherton E, Clive DLJ, Sheppard RC. J Chem Soc Perkin 1. 1981:529. [Google Scholar]; (b) Schnölzer M, Jones A, Alewood PF, Kent SBH. Anal Biochem. 1992;204:335. doi: 10.1016/0003-2697(92)90249-7. [DOI] [PubMed] [Google Scholar]; (c) Carpino LA, El-Faham A, Minor CA, Albericio F. J Chem Soc Chem Commun. 1994:201. [Google Scholar]
- 15.Gilchrist TL, Summersell RJ. J Chem Soc Perkin 1. 1988:2595. [Google Scholar]
- 16.Paquet A. Can J Chem. 1982;60:976. [Google Scholar]
- 17.For a general description of the syringe methodology see ref. 1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Additional experimental details for the synthesis of sulfone alcohols 11 and 17, full characterization data for all α- and β-Nsmoc amino acids and selected spectral data for these and other new compounds.