

Abstract
Gas-phase photoelectron spectroscopy and density functional theory have been used to investigate the electronic structures of open-shell bent vanadocene compounds with chelating dithiolate ligands, which are minimum molecular models of the active sites of pyranopterin Mo/W enzymes. The compounds Cp2V(dithiolate) [where dithiolate is 1,2-ethenedithiolate (S2C2H2) or 1,2-benzenedithiolate (bdt), and Cp is cyclopentadienyl] provide access to a 17-electron, d1 electron configuration at the metal center. Comparison with previously studied Cp2M(dithiolate) complexes, where M is Ti and Mo (respectively d0 and d2 electron configurations), allows evaluation of d0, d1 and d2 electronic configurations of the metal-center that are analogues for the metal oxidation states present throughout the catalytic cycle of these enzymes. A “dithiolate-folding effect” that involves an interaction between the vanadium d orbitals and sulfur p orbitals is shown to stabilize the d1 metal center, allowing the d1 electron configuration and geometry to act as a low energy electron pathway intermediate between the d0 and d2 electron configurations of the enzyme.
Introduction
The bent metallocene class of molecules (Cp2M(L)2, Cp = η5-cyclopentadienyl) provide access to many small molecules that contain metal-dithiolate ligation,1 and are therefore useful as models of the pyranopterin-dithiolate system of the mononuclear molybdenum-containing enzymes that catalyze a wide range of oxidation/reduction reactions in carbon, sulfur and nitrogen metabolism.2-6 This class of molecules allows access to d0 (M = Ti, Hf, Zr), d1 (M = V, Nb) and d2 (M = Mo, W) metal electron configurations, the same electron configurations as the metal center during the enzyme catalytic cycle. Bent metallocenes are known with a variety of different dithiolate ligands, many of which have been crystallographically characterized.1 A common structural motif of metallocene dithiolates is a folding along the S···S vector, as illustrated in Figure 1.7 Interestingly, the range of fold angles observed for metallocene dithiolates encompasses the range of fold angles that has also been observed in protein crystal structures of pyranopterin-dithiolate sites (7-30°).8-12 The exact role of the pyranopterindithiolate coordination in the overall catalytic cycle of molybdenum enzymes is not yet established,13 but the unusual ability of ene-1,2-dithiolate ligands to stabilize metals in multiple oxidation states has long been recognized.14,15 Proposed functions for the pyranopterin-dithiolate ligand include acting as an electron transfer conduit from the metal to other prosthetic groups16 and/or acting as a modulator of the oxidation/reduction potential of the metal site.16,17 Modeling of the active site of the mononuclear molybdenum enzymes with small molecules has revealed useful information about the activity of these enzymes.18
Figure 1.
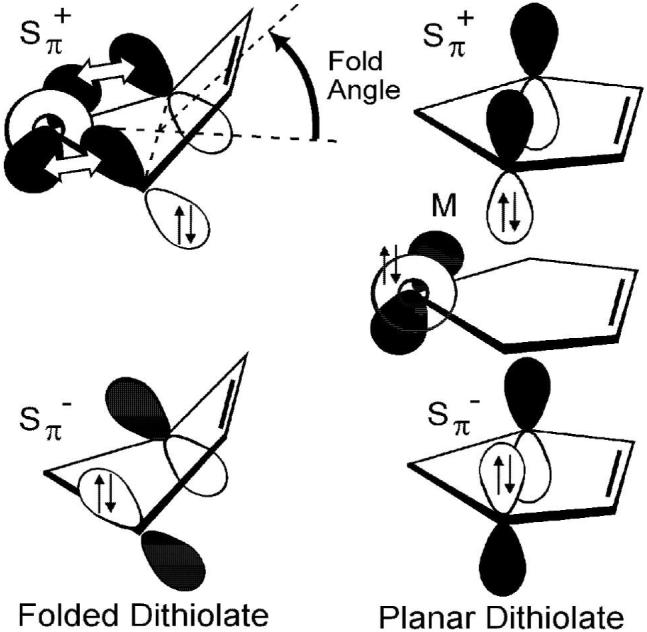
Representations of the valence orbitals of folded versus flat dithiolates. In the d0 case the metal orbital (M) is empty, and the Sπ+ orbital can interact upon folding. In the d2 case the metal orbital (M) is filled, and a planar orientation minimizes a filled-filled interaction. The Sπ- orbital does not have the correct symmetry to interact with the metal orbital.
A general bonding description of Cp2MX2 compounds is well understood,19-21 and Lauher and Hoffmann first explained the variation in fold angle for Cp2M(dithiolate) compounds as due to the occupancy of the metal d-orbital in the equatorial plane (or metal in-plane orbital, Mip) with respect to the dithiolate ligand. This orbital is empty for the d0 molecules, in which the metallocycle is folded along the S···S vector, and filled for the d2 molecules, in which the metallocycle is nearly planar. The observed folding for the d0 systems facilitates interaction of the filled Sπ orbitals with the empty Mip orbital, as shown in Figure 1. In the case of Cp2Ti(bdt), which has a formally Ti(IV) d0 metal center, the dithiolate ligand can be thought of as a six-electron donor. Each of the thiolate σ-orbitals provides two electrons, and two additional electrons come from the symmetric (Sπ+) orbital. Thus, folding the dithiolate ligand effectively stabilizes Cp2Ti(bdt) as an 18-electron complex.22,23 In contrast, for the d2 metal system, folding of the ligand would bring a filled Sπ orbital into close proximity with the filled Mip orbital; the observed nearly planar metallocycle minimizes the filled-filled interaction between the ligand and metal-based orbitals.12
Previously, we have investigated the electronic structures of the d0 and d2 metal centers of the metallocene dithiolates as active site models for molybdopterin enzymes.12,22 This study examines the d1 metal electron configuration that is observed for molybdopterin enzymes, and its role as an intermediate in facilitating electron transfer between the d0 and d2 electron configurations of the enzyme active site. The electronic structures of the compounds Cp2V(dithiolate) [where dithiolate is 1,2-ethenedithiolate (S2C2H2), 1,2-benzenedithiolate (bdt) and Cp is cyclopentadienyl] are examined by gas-phase photoelectron spectroscopy (PES) and density functional theory (DFT). This study fills in the connection between our previous studies of Cp2M(dithiolate) (M= Mo, Ti) that investigated the respective electronic structures of d2 and d0 metal centers. Investigation of Cp2V(dithiolate) as a model of the intermediate d1 electron configuration of pyranopterin Mo/W enzymes leads to a basic understanding of how the electronic and geometric structures of the d0, d1 and d2 electron configurations facilitate electron transfer to and from the enzyme active site.
Experimental methods
General
Electronic absorption (dichloromethane solutions on a modified Cary 14 with OLIS interface, 280-1280 nm) and mass spectrometry (direct ionization on a JEOL HX110) were used to identify the compounds. The compound Cp2V(bdt), was synthesized according to published procedures24 and under anaerobic conditions using a glove box. Cp2V (Strem), Cp2VCl2 (Aldrich), anhydrous THF (DriSolv) and anhydrous hexane (DriSolv) were used as supplied. The EPR spectrum of Cp2V(S2C2H2) was obtained at X-band at 77 K on a Bruker 300E spectrometer in the Electron Paramagnetic Resonance Facility at The University of Arizona. The X-ray crystallographic structure of Cp2V(S2C2H2) was determined by the Molecular Structure Laboratory at The University of Arizona.
Synthesis of Cp2V(S2C2H2)
To a suspension of freshly prepared Na2(S2C2H2)25 (0.068 g, 0.5 mmol) in THF (5 ml) was added a suspension of Cp2VCl2 (0.126 g, 0.5 mmol) in THF (8 ml). The mixture was stirred for 2 hrs and allowed to stand overnight at room temperature. The red/purple mixture was then filtered, evaporated to dryness under vacuum and washed with hexane (3 × 4 ml) to remove a blue impurity. The residue was dissolved in the minimum volume of THF (∼8 cm3), layered with hexane (∼10 ml) and allowed to stand overnight at room temperature, then refrigerated at -20 C for 3 days to yield dark purple block crystals suitable for crystallography. Yield: 0.052 g, 40%. UV/Vis/Near-IR: 10800 (ε = 400), 12900 (140) sh, 18400 (520), 21700 (310) sh, 23700 (210), 24900 cm-1 (400 M-1 cm-1) shoulder, and very intense UV transitions starting at 27500 cm-1. EPR: g1 = 2.007, g2 = 1.995, g3 = 1.995, A1 = 55.3, A2 = 25.1, A3 = 93.3 (10-4 cm-1). MS calculated m/z for VS2C12H12: 270.9820 (100), obs. 270.9822 (100); calc. 271.9850 (15.12), obs. 271.9863 (16.35); calc. 272.9789 (9.92), obs. 272.9784 (10.64 % normalized intensity).
X-ray Crystallographic Analysis
Data were collected for Cp2V(S2C2H2) on a Bruker SMART 1000 CCD detector X-ray diffractometer. The structure was solved using SIR92.26 Refinements were performed using SHELXL27 and illustrations were made using XP. Solution was achieved utilizing direct methods followed by Fourier synthesis. Hydrogen atoms were added at idealized positions, constrained to ride on the atom to which they are bonded and given thermal parameters equal to 1.2 Uiso of that bonded atom. A parameter describing extinction was included, but refined to zero and was removed prior to final refinement cycles. The final anisotropic full-matrix least squares refinement based on F2 of all reflections converged (maximum shift/esd = 0.000) at R1 = 0.0644, wR2 = 0.0986 and goodness-of-fit = 1.040. “Conventional” refinement indices using the 1972 reflections with F > 4 sigma(F) are R1 = 0.0389, wR2 = 0.0855. The model consisted of 136 variable parameters; no restraints were used. There were no correlation coefficients greater than 0.50. The highest peak on the final difference map was 0.495 e Å-3 located 0.82 Å from S1. The lowest peak on the final difference map was -0.343 e Å-3 located 0.72 Å from C2. Scattering factors and anomalous dispersion were taken from International Tables Vol. C Tables 4.2.6.8 and 6.1.1.4.
Photoelectron spectroscopy
Photoelectron spectra were recorded using an instrument, procedures and calibration that have been described in detail previously.28 The electron detection and instrument operation are interfaced to a National Instruments PCIe-6259 multi-function data acquisition card and custom software. During data collection the instrument resolution (measured using fwhm of the argon 2P3/2 peak) was 0.020-0.030 eV. The sublimation temperatures (at 10-5 Torr, monitored using a “K” type thermocouple passed through a vacuum feed-through and attached directly to the sample cell) were 190-200°C for Cp2V(S2C2H2) and 195-210°C for Cp2V(bdt). The samples sublimed cleanly without evidence of decomposition. In Figure 3, the vertical length of each data mark represents the experimental variance of that point. The valence ionization bands in the He I spectra are represented analytically with the best fit of asymmetric Gaussian peaks as described previously.29 The peak positions are reproducible to about ±0.02 eV (3). The analytical ionization bands that model the He I spectra are also used to model the ionizations in the He II spectra, with only the intensities of the analytical bands allowed to vary to account for the changes in photoelectron cross-sections with the change in excitation source energy from He I to He II.
Figure 3.
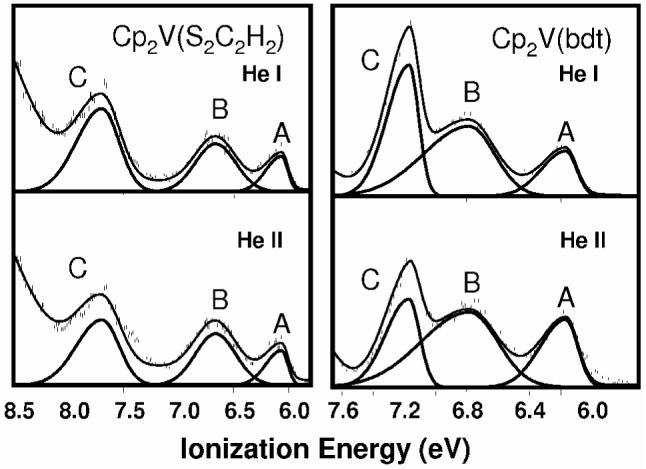
Low energy valence region of the gas-phase photoelectron spectra of Cp2V(S2C2H2) and Cp2V(bdt) with He I and He II excitation. Band C is due to the Sπ- orbital, B is the Sπ+/V bonding orbital, and A is the singly-occupied Sπ+ /V anti-bonding orbital.
Computational Methods
The Amsterdam Density Functional theory suite (ADF 2006.01b, using default parameters except for the options given in parentheses) was used to study the electronic structures of Cp2M(S2C2H2) and Cp2M(bdt), where M is V and Mo.30-34 The optimized geometries of Cp2V(S2C2H2), Cp2V(bdt) and Cp2Mo(bdt) (Tables S1 - S3, respectively, in Supporting Information) were obtained in Cs symmetry. Cp2V(S2C2H2), Cp2V(bdt) and Cp2Mo(bdt) were constructed such that the Cp rings were staggered with respect to each other, as similarly found in the crystal structure, and such that the Cs plane was coincident with the xy-plane. In Cp2V(S2C2H2), Cp2V(bdt) and Cp2Mo(bdt) the Cs plane bisects the metal atom and all three ligands. A generalized gradient approximation, with the correlation of Perdew, et al.35 and exchange correction of Handy and Cohen36 (GGA OPBE), was used for calculations of the optimized geometries. The calculations employed double- or triple-zeta basis sets with Slater type orbitals and polarization functions for all elements (DZP for C, H and S and TZP for V and Mo). Higher level basis sets did not give significantly different results for comparison of ionization energies. Calculations on the ground-state molecules were performed in the spin-unrestricted mode since each molecule contains one unpaired electron. Relativisitic effects were considered for all atoms by using the Zero Order Regular Approximation (relativistic scalar ZORA).37,38 The numerical integrals in ADF were evaluated to six significant figures (integration = 6.0) and the convergence criteria of the energy, gradients, and estimated coordinate uncertainty were tightened (converge E = 0.001, grad = 0.001, rad = 0.001). The self-consistent field (SCF) convergence limits were also tightened. (convergence 1e-6 1e-6). Analytical frequency calculations (AnalyticalFreq) were also carried out on the optimized geometry of Cp2Mo(bdt), and there were no imaginary frequencies.
ΔSCF calculations of the ionized states were performed at the fixed geometry of the neutral molecule, with one electron removed from the relevant orbital. The first positive ion of each molecule is a closed-shell singlet, but the higher positive ion states (corresponding to ionization of electrons from orbitals below the HOMO) contain two unpaired electrons and were calculated as both spin-unrestricted singlets and triplets. The ΔSCF estimate of the ionization energy is the difference between the calculated total energy of the ionized state and that of neutral ground state molecule.
Fold angle potential curves were created by utilizing the Linear Transit option in the Geometry block along with the Geovar keyword. Linear Transit calculations were run such that the fold angle was fixed every five or ten degrees through some range (e.g. 60° to -60°) and all geometric parameters, other than the fold angle, were allowed to fully optimize at each step.
Results and Discussion
Crystallography
There is one crystallographically independent molecule in the structure of Cp2V(S2C2H2). The structure (Figure 2, Table 1 and S4) is similar to that of Cp2V(bdt), as published by Stephan.24 Two cyclopentadienyl rings and two sulfur atoms complete a pseudo-tetrahedral coordination sphere of vanadium. The ene-dithiolate chelates to the V center through the two S atoms and shows folding along the S···S vector as illustrated in Figure 1, with a fold angle of 38.5°. The V-C and V-S distances average 2.302 and 2.425 Å, respectively. These compare with the V-C and V-S distances published for Cp2V(bdt) (2.30 and 2.431 Å).24 The S1-V-S2 angle of 81.14 (3)° is similar to that of 79.8° in Cp2 V(bdt).24 It is interesting to note that the bond lengths for the analogous titanium compounds are longer for M-C(Cp) (2.387 Å for Cp2Ti(S2C2H2) and 2.37 Å for Cp2Ti(bdt)) whereas the M-S bond lengths and S-M-S angles are very similar (2.417 Å and 83.23° for Cp2Ti(S2C2H2) and 2.416 Å and 82.2° for Cp2Ti(bdt)). The trend of Cl-M-Cl angles for Cp2MCl2 compounds is Ti/Zr > V/Nb > Mo.39,40 This trend is not echoed for Cp2M(bdt) (M= Ti, V and Mo) with the S-M-S angles of: 82.0, 79.8 and 82.1°, respectively. The effect of the electron density along the z-axis is overpowered, presumably, by the formation of the chelate ring.
Figure 2.
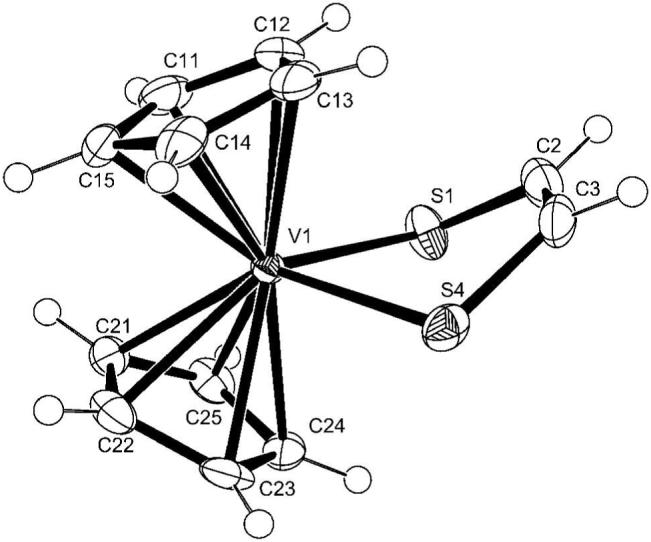
ORTEP representation of Cp2V(S2C2H2) with 50% thermal ellipsoids.
Table 1.
X-ray crystallographic parameters of Cp2V(S2C2H2)
| Empirical Formula | C12H12S2V |
| Formula weight | 271.28 |
| Temperature | 170(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P2(1)/c |
| Unit cell dimensions | A =1.3051(10) Å |
| B =7.5236(7) Å | |
| C =13.3767(12) Å | |
| β=104.849(2)° | |
| Volume | 1099.76(17) Å3 |
| Z | 4 |
| Density (calculated) | 1.638 Mg/m3 |
| Absorption coefficient | 1.240 mm-1 |
| Reflections utilized | 12814 |
| Independent reflections | 2675 [R(int) = 0.0497]a |
| Final R Indices | R1 = 0.0389, wR2 = 0.0855a |
| [I>2σ(I)] | |
| R Indices (all data) | R1 = 0.0644, wR2 = 0.0986a |
R(int) = Σ|Fo2-<Fo2>|/Σ[Fo2], R1 = Σ||Fo|-|Fc||/Σ|Fo|, wR2 = {Σ[w(Fo2-Fc2)2] / [Σw(Fo2)2]}½, w = 1/[σ2(Fo2)+(0.0466P)2] where P=(Fo2+2Fc2)/3
EPR
Previous studies of Cp2VX2 (where X is a monoanionic ligand, such as an alkyl or halide) have utilized electron paramagnetic resonance (EPR) to determine the molecular orbital character of the vanadium d1 electron. The EPR spectrum of Cp2V(S2C2H2) (Figure S2) is similar to that of Cp2VCl2 reported previously.39-42 The trend of the Cl-M-Cl angle to decrease with M, changing from Ti to V to Mo, and single crystal EPR studies led to the conclusion that the HOMO of the V and Mo compounds is primarily along an axis normal to the plane bisecting the Cl-M-Cl angle. Lowering the symmetry from C2v to Cs allows this axis to be labeled z, and the orbital was calculated as |Ψ0> = a|dz2> + b|dx2 - y2>, where a = -0.963 and b = 0.270 (Mulliken charges, a2:b2 = 20.5:1) for Cp2VCl2.40 This is close to the observed a = -0.976 and b = 0.218 (a2:b2 20.0:1) for (C5H4Me)2VCl2. These EPR parameters have been compared with those of Cp2VCl2, calculated as Mulliken charges.40 Using more current methods, a similar comparison can be made between the observed EPR and symmetry-adapted fragment orbital (SFO) parameters calculated by DFT. For Cp2VCl2, the singly-occupied, spin α orbital SFO component corresponding to dz2 is 0.801 and that to dx2 - y2 is -0.033, giving a ratio of a2:b2 = 24.1:1.43 This is in agreement with previous experiments (see above) concluding that the singly-occupied orbital is of primarily dz2 character.
The higher g values and lower A(51V) values of Cp2V(S2C2H2) reflect the softer coordination environment and covalent nature of the dithiolate ligand compared to the two chloride ligands in Cp2VCl2. Previous studies of Tp*MoOXY (where Tp* = hydrotris(3,5-dimethyl-1-pyrazolyl)borate; (X,Y) = Cl, or toluenedithiol (tdt)) exhibit a similar trend in the g and A(95,97Mo) values with change in coordination sphere.44 The higher g values and lower A(95,97Mo) values of Tp*MoO(tdt) compared to Tp*MoOCl2 are attributed to a low energy charge transfer transition in Tp*MoO(tdt) (9010 cm-1, ε = 520) that can mix sulfur character into the ground state.44 Tp*MoOCl2 does not exhibit a similar charge transfer transition, but has a d → d transition as the lowest energy band. Comparing the electronic spectra of Cp2VCl2 and Cp2V(S2C2H2) (Figure S3), there is a low energy ligand-to-metal charge transfer transition15 for Cp2V(S2C2H2) (10800 cm-1, ε = 400), but Cp2VCl2 lacks a similar low energy transition. The difference in the low energy charge transfer transitions between Cp2VCl2 and Cp2V(S2C2H2) would account for the greater g and lower A(51V) values exhibited by Cp2V(S2C2H2) compared to Cp2VCl2.45,46
Photoelectron Spectroscopy
Spectroscopic evidence supporting the Lauher and Hoffmann19 description of the electronic structure includes our recent PES study of the nearly planar metallocycle, Cp2Mo(bdt),12 and the folded metallocycle, Cp2Ti(bdt), for which ionizations from primarily metal based orbitals and primarily S pπ based orbitals can be experimentally distinguished from one another.12,22,47 The S pπ based orbitals of the two sulfur atoms form a symmetric and antisymmetric pair. The symmetric (Sπ+) orbital combination has the right symmetry and energy to match the Mip orbital upon folding of the dithiolate unit, whereas the antisymmetric sulfur (Sπ-) orbital combination does not (Figure 1). The substantial mixing of the out-of-plane Sπ+ orbital and the Mip orbital upon folding is shown experimentally in the PE spectra of Cp2Ti(bdt) by the increase in intensity of the Sπ+ ionization band relative to the Sπ- ionization band with change in ionization source energy from He I to He II. In order for this “dithiolate-folding effect” to be present there must be effective coupling between the S pπ orbitals and the C pπ orbitals derived from the ene-dithiolate ligand backbone.22 Such coupling raises the energy of the Sπ+ orbital above that of the Sπ- orbital, and poises the energy of the Sπ+ orbital for interaction with the metal-based in-plane orbital (Mip). This electronic structure facilitates the “dithiolate-folding effect” as only the Sπ+ orbital has the correct symmetry to overlap with the metal based in-plane orbital. For example, we have previously shown that Cp2Ti(bdt) and Cp2Ti(S2C2H2), which both have an unsaturated carbon-carbon bond linking the two S atoms, show a dithiolate-folding effect. However, if a saturated dithiolate ligand is present, such as in Cp2Ti(S2C3H6),22 the coupling between the pπ orbitals on the two S atoms is greatly reduced and the ordering of the Sπ orbitals is reversed in comparison to the unsaturated dithiolate.
The low energy valence regions of the gas-phase photoelectron spectra of Cp2V(S2C2H2) and Cp2V(bdt), collected with both He I and He II photon sources, are presented in Figure 3. Comparison of the He I spectra with their titanocene analogs22 in Figure 4 reveals a similar overall ionization profile in each case, except for the presence of the weak band A at low energy (6.06, and 6.17 eV for Cp2V(S2C2H2) and Cp2V(bdt), respectively), as expected for the additional electron in the vanadium complexes.
Figure 4.
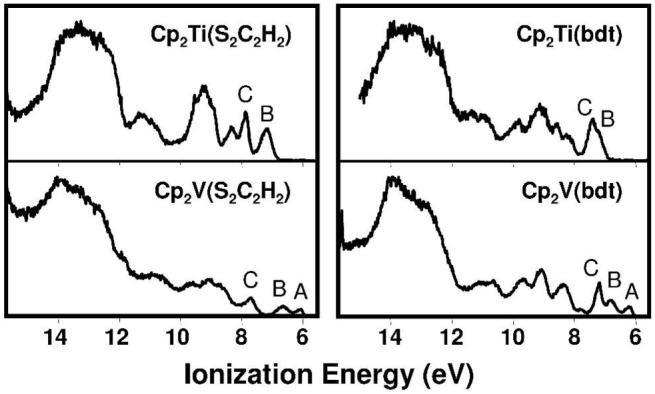
The spectra of Cp2V(S2C2H2) and Cp2V(bdt) have one more band (band A) than their respective titanocene analogs.
Table 2 lists the changes in areas of ionization bands B and C relative to the first ionization band A with change in ionization source from He I to He II for Cp2V(S2C2H2) and Cp2V(bdt). From previous experimental studies48-50 and calculations of atomic photoionization cross-sections,51 it is expected that ionizations from molecular orbitals with significant Ti 3d contributions will increase in intensity compared to ionizations of primarily S 3p character when data collected with a He II photon source are compared to data collected with a He I photon source (Table 2). The changes in relative areas of the bands in Cp2V(S2C2H2) and Cp2V(bdt) are similar, indicating that the atomic character of the molecular orbitals for the two molecules is similar. Band C in the titanocene dithiolate spectra is assigned to a Sπ- ionization, which does not mix significantly with the empty metal d acceptor orbital, and hence decreases in intensity significantly relative to the Sπ+ band (band B).22 The destabilization of band B in the vanadocene dithiolate spectra compared to the related ionization in the titanocene spectra can be attributed to the interaction of the additional vanadium d1 electron with the Sπ+ orbital, which decreases the fold angle and lowers the bonding interaction of Sπ+ with Mip. Band C in the spectra of Cp2V(S2C2H2) and Cp2V(bdt) decreases in intensity relative to both the first and second bands, and can therefore also be assigned to Sπ- (Table 2). Band C shows little change in ionization potential between the vanadocene and titanocene dithiolate spectra consistent with its non-bonding interaction with Mip. Band A increases slightly with respect to band B in Cp2V(bdt) and decreases slightly in Cp2V(S2C2H2), indicating mixing of metal and sulfur character and making absolute assignment difficult at this point. Comparison with the titanium analogs indicates that bands A and B both have mixed vanadium and Sπ+ character.
Table 2.
Ionization energies, band shapes, and He I and He II band areas for Cp2V(S2C2H2) and Cp2V(bdt)
| Cp2V(S2C2H2) | |||||
|---|---|---|---|---|---|
| Band | I.E.a (eV) | Band Width High (eV) | Band Width Low (eV) | Relative Areab |
|
| He I | He II | ||||
| A | 6.06 | 0.34 | 0.14 | 1 | 1 |
| B | 6.67 | 0.43 | 0.43 | 2.46 | 2.69 |
| C | 7.70 | 0.54 | 0.36 | 4.41 | 3.58 |
| Cp2V(bdt) | |||||
|---|---|---|---|---|---|
| Band | I.E. (eV) | Band Width High (eV) | Band Width Low (eV) | Relative Area |
|
| He I | He II | ||||
| A | 6.17 | 0.36 | 0.16 | 1 | 1 |
| B | 6.78 | 0.69 | 0.38 | 3.15 | 2.28 |
| C | 7.17 | 0.34 | 0.14 | 2.65 | 1.20 |
Vertical ionization energy
Areas are relative to Band A.
Computations
DFT calculations provide additional insight into the metal-ligand interactions for Cp2V(S2C2H2) and Cp2V(bdt) indicating that upon dithiolate folding the mixing of metal d and sulfur pπ orbitals can be favored by their energy and symmetry match. The calculated molecular structures of Cp2V(S2C2H2) and Cp2V(bdt) in general agree with the reported structures determined from X-ray crystallography (Table S4).24,52 The Cp rings were staggered with respect to each other in the initial input guess geometries similar to the crystal structure of the ene-dithiolate complex and as determined to be a true minimum from the frequency analysis of the related Mo molecule. Rotation of the two Cp ligand rings has been shown to be a low energy process53 and therefore additional Cp orientations were not explored. The calculated fold angles for the coordinated dithiolates are smaller than those determined crystallographically. For Cp2V(S2C2H2) the calculated angle is 29.9° and the experimental angle is 38.5°, and for Cp2V(bdt) the calculated angle is 36.6° and the experimental angle is 41.0 and 40.1°.24 Solid state effects likely contribute to the differences between the calculated and crystallographic fold angles.54
The energies of the ionizations observed for Cp2V(S2C2H2) and Cp2V(bdt) by photoelectron spectroscopy are compared with those calculated by the ΔSCF method in Table 3. The ground states of the neutral molecules have s = ½ (V4+, d1, 2A’); ionization from the SOMO will lead to a singlet state, but ionization from the doubly-occupied orbitals leads to either singlet or triplet state configurations with singlet:triplet band relative intensities approximately 1:3. Table 3 shows the calculated values for the singlet and triplet states of Cp2V(S2C2H2) and Cp2V(bdt) using the ΔSCF method. For Cp2V(S2C2H2), the calculated singlet/triplet state separation for band B is 0.53 eV and should be discernable in the experiment, although the band maximum will be dominated by the triplet state. The calculated energy for the formation of a triplet state (6.57 eV) agrees well with the observed ionization energy maximum for band B (6.67 eV), and no distinct ionization is observed for the singlet state. For band C, the calculated singlet/triplet state separation is 0.14 eV, which is less than the width of the vibrational manifolds associated with these ionizations, and a singlet state shoulder on the triplet state ionization is not observable. Again, the calculated vertical ionization for the triplet state (7.77 eV) agrees well with the observed band maximum for band C (7.70 eV).
Table 3.
Experimental and calculated orbital ionization energies (eV) for bands A, B and C of Cp2V(S2C2H2) and Cp2V(bdt)
| Band | I.E.a | Labelb | Kohn-Sham Orbital Energyc | ΔSCFd | ||
|---|---|---|---|---|---|---|
| α-spin | β-spin | Singlet | Triplet | |||
| Cp2V(S2C2H2) | ||||||
| A | 6.06 | V d1/Sπ+ | -3.87 | 6.12 | ||
| B | 6.67 | V d1/Sπ+ | -4.91 | -4.40 | 7.10 | 6.57 |
| C | 7.70 | Sπ- | -5.68 | -5.50 | 7.91 | 7.77 |
| Cp2V(bdt) | ||||||
| A | 6.17 | V d1/Sπ+ | -4.21 | 6.22 | ||
| B | 6.78 | V d1/Sπ+ | -5.21 | -4.83 | 7.18 | 6.77 |
| C | 7.17 | Sπ- | -5.36 | -5.11 | 7.27 | 7.10 |
Experimental vertical ionization energy.
Primary character.
The calculated orbital energy for the ground state.
The difference in total energy between molecular ground state, and the molecule with an electron removed from the specified orbital.
A similar assignment is given for Cp2V(bdt) based on the ΔSCF calculations. The increase in ionization energies for bands A and B and the decrease in ionization energy for band C from Cp2V(S2C2H2) to Cp2V(bdt) observed in the photoelectron spectra also is obtained from the calculations. The observed separation in energy between bands B and C is different for Cp2V(S2C2H2) and Cp2V(bdt) (1.03 and 0.39 eV, respectively) correlating with the separation in energy between the Sπ+ and Sπ- for their titanocene analogues (0.70 and 0.22 eV, for Cp2Ti(S2C2H2) and Cp2Ti(bdt), respectively). This trend, which is reproduced computationally, is presumably due to different inductive effects of the ligands and the greater involvement of the benzene ring in the π-system of the ligand for Cp2V(bdt) and Cp2Ti(bdt). In general, the calculated singlet/triplet separations for Cp2V(S2C2H2) and Cp2V(bdt) are small and within the experimental band widths for the ionizations, which is in agreement with previous studies of vanadium(IV) systems.55,56
Also included for comparison in Table 3 are the Kohn-Sham orbital energies for the α and β spin electrons. Early calculations of metal complexes with a partially-filled metal d orbital bury the partially-filled orbital below the filled ligand orbitals,57-61 but in the present calculations the orbital ordering agrees with the calculated and observed sequence of ionizations. Contour plots of the spin α and β orbitals for Cp2V(S2C2H2) and Cp2V(bdt) that correspond to the ionizations evaluated by photoelectron spectroscopy are shown in Figure 5. The contour plots show that for Cp2V(S2C2H2) and Cp2V(bdt) the SOMOs are substantially delocalized with greater sulfur 3p character than vanadium dz2 character, in agreement with the greater g and lower A values observed in the EPR experiment. The contour plots for Cp2V(bdt) also show that the orbital character is extended onto the benzene π-system (the orbital percent contributions shown in Figure 5 are given in Table S5).
Figure 5.

Spin correlation diagrams for Cp2V(S2C2H2) and Cp2V(bdt) showing α- and β-spin eigenvalues, molecular orbital character and electron occupations based on ground state configuration.
The description of these orbitals is consistent with the Sπ+ and V dz2 orbitals forming a bonding and anti-bonding pair of orbitals, as shown in Figure 5, where the SOMO is the anti-bonding combination and the next orbital is the doubly-occupied bonding combination. This bonding/anti-bonding combination explains the strong vanadium and sulfur character mixing observed for bands A and B in the photoelectron spectra. Overall, the orbital picture corresponds to the bonding interaction proposed by Lauher and Hoffman.19
DFT calculations also offer insight into how the dithiolate-folding effect and the fold angle influence the oxidation/reduction and electron transfer properties of the active site metal center. Using the linear transit option in the ADF package, potential energy curves for Cp2V(bdt) as neutral and cation molecules can be constructed to show how the fold angle changes with oxidation of the d1 Mip orbital. Figure 6 shows a fold angle potential energy curve plot for Cp2V(bdt) and [Cp2V(bdt)]+, where the change in relative energy with fold angle is compared. The potential energy curve of the neutral Cp2V(bdt) molecule shows that the energy minimum lies at approximately 37°. Oxidation of the Mip orbital leads to an increase in the fold angle from 37° to 40°, resulting in a deeper potential well for the cation. The potential energy curve for the d1 and d0 electronic configurations of Cp2V(S2C2H2) is similar (Figure S1).
Figure 6.
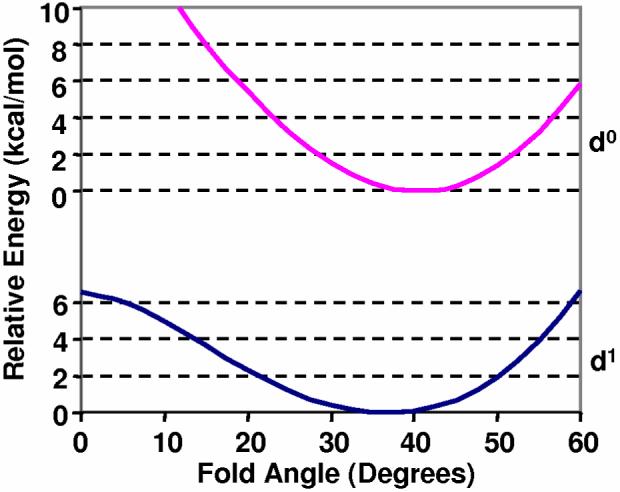
Potential energy diagrams showing the calculated total energy with change in fold angle for the neutral (d1) and cation (d0) of Cp2V(bdt).
Figure 7 presents the potential energy curves of the neutral (d2), cation (d1) and dication (d0) of Cp2Mo(bdt) obtained from the gas-phase linear transit calculations with varying fold angle. The figure shows the calculated relative total energies between the optimized geometries of each electron configuration, along with the relative energy perturbation caused by dithiolate-folding for each electronic configuration (d2, d1, and d0).62 For Cp2Mo(bdt) the calculated fold angle for the neutral (d2) molecule exhibits a potential minimum at 0°. Upon oxidation from Mip the fold angle increases to 25° for the cation (d1), and to 35° for the dication (d0). This range of fold angles is consistent with the DFT analysis by Domercq et al.63 The analytical frequency calculations for Cp2Mo(bdt) show that the folding vibration is the lowest energy vibration in the molecule at 28 cm-1, and a nearly classical description of the vibronic states is appropriate for these curves and thermal populations will be significant during electron transfer. This trend in the calculated fold angles for the Cp2Mo(bdt) series in Figure 7 follows that of the neutral d2, d1 and d0 metallocene dithiolate crystal structures (vida supra).
Figure 7.
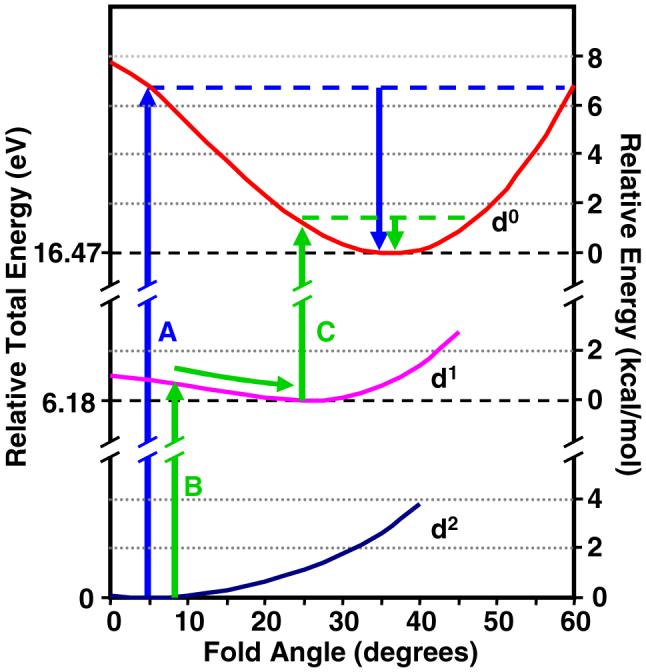
The gas-phase potential energy curve plots of the neutral (d0), cation (d1) and dication (d2) of Cp2Mo(bdt) are shown with relative total energies (eV, left-hand side) and relative potential energies with change in fold angle (kcal/mol, right-hand side). The oxidation along Path A (blue arrows and line) from the d2 to d0 electron configurations involves a large energy barrier to oxidation and larger reorganization energies with change in fold angle. Paths B and C (green arrows and lines) show the two-step two-electron oxidation of the d2 to d1 to d0 electron configurations. Utilization of the shallow d1 potential curve poises oxidation to occur with minimal reorganization energies with change in fold angle.
Figure 7 not only shows the increase in fold angle with oxidation, but also gives insight into the role of the d1 electron configuration in molybdopterin enzymes during substrate turnover. The relative energies between the d2, d1, and d0 configurations will be reduced from the gas-phase values in Figure 7 by stabilization of the cations in the molybdopterin enzyme environment. Oxidation starting from the d2 configuration and proceeding to the d0 electron configuration without geometry reorganization along path A, followed by reorganization of the d0 configuration to the minimum of the potential well (blue arrows and line in Figure 7), is a high energy process with a large reorganization energy. The reorganization energy is approximately 0.26 eV (7 kcal mol-1). Alternatively, oxidation from the d2 to d1 electron configurations allows access to the shallow d1 potential with little reorganization energy investment upon changing the fold angle from 5° to 25° (arrow B). Likewise, oxidation from the folded geometry of the d1 configuration allows a low energy investment to the folded structure of the d0 electronic configuration (arrow C). The coupling of paths B and C (green arrows and line) through the d1 electron configuration results in a two-step two-electron oxidation of the d2 to d0 electron configuration with smaller barrier and reorganization energies. The shallow potential surface calculated for the d1 electron configuration and the low frequency of the dithiolate folding motion provide an intermediate that can effectively link the d2 and d0 electron configurations and geometries. Thus, the potential curves of Figure 7 show how sequential one-electron transfers coupled to fold angle variation offer a reaction pathway for reoxidation of molybdenum enzymes with a lower overall energy barrier than two-electron oxidation without the d1 configuration or the dithiolate-folding effect. These smaller energy barriers allow the reoxidation of the metal center to occur with concomitant electron transfer and change in the fold angle.
Conclusions
The vanadocene dithiolate systems compare well with previous studies of the titanocene and molybdocene dithiolate electronic structures,22,47 thus completing the d0, d1, and d2 configurations found in mononuclear molybdenum protein active sites. The crystallographic and computational data also supports the change in fold-angle upon filling the metal in-plane orbital.
Comparison of the PES of vanadocene systems with their respective titanocene analogs shows that their electronic structures are similar, with the exception of the extra electron in the vanadium complexes. For Cp2V(S2C2H2) and Cp2V(bdt) both the SOMO and the next lowest energy orbital show considerable vanadium and sulfur character, forming a bonding and anti-bonding pair. The SOMO is only half-filled, so the resulting contribution to the bond order from this interaction is one half. The ΔSCF calculations compare well with the PES spectra of Cp2V(S2C2H2) and Cp2V(bdt) and place the SOMO as the lowest energy orbital.
The use of DFT calculations further supports the importance of the d1 electronic configuration as a low energy pathway for reoxidation of the metal center during catalysis. Two-electron oxidation of the d2 electronic configuration to the d0 configuration requires a larger relative energy investment for the removal of both electrons and geometric reorganization of the fold angle. The intermediate d1 electronic configuration offers a low energy pathway where a minimal amount of energy is required for reoxidation and geometric reorganization of the fold angle bridging the d2 and d0 electronic configurations.
The variation in dithiolate fold angle with formal d electron count observed for Cp2M(dithiolates) supports the hypothesis that the change in the dithiolate fold angle effectively stabilizes d0 and d1 metal centers via π-donation from Sπ+ to the Mip orbital. This “dithiolate-folding effect” has implications for Mo/W enzymes since folding of the pyranopterindithiolate cofactor poises the metal center for reoxidation and geometric reorganization, modulating the reactivity of the enzyme active site as the metal center passes through the M(IV), M(V), and M(VI) formal oxidation states during electron and oxygen atom transfer.
Supplementary Material
Acknowledgments
We gratefully acknowledge support of this research by the National Institutes of Health (Grant GM-37773 to J.H.E.), a Galileo Circle Fellowship (M.A.C.), and the National Science Foundation (Grant CHE-0416004 to D.L.L. and Grant CHE-9601809 for the mass spectrometer). We thank Dr. A. Somogyi for mass spectrometry and Dr. Andrei Astashkin for the EPR spectra.
References
- (1).Fourmigué M. Coord. Chem. Rev. 1998;178-180:823–864. [Google Scholar]
- (2).Rajagopalan KV, Johnson JL. J. Biol. Chem. 1992;267:10199–10202. [PubMed] [Google Scholar]
- (3).Hille R. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- (4).Sigel A, Sigel H. Molybdenum and Tungsten: Their Roles in Biological Processes. Vol. 39. Metal Ions in Biological Systems; Dekker: New York: 2002. [Google Scholar]
- (5).Garner DC, Banham R, Cooper SJ, Davies ES, Stewart LJ. Handbook on Metalloproteins. 2001. pp. 1023–1090. [Google Scholar]
- (6).Burgmayer SJN. Prog. Inorg. Chem. 2004;52:491–537. [Google Scholar]
- (7).The fold angle is the acute angle between the vector from the metal to the centroid of the two sulfur atoms and the vector from this centroid to that of the two carbon atoms of the metallacycle. .
- (8).Kisker C, Schindelin H, Pacheco A, Wehbi W, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. Cell. 1997;91:973–983. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- (9).Rebelo JM, Dias JM, Huber R, Moura JJG, Romão MJ. J. Biol. Inorg. Chem. 2001;6:791–800. doi: 10.1007/s007750100255. [DOI] [PubMed] [Google Scholar]
- (10).Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li H, Temple C, Rajagopalan KV, Schindelin H. J. Am. Chem. Soc. 2000;122:7673–7680. [Google Scholar]
- (12).Joshi HK, Cooney JJA, Inscore FE, Gruhn NE, Lichtenberger DL, Enemark JH. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3719–3724. doi: 10.1073/pnas.0636832100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schindelin H, Kisker C, Rees DC. J. Biol. Inorg. Chem. 1997;2:773–781. [Google Scholar]
- (14).Eisenberg R. Prog. Inorg. Chem. 1970;12:295–369. [Google Scholar]
- (15).Stiefel EI, Karlin KD, editors. Progress in Inorganic Chemistry. Vol. 52. John Wiley and Sons, Inc; Hoboken, New Jersey: 2004. Dithiolene Chemistry: Synthesis, Properties, and Applications; p. 738. [Google Scholar]
- (16).Inscore FE, McNaughton R, Westcott B, Helton ME, Jones R, Dhawan IK, Enemark JH, Kirk ML. Inorg. Chem. 1999;38:1401–1410. [Google Scholar]
- (17).Westcott BL, Gruhn NE, Enemark JH. J. Am. Chem. Soc. 1998;120:3382–3386. [Google Scholar]
- (18).Enemark JH, Cooney JJA, Wang JJ, Holm RH. Chem. Rev. 2004;104:1175–1200. doi: 10.1021/cr020609d. [DOI] [PubMed] [Google Scholar]
- (19).Lauher JW, Hoffmann R. J. Am. Chem. Soc. 1976;98:1729–1742. [Google Scholar]
- (20).Green JC. Chem. Soc. Rev. 1998;27:263–272. [Google Scholar]
- (21).Green JC. Struct.Bonding. 1981;43:37–112. [Google Scholar]
- (22).Cooney JJA, Cranswick MA, Gruhn NE, Joshi HK, Enemark JH. Inorg. Chem. 2004;43:8110–8118. doi: 10.1021/ic049207+. [DOI] [PubMed] [Google Scholar]
- (23).Pyykko P. J. Organometal. Chem. 2006;691:4336–4340. [Google Scholar]
- (24).Stephan DW. Inorg. Chem. 1992;31:4218–4223. [Google Scholar]
- (25).King RB, Eggers CA. Inorg. Chem. 1968;7:340–345. [Google Scholar]
- (26).Altomare A, Cascarano G, Giacovazzo C, Guagliardi A. Journal of Applied Crystallography. 1993;26:343–350. [Google Scholar]
- (27).Sheldrick GM. SHELXL97. University of Göttingen; Germany: 1997. [PubMed] [Google Scholar]
- (28).Lichtenberger DL, Kellogg GE, Kristofzski JG, Page D, Turner S, Klinger G, Lorenzen J. Rev. Sci. Instrum. 1986;57:2366. [Google Scholar]
- (29).Lichtenberger DL, Copenhaver AS. J. Elec. Spec. and Rel. Phenom. 1990;50:335–352. [Google Scholar]
- (30).Baerends EJ, Ellis DE, Ros P. Chem. Phys. 1973;2:41–51. [Google Scholar]
- (31).Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ. Theor. Chem. Acc. 1998;99:391–399. [Google Scholar]
- (32).te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, Van Gisbergen SJA, Snijders JG, Ziegler T. J. Comput. Chem. 2001;22:931–967. [Google Scholar]
- (33).te Velde G, Baerends EJ. J. Comput. Phys. 1992;99:84–98. [Google Scholar]
- (34).Versluis L, Ziegler T. J. Chem. Phys. 1988;88:322–329. [Google Scholar]
- (35).Perdew JP, Burke K, Wang Y. Phys. Rev. B. 1996;54:16533–16539. doi: 10.1103/physrevb.54.16533. [DOI] [PubMed] [Google Scholar]
- (36).Handy NC, Cohen AJ. Mol. Phys. 2001;99:403–412. [Google Scholar]
- (37).Vanlenthe E, Baerends EJ, Snijders JG. J. Chem. Phys. 1993;99:4597–4610. [Google Scholar]
- (38).Vanlenthe E, Baerends EJ, Snijders JG. J. Chem. Phys. 1994;101:9783–9792. [Google Scholar]
- (39).Petersen JL, Dahl LF. J. Am. Chem. Soc. 1974;96:2248–2250. [Google Scholar]
- (40).Petersen JL, Lichtenberger DL, Fenske RF, Dahl LF. J. Am. Chem. Soc. 1975;97:6433–6441. [Google Scholar]
- (41).Petersen JL, Dahl LF. J. Am. Chem. Soc. 1975;97:6416–6422. [Google Scholar]
- (42).Petersen JL, Dahl LF. J. Am. Chem. Soc. 1975;97:6422–6433. [Google Scholar]
- (43).Cranswick MA, Gruhn NE, Enemark JH, Lichtenberger DL. Manuscript in preparation. [Google Scholar]
- (44).Cleland WE, Jr., Barnhart KM, Yamanouchi K, Collison D, Mabbs FE, Ortega RB, Enemark JH. Inorg. Chem. 1987;26:1017–1025. [Google Scholar]
- (45).Stewart CP, Porte AL. Dalt. Trans. 1973;7:722–729. [Google Scholar]
- (46).Bakalik DP, Hayes RG. Inorg. Chem. 1972;11:1734–1738. [Google Scholar]
- (47).Joshi HK. The University of Arizona; 2003. [Google Scholar]
- (48).Glass RS, Gruhn NE, Lichtenberger DL, Lorance E, Pollard JR, Birringer M, Block E, DeOrazio R, He C, Shan Z, Zhang X. J. Am. Chem. Soc. 2000;122:5065–5074. [Google Scholar]
- (49).Green JC. Acc. Chem. Res. 1994;27:131–137. [Google Scholar]
- (50).Gelius U. J. Elec. Spec. and Rel. Phenom. 1974;5:985–1057. [Google Scholar]
- (51).Yeh JJ, Lindau I. At.Data Nucl.Data Tables. 1985;32:1–155. [Google Scholar]
- (52).Tzavellas N, Klouras N, Raptopoulou CP. Zeitschrift fuer Anorganische und Allgemeine Chemie. 1996;622:898–902. [Google Scholar]
- (53).Cacelli I, Keogh DW, Poli R, Rizzo A. J Phys Chem A. 1997;101:9801–9812. [Google Scholar]
- (54).Fourmigue M. Acc. Chem. Res. 2004;37:179–186. doi: 10.1021/ar030214w. [DOI] [PubMed] [Google Scholar]
- (55).Wescott BL, Gruhn NE, Michelsen LJ, Lichtenberger DL. J. Am. Chem. Soc. 2000;122:8083–8084. [Google Scholar]
- (56).Gruhn NE, Michelsen LJ, Westcott BL. Inorg. Chem. 2002;41:5907–5911. doi: 10.1021/ic0256056. [DOI] [PubMed] [Google Scholar]
- (57).Doran M, Hillier IH, Seddon EA, Seddon KR, Thomas VH, Guest MF. Chemical Physics Letters. 1979;63:612–614. [Google Scholar]
- (58).Berry M, Garner CD, Hillier IH, Macdowell AA. Inorg. Chem. 1981;20:1962–1965. [Google Scholar]
- (59).Dibella S, Gulino A, Lanza G, Fragala IL, Marks TJ. J. Phys. Chem. 1993;97:11673–11676. [Google Scholar]
- (60).Di Bella S, Lanza G, Gulino A, Fragala I. Inorg. Chem. 1996;35:3885–3890. doi: 10.1021/ic951457q. [DOI] [PubMed] [Google Scholar]
- (61).DiBella S, Lanza G, Fragala IL, Marks TJ. Organometallics. 1996;15:205–208. [Google Scholar]
- (62).A reviewer commented on the 5 kcal mol-1 difference in the barrier energies of the Mo and V d1 configurations at 25°. Our calculated fragment wavefunction analysis suggests that this difference cannot be specifically ascribed to fragment energy matching and/or overlap effects of the fragment orbitals and that this energy difference is due to subtle differences between the Mo and V cases. In the d0 electron configurations a larger barrier energy for V is also observed. .
- (63).Domercq B, Coulon C, Fourmigué M. Inorg. Chem. 2001;40:371–378. doi: 10.1021/ic000550y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.