

Abstract
We chose crambin, a hydrophobic and water-insoluble protein originally isolated from the seeds of the plant Crambe abyssinica, as a model for NMR investigations of membrane-associated proteins. We produced isotopically labeled crambin(P22,L25) as a cleavable fusion with staphylococcal nuclease and refolded the protein by an approach that has proved successful for the production of proteins with multiple disulfide bonds. We used NMR spectroscopy to determine the three-dimensional structure of the protein in two membrane-mimetic environments: in a mixed aqueous-organic solvent (75%/25%, acetone/water) and in DPC micelles. With the sample in the mixed solvent, it was possible to determine (>NH···OC<) hydrogen bonds directly by the detection of h3JNC′ couplings. H-bonds determined in this manner were utilized in the refinement of the NMR-derived protein structures. With the protein in DPC micelles, we used manganous ion as an aqueous paramagnetic probe to determine the surface of crambin that is shielded by the detergent. With the exception of the aqueous solvent exposed loop containing residues 20 and 21, the protein surface was protected by DPC. This suggests that the protein may be similarly embedded in physiological membranes. The strategy described here for the expression and structure determination of crambin should be applicable to structural and functional studies of membrane active toxins and small membrane proteins.
Keywords: membrane associated protein, protein-lipid interactions, thionin, crambin
Introduction
Although methodological advances, such as TROSY1 and isotope labeling approaches,2 have enabled recent NMR investigations of β-barrel membrane proteins,3–5 solution-state structural investigations of membrane proteins have lagged behind those of water-soluble proteins. In general, membrane proteins have proven to be more difficult than water soluble proteins to express, purify, and refold. Additionally, structural and functional studies of membrane proteins require a membrane-mimetic environment – usually, detergent micelles, bicelles, lipid bilayers, or lipid vesicles.6–8 These requirements have hampered structural investigation of membrane proteins by X-ray crystallography and NMR spectroscopy.
Crambin, a highly hydrophobic plant protein first isolated from the seeds of the plant Crambe abyssinica,9 is a member of the thionin family of membrane-active plant toxins.10, 11 Thionins were first isolated from cereal grains as protein-lipid complexes. The presence of amphipathic helices suggested that crambin might form a complex with lipid molecules, and the protein has been successfully incorporated into lipid vesicles.12
The three-dimensional structure of crambin has been determined by X-ray crystallography from crystals grown in 50% ethanol13 as well as by 1H NMR spectroscopy in a mixed organic-aqueous solvent mixture consisting of 75% acetone/25% water.14 An X-ray structure of crambin(P22,L25/S22,I25) has been determined at exceptionally high resolution (0.54 Å),15 and thus the molecule is of considerable interest for model studies. Crambin has been also used widely in developing methodology for the determination of protein structure from NMR data.14, 16–19
Here we report the system we developed for producing crambin in good yield from Escherichia coli. This system enabled us to prepare the first samples of this protein labeled with stable isotopes (15N and 13C) for multinuclear magnetic resonance investigations. Whereas naturally occurring crambin shows recognizable sequence micro heterogeneity at positions 22 (Pro/Ser) and 25 (Leu/Ile), recombinant crambin can be produced as a homogeneous peptide. We used NMR spectroscopy to determine the three-dimensional structure of crambin(P22,L25) under two conditions: in 75% acetone/25% water and in DPC micelles in aqueous solution. Although solution structures of proteins have been determined in organic/aqueous solvent mixtures 20, 21 and in detergent micelles,3–5 to our knowledge, this is the first comparison of the structure of the same protein determined under both conditions. With the protein in DPC micelles, we determined, by the use of manganous ion as an aqueous paramagnetic probe, the surface of crambin that is shielded by the detergent. This provided basic information about the possible interaction between this membrane-associated protein and a lipid bilayer.
Experimental Section
Cloning, expression and purification of crambin
Profs. C. M. Rienstra and M. M. Teeter generously supplied a pET23 plasmid containing DNA coding for crambin(P22,L25). We isolated the NdeI/BamHI fragment of the plasmid DNA, which codes for the full sequence of crambin(P22,L25), and inserted it into pET3a/SNase, a construct designed to produce a protein fused to the C-terminus of staphylococcal nuclease (SNase)22 with an engineered methionine residue to serve as a cyanogen bromide (CNBr) cleavage site and with the SNase methionines mutated to alanines to simplify the cleavage products.23 The plasmid pET3a/SNase-crambin was transformed to E. coli strain BL21 (DE3) pLysS. The procedures used to produce crambin were analogous to those described for the production of brazzein23 with minor modification (Figure 1). Briefly, cells were harvested and disrupted by three freeze/thaw cycles. Most of the fusion protein was expressed in inclusion bodies. The inclusion bodies were solubilized in 6 M guanidinium chloride, and the resulting solution was dialyzed against 0.1% acetic acid with several changes. The SNase-crambin fusion protein was cleaved by CNBr in 0.1 M HCl.24 The cleavage mixture was dissolved in the refolding solution (2 M urea, 100 mM Tris, 8 mM cysteine, 1 mM cystine, pH 8.0)25 and loaded onto an SP-Sepharose column. Most of crambin remained in the flow-through, whereas SNase and uncleaved SNase-crambin were retarded on the SP-Sepharose column. Further purification was performed by reversed-phase HPLC (RP-HPLC) using a Vydec C4 column (250 × 10 mm) with an acetonitrile gradient from 0 % to 70 % in the presence of 0.1 % trifluoroacetic acid. Crambin began to elute at the acetonitrile concentration of ~37 %. Fractions containing crambin mixed with uncleaved fusion protein and SNase were re-chromatographed on RP-HPLC. The fractions containing crambin were pooled and lyophilized, and the protein powder was stored at −20 °C. The total yield of purified crambin from 1 L culture was about 5 mg.
Figure 1.
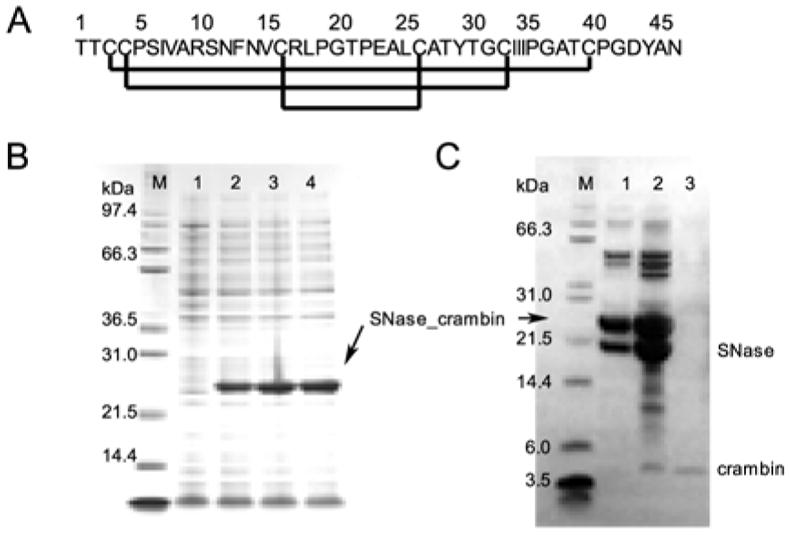
Sequence, expression, and purification of crambin(P22,L25). (A) Sequence of crambin showing the conserved disulfide bonds found in thionins. (B) SDS-PAGE showing expression of the fusion protein following induction with IPTC: (Lane M) molecular weight markers; (lane 1) before induction, (lane 2) 1.5 h post induction; (lane 3) 3 h post induction; (lane 4) 4.5 h post induction. (C) SDS-PAGE of fractions eluted from RP-HPLC following CNBr cleavage: (Lane M) molecular weight markers; (lanes 1 and 2) two fractions eluted in the time course near the acetonitrile gradient level of 37% (these fractions were re-chromatographed) ; (lane 3) purified crambin.
NMR sample preparation
To prepare isotope-labeled crambin, cells were grown in M9 minimal medium containing 15NH4Cl (1 g/L) and/or 13C glucose (2 g/L). The lyophilized crambin powder was dissolved in 75 % acetone-D6 (Fluka)/15 % H2O/10 % D2O (referred to as the mixed solvent) as in an earlier NMR study.14 Buffers used for crambin in detergent micelles contained 300 mM DPC-D38 or DHPC-D40 (Cambridge Isotope laboratories, Inc, MA), and 20 mM potassium phosphate, pH 6.0. The protein concentrations were 1 mM for crambin in the mixed solvent and 0.5 mM for crambin in detergent micelles. At this concentration, crambin in DPC micelles was stable for more than 6 months at room temperature, whereas crambin in DHPC micelles was stable for only a few hours at 25 °C, with a white precipitate forming irreversibly.
NMR spectroscopy
NMR experiments were performed at 25 °C on Varian Inova 600 and 800 MHz spectrometers, each equipped with a triple-resonance ColdProbe™ and on a Bruker DMX 500 and 750 MHz spectrometer with a conventional triple-resonance probe or with CryoProbe™. All spectrometers were equipped with pulsed field gradients. Backbone assignments were deduced from HSQC, HNCO, HNCA, CBCA(CO)NH, and HNCACB spectra. Side chains were assigned from HCCH-TOCSY, C(CO)NH, and HC(CO)NH spectra. Distance constraints for crambin in the mixed solvent and in DPC micelles were obtained from three-dimensional (3D) 15N- and 13C- NOESY-HSQC spectra with 150 ms mixing times. It was possible to resolve h3JNC′ couplings only with the double-labeled crambin sample in mixed solvent (Table 1). Coupling data were acquired on a Bruker 600 MHz spectrometer (at the Mayo Clinic and Foundation, MN) equipped with a CryoProbe™. Details of the 3D HNCO-J pulse sequence used have been published.26 All 1H dimensions were referenced to the methyl signal of DSS at 25 °C. 13C and 15N nuclei were referenced to indirectly to DSS. Spectra were processed with nmrPipe27 and analyzed with NMRView.28 The assigned chemical shift values for crambin(P22,L25) in the mixed solvent and in DPC micelles were deposited at BioMagResBank as entries 6455 and 6504, respectively; and h3JNC′ values for crambin(P22,L25) in the mixed solvent were deposited under BMRB 6455.
Table 1.
h3JNC′ couplings for H-bond donor and acceptors in crambin.
| Donor (N-H) | Acceptor (C=O) | h3JNC′ Hz) |
|---|---|---|
| 3 | 33 | 0.60 ± 0.20a |
| 4 | 46 | 0.46 ± 0.05 |
| 10 | 6 | 0.31 ± 0.05 |
| 11 | 7 | 0.60 ± 0.10 |
| 12 | 8 | 0.58 ± 0.05 |
| 13 | 9 | 0.63 ± 0.05 |
| 14 | 10 | 0.59 ± 0.05 |
| 15 | 11 | 0.46 ± 0.10 |
| 16 | 12 | 0.50± 0.05 |
| 17 | 13 | 0.37 ± 0.05 |
| 26 | 22 | 0.36 ± 0.06 |
| 27 | 23 | 0.40 ± 0.10 |
| 28 | 24 | 0.20 ± 0.10 |
| 29 | 25 | 0.27 ± 0.06 |
| 31 | 27 | 0.46 ± 0.05 |
| 33 | 3 | 0.80 ± 0.20a |
| 35 | 1 | 0.66 ± 0.05 |
| 45 | 40 | 0.18 ± 0.09 |
| 46 | 4 | 0.39 ± 0.05 |
Errors are large because of overlap with the intra-residue 2JNC′ couplings.
For crambin in the mixed solvent, relaxation data were acquired on a Bruker 500 MHz spectrometer with: the 15N T1 relaxation delays were 10, 100, 300, 550, 800, 1100, 1400, and 1800 ms, and the 15N T2 relaxation delays were 8, 24, 48, 80, 112, 152, 200, and 240 ms. For crambin in DPC micelles, relaxation data were acquired on a Varian 600 MHz spectrometer: the 15N T1data relaxation delays were 10, 100, 300, 500, 700, 900, 1200, 1500, and 2000 ms, and the 15N T2. delays were 10, 30, 50, 70, 90, 130, and 150 ms. For both samples, 1H-15N NOE data were acquired with and without a 3-s proton saturation period. The Rate Analysis protocol in the NMRView software was used to derive T1 and T2 values from the fitted data, and HetNOE protocol in the NMRView software was used to calculate the 1H-15N NOE values.
Structure determination
The structure of crambin(P22,L25) in the mixed solvent was calculated from 539 distance constraints, 40 torsion angle constraints (20 φ and 20 ψ), and 38 H-bond constraints. The structure of crambin(P22,L25) in DPC micelles was calculated from 637 distance constraints, 38 torsion angle constraints (19 φ and 19 ψ), and 38 H-bond constraints (Tables 1 and 2). With each NOESY data set, distance boundaries were calibrated by use of the NoeAnalysis tool in the NMRView software and grouped into three distance regions, 1.8–3.0 Å, 1.8–4.0 Å, and 1.8–5.0 Å, corresponding to strong, medium, and weak NOE intensities. The TALOS software29 was used to determine torsion angle constraints from the assigned chemical shift values. H-bond constraints identified from h3JNC′ values determined for crambin(P22,L25) in the mixed solvent were used in both structure calculations: assumed distances were 2.0 Å for HN–O and 3.0 Å for N–O. Although these constraints were not observable for crambin(P22,L25) in DPC micelles, TALOS analysis of chemical shifts indicated the same secondary structure and justified our use of these constraints in this structural model. The structures were calculated on a SGI Altix 3300 Linux workstation by using the simulated annealing protocols in Xplor-NIH (Version 2.9.3).30 The best 20 conformers from 100 simulated annealing structures were selected on the basis of energy to represent the structure of crambin(P22,L25) in the two environments. These were then subjected to water refinement (DPC micelle conformers) and DMSO refinement (mixed solvent conformers). The OPLS force field was used for the refinements according to protocols19 incorporated in the ARIA1.2 software.31 These refinements improved the quality of both structures as judged from improvements in Ramachandran plots (see Supporting Information). Atomic coordinates for these conformers were deposited in the Protein Data Bank: accession codes for crambin(P22,L25) in the mixed solvent were 1YV8 (with simulated annealing refinement from Xplor-NIH), 2EYA (with DMSO solvent refinement), and 2EYB (with water refinement), and for crambin(P22,L25) in DPC micelles 1YVA (with simulated annealing refinement from Xplor-NIH), 2EYC (with DMSO solvent refinement) and 2EYD (with water refinement).
Table 2.
Statistics for the structural models of crambin(P22,L25) in the mixed solvent and in DPC micelles.
| Parameter | Crambin in the mixed solventa | Crambin in DPC micellesa |
|---|---|---|
| Number of experimental constraints | ||
| NOE distance constraints | ||
| All | 539 | 637 |
| Intraresidue (i = j) | 139 | 158 |
| Sequential (|i – j| = 1) | 180 | 211 |
| Medium range (1 < |i – j| < 5) | 97 | 154 |
| Long range (|i – j| ≥ 5) | 123 | 114 |
| Torsion angle constraints (ϕ and ψ) | 40 | 38 |
| Hydrogen bond constraints | 38 | 38 |
| Rmsd from experimental distance restrains (Å)b | 0.014 ± 0.001 | 0.029 ± 0.001 |
| Rmsd from torsion angle constraints (°)b | 0.044 ± 0.055 | 0.027 ± 0.056 |
| Rmsd from idealized covalent geometry | ||
| Bonds (Å) | 0.002 ± 0.0001 | 0.003 ± 0.0001 |
| Angles (°) | 0.550 ± 0.003 | 0.625 ± 0.010 |
| Improper (°) | 0.381 ± 0.005 | 0.439 ± 0.012 |
| Ramachandran plotc | ||
| Most favored regions | 88.7 | 79.6 |
| Additionally allowed | 10.3 | 17.7 |
| Generously allowed | 1.0 | 2.0 |
| Disallowed | 0.0 | 0.7 |
| Coordinate precision (Å) | ||
| Rmsd of backbone atoms to the mean (Å) | 0.79 | 0.69 |
| Rmsd of all heavy atoms to the mean (Å) | 1.03 | 1.02 |
| Comparison with the previous structuresd | ||
| Rmsd with the crystal structure, 1EJG | 0.77 (0.60) | 0.80 (0.72) |
| Rmsd with the solution structure, 1CCN | 1.15 (0.60) | 1.23 (0.67) |
The SA ensemble corresponds to the final 20 structures from 100 simulated annealing structures.
None of the structures exhibited interproton distance violations of > 0.5 Å or torsion angle violations of >5°.
PROCHECK NMR40 was used for the calculation of Ramachandran statistics.
Numbers outside parentheses are backbone rmsd values for the entire molecule and numbers inside parentheses are backbone rmsd values over regions of defined secondary structure (residues 2–3, 7–17, 23–30, 33–34)
Solvent accessibility of crambin in DPC micelles
Aliquots of a concentrated solution of MnCl2 were added to 15N labeled crambin in DPC micelles up to final concentrations of 1, 5, and 10 mM. 1H-15N HSQC spectra were recorded, and the intensities of the signals were compared to those from the spectrum without Mn2+.
Results
Expression and purification of isotope labeled crambin
Previous attempts to express crambin alone were unsuccessful, probably because of its hydrophobic nature and large number of cysteine residues. Crambin has been expressed as a fusion with maltose binding protein, but the final yield was low.32 We found that crambin(P22,L25) could be expressed well in E. coli as a C-terminal fusion with modified staphylococcal nuclease (SNase), an approach that has been used successfully with other proteins containing multiple disulfide bonds.22, 23 The expression and purification of SNase-crambin(P22,L25) fusion protein was followed by SDS-PAGE analysis (Figure 1). The yield of the crude fusion protein from 1 L unlabeled medium was more than 100 mg.
About 50% of the fusion protein was cleaved after incubation with CNBr for 24 h. The limited yield probably resulted from the formation of an uncleavable byproduct at the Met–Thr cleavage site during the CNBr reaction. SP-Sepharose column chromatography was performed in a buffer containing 2 M urea, 8 mM cysteine and 1 mM cystine; under these conditions crambin was solubilized and refolded, but SNase was partly unfolded. As a result, the SP-Sepharose chromatography step did not separate crambin cleanly from SNase and the remaining fusion protein. RP-HPLC of the protein mixture eluted from the SP-Sepharose column gave overlapped peaks at about 37% acetonitrile (Figure 1C, lanes 1 and 2) and a small crambin peak at higher acetonitrile (Figure 1C, lane 3). The RP-HPLC chromatography step also separated misfolded and folded protein: misfolded crambin(P22,L25) eluted at a lower acetonitrile concentration than folded crambin(P22,L25).25 Fractions containing crambin mixed with SNase and uncleaved fusion protein were re-chromatographed on RP-HPLC to recover additional folded crambin. The yield of labeled protein from 1 L culture was sufficient for the subsequent NMR studies.
Comparison of the NMR chemical shifts of crambin(P22,L25) in the different environments
We used conventional triple resonance methods to determine nearly complete chemical shift assignments for crambin(P22,L25) in both the mixed solvent and DPC micelle environments. The participation of all cysteines in disulfide bonds was confirmed from their 13Cβ chemical shift values (all between 38 and 49 ppm). We observed large changes in the positions of the backbone 1HN and 15N signals between the two solvents, but they were not confined to particular parts of the protein (Figure 2). Previous 1H NMR chemical shift values are available (BMRB 4509) for crambin(S22,I25) (variant isolated by chromatography from plant-derived protein) in the mixed solvent;17 our corresponding 1H chemical shift values for crambin(P22,L25) are very similar with the exception of signals from the variant residues 22 and 25.
Figure 2.

1H-15N HSQC spectra of crambin(P22,L25): (A) in the mixed solvent; (B) in DPC micelles. Backbone assignments are indicated by residue numbers and amino acid code. (C) Overlaid 1H-15N HSQC spectra of crambin in DPC micelles (black) and in DHPC micelles (red). Residues showing significant changes are labeled.
Crambin was also incorporated into DHPC micelles. Overlay of the 1H-15N HSQC spectra (Figure 2C) of DHPC-solubilized crambin (red peaks) and DPC-solubilized crambin (black peaks) shows that most of the amide 1H-15N resonances have very similar chemical shifts. This result suggests that the structures of crambin in the two detergents are very similar. Residues exhibiting chemical shift differences greater than 0.05 ppm for 1H or 0.3 ppm for 15N are labeled in Figure 2C. These comprise residues in the first (I7–L18) and second (E23–T30) α-helices and in the loop connecting them (P19–P22). Of these, the largest differences are in residues L18, G20, and T21 (in or near the loop) and residue Y29. It was not possible to carry out more detailed studies of crambin in DHPC micelles, because the protein precipitated after a few hours.
Dynamics of crambin in the two solvent systems
We determined 15N T1 and T2 relaxation values for the backbone residues of crambin in the two membrane-mimetic environments (Supporting Information). The correlation times for the protein, as estimated from T1/T2 ratios,33 were 3.1 ns for crambin in the mixed solvent and 9.8 ns for crambin in DPC micelles. The value of 3.1 is that expected for a monomeric protein of 46 amino acids in a solvent of low viscosity. The value of 9.8 corresponds to a spherical particle of about 20 kD in an aqueous environment. Under the assumption that crambin and DPC form a tight complex, one estimates from this that 50–60 DPC molecules (each with MW 352) are associated with each crambin molecule.
Detection of H-bonds
With the crambin sample dissolved in 75% acetone/25% water it was possible to detect h3JNC′ couplings that reported the presence of 19 H-bonds (Table 1). On the basis of the X-ray crystal-structure (1EJG), the crambin backbone has 22 H-bonds shorter than 2.3 Å. The undetected H-bonds (18 →15, 20 → 17, 44 → 41) in the X-ray structure are highly bent (H···O–C angle ~130º). The H-bonds identified in this way were incorporated as distance constraints into the structure determinations.
Solution structures of crambin in the two membrane mimetic environments
The backbone rmsd value of the 20 structure ensemble to the mean structure was 0.79 Å for crambin(P22,L25) in the mixed solvent and 0.69 Å for the protein in DPC micelles (Table 2). The structures (Figure 3) show two well defined α-helices, I7–L18 and E23–T30, and two short β-sheets, T2–C3 and I33–I34. The slightly larger rmsd value for the family of conformers in the mixed solvent can be attributed to fewer NOE constraints in loop region (P19–P22) than in the crambin in DPC micelles. This suggests that the flexibility of the loop is restricted by interaction with the detergent micelle. The C-terminal loop (P36–T39) is somewhat disordered in both structures; however, the C-terminus itself is ordered in both, probably because of the presence of a disulfide bond (C3–C40) and an H-bond between the amide proton of C4 and the carboxyl oxygen of N46.
Figure 3.
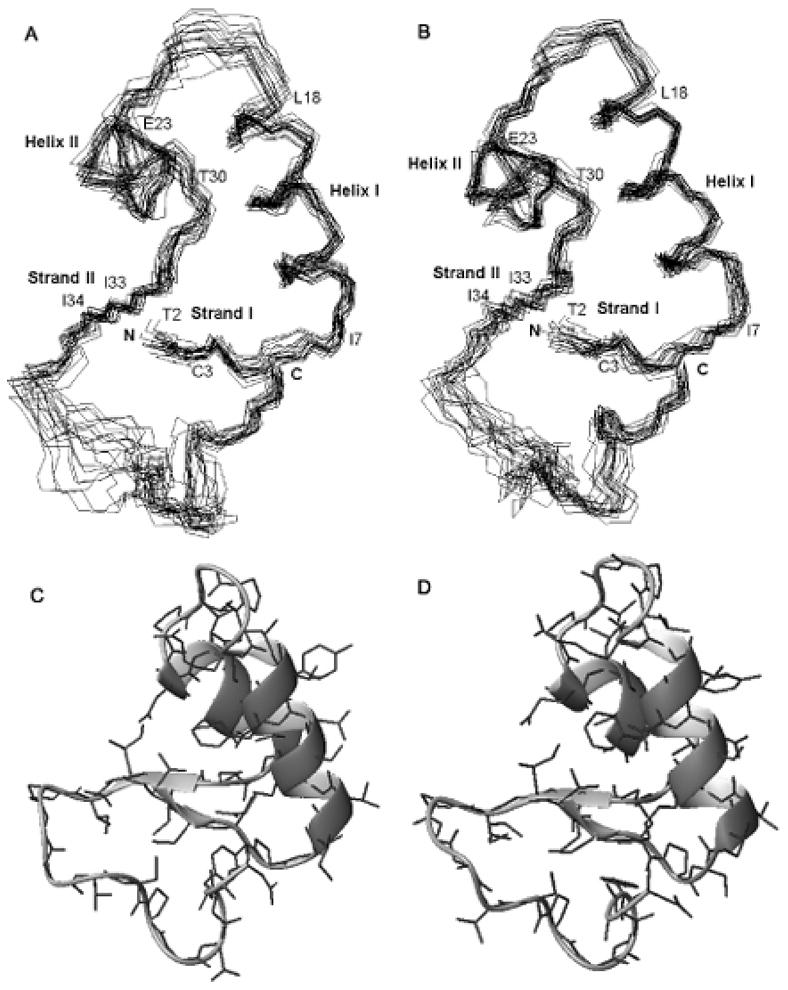
Ensembles of 20 conformers representing the structure of crambin(P22,L25): (A) in the mixed solvent; (B) in DPC micelles. Ribbon representations with side chains of the lowest energy conformers: (C) in the mixed solvent; (D) in DPC micelles. Secondary structural elements are indicated: strand I (T2–C3), helix I (I7–L18), helix II (E23–T30), and strand II (I33–I34).
In comparing the solution structures of structures of crambin(P22,L25) in the mixed solvent and in DPC micelles, in regions of secondary structure the all heavy atom rmsd value was 0.95 Å and the backbone heavy atom rmsd value was 0.61 Å. Thus the two structures are very similar despite the fact that the input data for the model in DPC micelles contained several NOEs between secondary structure elements not observed for crambin in the mixed solvent. For example, in DPC micelles 1Hδ3 of I34 showed NOE cross-peaks with amide protons from all residues E23–T28, whereas in the mixed solvent 1Hδ3 of I34 exhibited cross-peaks only with amide protons of A24 and A27 (Figure 4). Similarly, in DPC micelles the side-chain protons of F13 showed cross-peaks not only with the amide proton of A27 but also with the amide protons of C26 and T28. F13 is situated in the middle of helix I and C26–T28 are in helix II; thus, NOE cross-peaks were observed between two α-helices in the DPC micelles but not in the mixed solvent. In addition, NOE cross-peaks between 1Hδ3 of I34 and amide protons from E23–T28, which were observed in DPC micelles but not in mixed solvent, indicate a closer apposition of helix II and strand II in the micellar environment. The additional NOEs observed for crambin(P22,L25) in DPC micelles were fully consistent with available restraints for the protein in the mixed solvent, and thus did not support any structural difference.
Figure 4.

2D strips at the indicated 15N chemical shift from 3D 1H-15N NOE-HSQC data sets. The results show different patterns of 1H-1H NOE cross-peaks from crambin(P22,L25) in (A) in the mixed solvent and (B) in DPC micelles. Cross-peaks of interest are asterisked, and the corresponding atoms depicted to the right. Experiments were done with 150 ms mixing times.
The structures of crambin(P22,L25) in the mixed solvent and in DPC micelles determined here are each very similar (Figure 3) to the high-resolution crystal structure model for crambin(P22,L25) (1EJG: 15). We also compared these NMR structures with an earlier NMR structure (1CCN) of crambin(P22,L25) (unlabeled protein isolated by chromatography from plant-derived protein)14 in the same mixed solvent. The structures are similar in regions of secondary structure, but the P19–P22 loop is less ordered in 1CCN than in the X-ray structure (1EJG) or present NMR structures (1YV8, 1YVA). Rmsd values between the structures determined here and previous structures are presented in Table 2.
Residues of crambin shielded by the detergent
The accessibility of crambin in DPC micelles to aqueous solvent was deduced from perturbations of amide 1H-15N resonances caused by the addition of the paramagnetic ion Mn2+ (Figure 5A). Residues 20 and 21 were the most sensitive and disappeared upon addition of 5 mM Mn2+. This result suggests that the loop containing these residues is highly solvent exposed; signals were not detected from the adjacent prolines (P19 and P22) because of their lack of an HN atom. Residues showing the next level of sensitivity were N12, T30, G31, and D43, which are mostly hydrophilic amino acids. These surface residues appear to be only slightly shielded by the detergent and may be located at or just under the shell structure of the micelles. Residues most protected from the effects of added Mn2+ are located in the two-stranded β-sheet and adjacent regions; this suggests that these residues are fully covered by detergent in the DPC micelles. Residues of crambin in DPC micelles that exhibited the highest aqueous solvent exposure (< 35% original intensity upon addition of 5 mM Mn2+) are highlighted (dark gray) on the 3D structure in Figure 5B.
Figure 5.
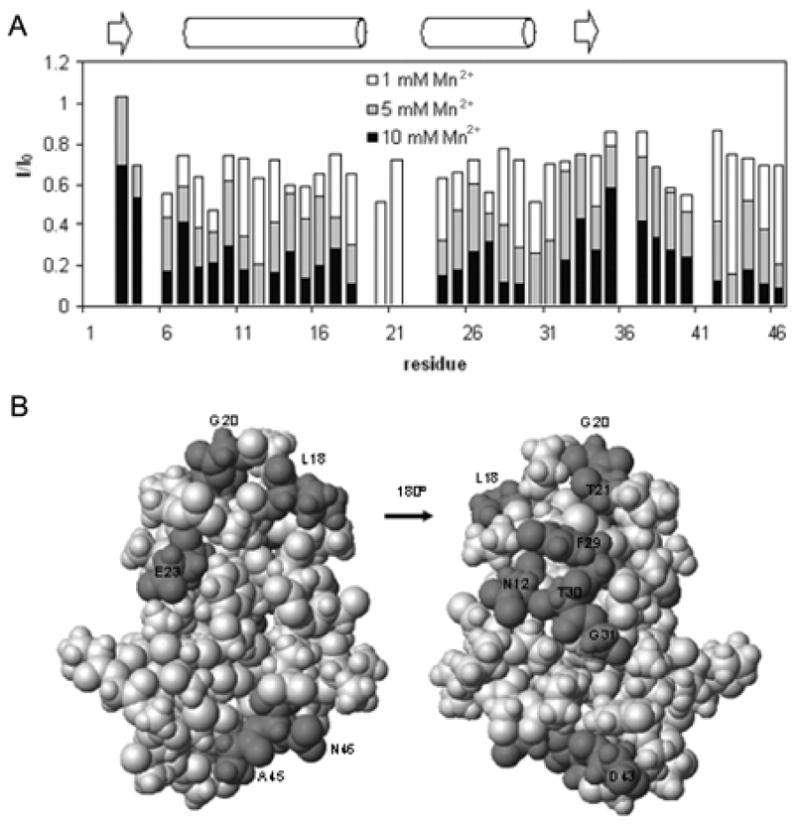
Solvent accessibility of crambin in DPC micelles. (A) Attenuation of HSQC 1H-15N signal intensities by the addition of Mn2+. The bar graph shows the intensity ratio I/I0, where I0 is the peak height in the absence of Mn2+ and where I is the intensity of the signal at a given concentration of added Mn2+. Bars are shaded to indicate different concentrations of Mn2+ as indicated in the key; the tops of each bar represent the I/I0 value at that Mn2+ concentration. The secondary structure of crambin is shown for reference. (B) Darker gray highlights residues of crambin in DPC micelles that exhibited the highest aqueous solvent accessibility (< 35% original intensity upon addition of 5 mM Mn2+) mapped onto the 3D structure.
Discussion
Thionins play an important role in the defense mechanism of plants.34 Although it has been reported that β-purothionin forms ion channels in lipid membranes,35 the mechanism of the membrane activity of thionins is still unclear. In the absence of efficient expression and isotope labeling procedures, all prior solution structures of thionins, including crambin, were determined by homonuclear 1H NMR spectroscopy.14, 36–39 Previous efforts to express crambin alone or as a MBP fusion protein in E. coli were unsuccessful because of the toxicity during the expression or low yield of production. In this work, we successfully expressed large quantity of crambin in E. coli as a fusion protein with staphylococcal nuclease. This enabled the production of stable isotope labeled protein for investigations of its solution structure in two membrane-mimetic environments and its interaction surface with DPC. The strategy reported here for crambin should be applicable to a wide range of thionins and should enable multinuclear NMR studies aimed at understanding the mechanism their membrane activity.
For the structure calculation of crambin we used H-bond restraints derived from h3JNC′ values determined for crambin(P22,L25) in the mixed solvent. Because of the shorter correlation time, it was not possible to detect these couplings with protein in DPC micelles. The similarity of the backbone chemical shifts for crambin(P22,L25) in DPC micelles to those of the protein in the mixed solvent provided similar TALOS-derived constraints and justified our use of these H-bonds in the second structure.
Even though the structures of the protein in the two membrane-mimetic solvents are similar, comparison of interhelical distances suggest that the two α-helices may be more tightly packed in crambin(P22,L25) in DPC micelles than in the mixed solvent (Table 3). The helices may also be more tightly packed in DPC micelles than in the crystal structure (Table 3). We suggest that the driving force for the observed helical rearrangement may be stress from the micelle molecules. This stress would lead to different effects along the helices. The helical ends located toward the center of crambin (represented by the 9–30 distance) are similar in all three structures, because they are constrained by the flanking β-sheets; they simply serve as a pivot. By contrast, the midpoints of the helices and the ends close to the solvent-exposed loop are less constrained and are expected to be influenced by the charged groups of the DPC micelle. Thus, incorporation of crambin into detergent micelles may introduce stress on the two helical ends that lie beneath the detergent shell (Figure 5) and may lead to the observed helical rearrangement.
Table 3.
Interhelical distances of crambin in each condition.
| residues | crambin in the mixed solvent (this work) | crambin in DPC micelles (this work) | PDB1EJG | |
|---|---|---|---|---|
| Cα-Cα distancesa | 9–30 | 5.5 ± 0.5 | 5.3 ± 0.6 | 5.4 |
| 13–26 | 5.6 ± 0.4 | 4.7 ± 0.4 | 5.7 | |
| 17–23 | 6.5 ± 0.5 | 5.9 ± 0.4 | 7.1 |
Distances (Å) between the Cα atoms of the residues specified in the various structural models. Distances between chosen atoms were calculated using ‘CalcDist’ macro in MOLMOL.41
Interestingly the major chemical shift differences between crambin in DPC and DHPC micelles was found in the solvent-accessible loop. The DHPC molecule has two C6 hydrophobic tails, whereas DPC has a single, longer C12 hydrophobic tail. These results suggest that this loop is more solvent exposed in DHPC micelles than in DPC micelles. This greater exposure may account for the poor stability of crambin in DHPC micelles.
In conclusion, the expression and isotope labeling of crambin enabled the determination of the structure of the membrane-associated protein, crambin, in two membrane-mimetic environments: a mixed aqueous/organic solvent and detergent micelles. With crambin in the mixed solvent, hydrogen bonds were detected easily and precisely from J-couplings across H-bonds. These were incorporated to the structure calculation of the protein in both solvent systems. The structures of crambin in the mixed solvent and detergent micelles were almost identical. This result suggests that the structures of other membrane-active proteins stabilized by disulfide bridges could be investigated in such a mixed solvent. The paramagnetic perturbation study of crambin in DPC micelles revealed that most of the protein surface is poorly accessible to aqueous solution. Thus one can model the protein as sitting largely in the center of a membrane bilayer with only the hydrophilic loop sticking out (see the table of contents figure).
Supplementary Material
Ramachandran plots for structures of crambin in the mixed solvent and in DPC micelles with water (or DMSO) refinements, and 15N relaxation parameters (T1, T2, and 1H-15N NOE) for crambin in both solvent systems. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Professors Martha M. Teeter (Boston College) and Chad M. Rienstra (University of Illinois) for generously supplying the clone for crambin(P22,L25), Dr. Marco Tonelli for assistance with NMR experiments, and Dr. Fariba M. Assadi-Porter for helpful discussions on the cloning and purification of crambin(P22,L25). This work was supported by NIH grant RR02301.
Abbreviations
- crambin(P22, L25)
variant of crambin containing Pro22 and Leu25
- crambin(S22
I25), variant of crambin containing Ser22 and Ile25
- crambin(S22
I25/P22,L25), mixture of crambin(S22,I25) and crambin(P22,L25)
- DHPC
dihexanoylphosphatidylcholine
- DPC
dodecylphosphocholine
- mixed solvent used for NMR spectroscopy
75 % acetone-D6/15 % H2O/1 0 % D2O
- rmsd
root mean squared deviation
- SNase
staphylococcal nuclease
Footnotes
Data deposition: The assigned chemical shift values were deposited at BioMagResBank as entries 6455 and 6504 for crambin(P22,L25) in the mixed solvent and in DPC micelles, respectively; trans H-bond couplings for crambin(P22,L25) in the mixed solvent were deposited as entry (6455). Atomic coordinates for these conformers were deposited in the Protein Data Bank: accession codes for crambin(P22,L25) in the mixed solvent were 1YV8 (with simulated annealing refinement from Xplor-NIH), 2EYA (with DMSO solvent refinement), and 2EYB (with water refinement), and for crambin(P22,L25) in DPC micelles 1YVA (with simulated annealing refinement from Xplor-NIH), 2EYC (with DMSO solvent refinement) and 2EYD (with water refinement).
References
- 1.Pervushin K, Riek R, Wider G, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner KH, Kay LE. Annu Rev Biophys Biomol Struct. 1998;27:357–406. doi: 10.1146/annurev.biophys.27.1.357. [DOI] [PubMed] [Google Scholar]
- 3.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Nat Struct Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 4.Fernández C, Adeishvili K, Wüthrich K. Proc Natl Acad Sci USA. 2001;98:2358–2363. doi: 10.1073/pnas.051629298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang PM, Choy W, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Privé GG, Bishop RE, Kay LE. Proc Natl Acad Sci USA. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marassi FM, Opella SJ. Curr Opin Struct Biol. 1998;8:640–648. doi: 10.1016/s0959-440x(98)80157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arora A, Tamm LK. Curr Opin Struct Biol. 2001;11:540–547. doi: 10.1016/s0959-440x(00)00246-3. [DOI] [PubMed] [Google Scholar]
- 8.Fernández C, Wüthrich L. FEBS Lett. 2003;555:144–150. doi: 10.1016/s0014-5793(03)01155-4. [DOI] [PubMed] [Google Scholar]
- 9.Van Etten CH, Nielson HC, Peters JE. Phytochemistry. 1965;4:467–473. [Google Scholar]
- 10.Bohlmann H, Apel K. Annu Rev Plant Physiol Plant Mol Biol. 1991;42:227–240. [Google Scholar]
- 11.Florack DEA, Stiekema WJ. Plant Mol Biol. 1994;26:25–37. doi: 10.1007/BF00039517. [DOI] [PubMed] [Google Scholar]
- 12.Wallace BA, Kohl N, Teeter MM. Proc Natl Acad Sci USA. 1984;81:1406–1410. doi: 10.1073/pnas.81.5.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendrickson WA, Teeter MM. Nature. 1981;290:107–113. doi: 10.1038/290107a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonvin AM, Rullmann JA, Lamerichs RM, Boelens R, Kaptein R. Proteins. 1993;15:385–400. doi: 10.1002/prot.340150406. [DOI] [PubMed] [Google Scholar]
- 15.Jelsch C, Teeter MM, Lamzin V, Pichon-Lesme V, Blessing B, Lecomte C. Proc Natl Acad Sci USA. 2000;97:3171–3176. doi: 10.1073/pnas.97.7.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nilges M, Clore GM, Gronenborn AM. FEBS Lett. 1988;239:129–136. doi: 10.1016/0014-5793(88)80559-3. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Wu J, Gorenstein D, Braun W. J Magn Reson. 1999;136:76–85. doi: 10.1006/jmre.1998.1616. [DOI] [PubMed] [Google Scholar]
- 18.Spronk CA, Linge JP, Hibers CW, Vuister GW. J Biomol NMR. 2002;22:281–289. doi: 10.1023/a:1014971029663. [DOI] [PubMed] [Google Scholar]
- 19.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 20.Girvin ME, Rastogi VK, Abildgaard F, Markley JL, Fillingame RH. Biochemistry. 1998;37:8817–8824. doi: 10.1021/bi980511m. [DOI] [PubMed] [Google Scholar]
- 21.Dmitriev OY, Abildgaard F, Markley JL, Fillingame RH. Biochemistry. 2002;41:5537–5547. doi: 10.1021/bi012198l. [DOI] [PubMed] [Google Scholar]
- 22.Hinck AP, Walkenhorst WF, Westler WM, Choe H, Markley JL. Protein Eng. 1993;6:221–227. doi: 10.1093/protein/6.2.221. [DOI] [PubMed] [Google Scholar]
- 23.Assadi-Porter FM, Aceti DJ, Cheng H, Markley JL. Arch Biochem Biophys. 2000;376:252–258. doi: 10.1006/abbi.2000.1725. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Dillen L, Vanhoutte K, Van Dongen W, Esmans E, Claeys M. Anal Chem. 1996;68:3422–3430. doi: 10.1021/ac9602229. [DOI] [PubMed] [Google Scholar]
- 25.Bang D, Chopra N, Kent SB. J Am Chem Soc. 2004;126:1377–1383. doi: 10.1021/ja0385078. [DOI] [PubMed] [Google Scholar]
- 26.Juranic N, Moncrieffe MC, Likic VA, Prendergast FG, Macura S. J Am Chem Soc. 2002;124:4221–14226. doi: 10.1021/ja0273288. [DOI] [PubMed] [Google Scholar]
- 27.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 28.Johnson BA, Blevins RA. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 29.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 30.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. J Magn Res. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 31.Linge JP, Habeck H, Rieping W, Nilges M. Bioinformatics. 2003;19:315–316. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- 32.Lobb L, Stec B, Kantrowitz EK, Yamato A, Stojanoff V, Markman O, Teeter MM. Protein Eng. 1996;9:1233–1239. doi: 10.1093/protein/9.12.1233. [DOI] [PubMed] [Google Scholar]
- 33.Kay LE, Torchia DA, Bax A. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 34.Stec B, Markman O, Rao U, Heffron G, Henderson S, Vernon LP, Brumfeld V, Teeter MM. J Pept Res. 2004;64:210–224. doi: 10.1111/j.1399-3011.2004.00187.x. [DOI] [PubMed] [Google Scholar]
- 35.Hughes P, Dennis E, Whiteross M, Llewellyn D, Gage P. J Biol Chem. 2000;275:823–827. doi: 10.1074/jbc.275.2.823. [DOI] [PubMed] [Google Scholar]
- 36.Bloch C, Jr, Patel SU, Baud F, Zvelebil MJ, Carr MD, Sadler PJ, Thornton JM. Proteins. 1998;32:334–349. doi: 10.1002/(sici)1097-0134(19980815)32:3<334::aid-prot9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 37.Bruix M, Gonzalez C, Santoro J, Soriano F, Rocher A, Mendez E, Rico M. Biopolymers. 1995;36:751–763. doi: 10.1002/bip.360360608. [DOI] [PubMed] [Google Scholar]
- 38.Fant F, Vranken W, Broekaert W, Borresmans F. J Mol Biol. 1998;279:257–270. doi: 10.1006/jmbi.1998.1767. [DOI] [PubMed] [Google Scholar]
- 39.Romagnoli S, Fogolari F, Catalano M, Zetta L, Schaller G, Urech K, Giaanttasio M, Ragona L, Molinari H. Biochemistry. 2003;42:12503–12510. doi: 10.1021/bi034762t. [DOI] [PubMed] [Google Scholar]
- 40.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 41.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ramachandran plots for structures of crambin in the mixed solvent and in DPC micelles with water (or DMSO) refinements, and 15N relaxation parameters (T1, T2, and 1H-15N NOE) for crambin in both solvent systems. This material is available free of charge via the Internet at http://pubs.acs.org.