

Abstract
Thiacetazone (TAZ) and ethionamide (ETA) are, respectively, thiourea and thioamide-containing second line antitubercular prodrugs for which there is an extensive clinical history of cross-resistance in Mycobacterium tuberculosis. EtaA, a recently identified flavin-containing monooxygenase (FMO), is responsible for the oxidative activation of ETA in M. tuberculosis. We report here that EtaA also oxidizes TAZ and identify a sulfinic acid and a carbodiimide as the isolable metabolites. Both of these metabolites are derived from an initial sulfenic acid intermediate. Oxidation of TAZ by EtaA at basic pH favors formation of the carbodiimide, whereas neutral or acidic conditions favor formation of the sulfinic acid. The same metabolites are formed from TAZ by human FMO1 and FMO3. The sulfenic acid and carbodiimide metabolites, but not the sulfinic acid product, readily react with glutathione, the first to regenerate the parent drug and the second to give a glutathione adduct. These reactions that may contribute to the antitubercular activity and/or toxicity of TAZ.
Keywords: antituberculosis drug, drug metabolism, flavin-containing monooxygenase, thiacetazone, ethionamide, thioamide metabolism, thiourea metabolism
Introduction
Tuberculosis (TB) is the most serious infectious lung disease in the world. It claims over two million lives worldwide each year and dwells, hidden, in as many as two billion people (1). It is estimated that between 2005 and 2020, one billion people will be newly infected, over 125 million people will get sick, and 30 million will die of TB if control is not further strengthened (2). The worsening situation has prompted the World Health Organization to declare TB a global public health crisis. In the United States, despite the fact that the incidence of newly reported cases of tuberculosis has fallen steadily since 1992, TB still remains a growing concern, primarily because of the growing incidence of antibiotic-resistant strains of Mycobacterium tuberculosis (3). It was reported that around 13% of all the TB cases in the United States exhibited resistance to at least one of the first-line drugs (isoniazid, rifampin, pyrazinamide, streptomycin and ethambutol), and the extent of resistance rose significantly for patients with a prior episode of TB (4, 5). Of even greater concern is the appearance of multidrug-resistant (MDR) organisms that are resistant to at least isoniazid and rifampin. Patients with MDR-TB must fall back on a regimen of less-active, more expensive and/or more toxic second-line drugs (6–8).
The thioamide ethionamide (ETA) and the thiourea thiacetazone (TAZ) are two second line drugs for the treatment of tuberculosis (Figure 1). ETA is used for the treatment of patients infected with multidrug-resistant TB in the United States, while TAZ is widely used for the same purpose in developing countries (7, 9, 10). These drugs are associated with gastrointestinal disturbances, hepatotoxicity, and dermal hypersensitivity that can result in life-threatening skin reactions (11–13). It has long been known that both ETA and TAZ are pro-drugs that have to be converted to their active forms by mycobacterial enzymes (14).
Figure 1.
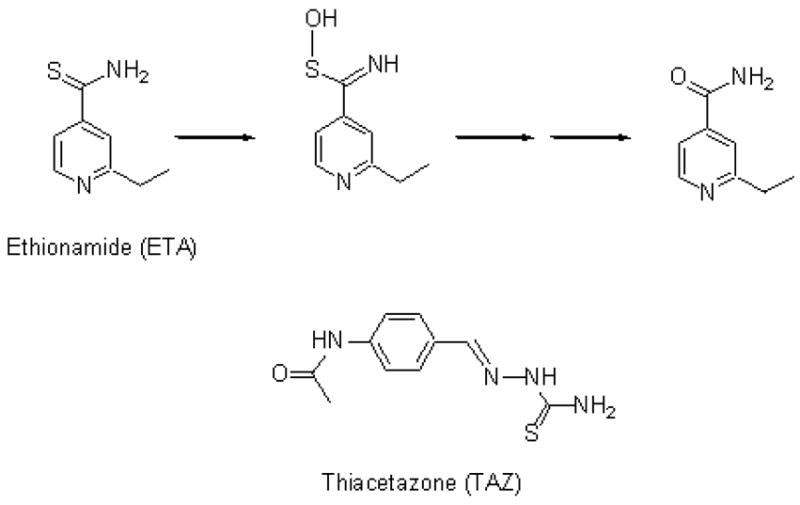
Chemical structures of the second line antitubercular drugs ETA and TAZ, and the known metabolic transformation of ETA.
In 2000, two laboratories (14, 15) independently showed that the M. tuberculosis gene etaA encodes a putative flavin-containing monooxygenase EtaA that oxidizes ETA, and that activation of ETA by EtaA is required for the antimycobacterial activity of the drug. Furthermore, the genetic evidence indicates that resistance to ETA and related drugs involves mutations that lower or eliminat the activity of EtaA (14, 15). The role of EtaA in activation of ETA was confirmed by the finding that overexpression of EtaA in Mycobacterium smegmatis resulted in ETA-hypersensitive mycobacteria. In 2002, Vannelli et al. successfully expressed, purified, and characterized the EtaA protein (16). These authors demonstrated in vitro that EtaA, in the presence of NADPH, catalyzes two oxidations of ETA, first to the thioamide S-oxide and subsequently to 2-ethyl-4-carboxamidopyridine (Figure 1) (16). Intensive efforts to characterize an intermediate in the oxidative conversion of the thioamide S-oxide to 2-ethyl-4-carboxamidopyridine have been unsuccessful due to the instability of the intermediate (T. Vannelli, L. Qian, unpublished results).
Very limited information is available concerning the metabolism and biological action of TAZ. There is an impressive clinical history of cross-resistance between ETA and TAZ not only in M. tuberculosis but also in the related pathogen Mycobacterium. leprae (17, 18). Indeed, genome analysis shows that an etaA gene ortholog is conserved in M. leprae despite the dramatically reduced genome size of that organism (19, 20). This suggests that EtaA is involved in the activation of thiocarbonyl drugs in both M. tuberculosis and M. leprae, a suggestion consistent with the observation that resistance of M. tuberculosis to both ETA and TAZ involves mutations in EtaA (14, 15).
Here we report the identification of two metabolites generated in vitro by the EtaA-or human FMO1-catalyzed oxidation of TAZ. The results not only establish that EtaA is responsible for TAZ activation but also identify two electrophilic metabolites that can react with glutathione (GSH) and thus may contribute to the cytotoxicity of this drug.
Experimental Procedures
Materials
TAZ, NADPH, trifluoroacetic acid, formic acid, H2O2, peracetic acid, sodium chlorite and cuprous chloride were purchased from Sigma-Aldrich. Human FMO1 and FMO3 were obtained from BD Biosciences (San Jose, CA). Buffer salts and CH3CN (HPLC grade) were purchased from Fisher Scientific. All other reagents and biochemicals, unless otherwise specified, were of the highest commercially available grade.
Expression and Purification of EtaA
The expression and purification of EtaA were performed as previously reported (16). Transformation of the Rv3854c gene into commercial (Invitrogen) competent DH5α Escherichia coli cells resulted in overexpression of the recombinant EtaA with a poly-His tag that does not interfere with its catalytic function. After Ni-NTA affinity chromatography, the heterologous expression of EtaA yielded 15 mg of purified protein/liter of medium.
Enzyme Incubations with TAZ
The enzyme reactions in 100 mM potassium phosphate buffer (pH 7.4) contained 150 mM NaCl, catalase (100 units/mL), superoxide dismutase (100 units/mL), bovine serum albumin (0.1 mg/mL), NADPH (1 mM) and an NADPH-regenerating system consisting of glucose-6-phosphate dehydrogenase (2 units/mL) and glucose-6-phosphate (2.5 mM). For the steady-state kinetic studies, the concentration of EtaA was 1 μM. Stock TAZ solutions (1, 2, 5, 10, 25, 50, and 100 mM) in DMSO were prepared and the incubations were initiated by adding TAZ at final concentrations from 10 to 500 μM. The final organic solvent concentration was held to 1% (v/v). After incubation at 37 °C for 10 min, the reactions were stopped by the addition of 100 μL of ice-cold CH3CN. The mixtures were diluted and transferred to Microcon centrifugal filters (Millipore, YM-10), which were then centrifuged at 10,000 g for 30 min at 4 °C. The filtrates were analyzed by HPLC and the kinetic parameters (Km and kcat) were determined using nonlinear analysis with Graph-Pad Prism software (Graph-Pad, San Diego, CA). For studies of the effect of pH on the reaction, a 300 nM concentration of the enzyme (EtaA, FMO1, or FMO3) was employed and the incubation time was 1 h at 37 °C. The oxidation of methimazole was used as a positive control for the activities of both FMO1 and FMO3. The pH effect was examined using the following buffers: 100 mM citrate (pH 5), 100 mM phosphate (pH 6.5, 7.5), 100 mM tricine (pH 9), and ethanolamine (pH 10). The ionic strength was held constant at 0.15 M by addition of NaCl. For the GSH capture experiments, the concentration of EtaA was 15 μM and that of TAZ 1 mM. Incubations were carried out in which GSH was added at concentrations ranging from 0.025 to 5 mM at the beginning of the incubation, after 30 min of the incubation, or after the enzyme incubation was quenched. The samples were analyzed as described below.
Analyses by High Pressure Liquid Chromatography (HPLC)
HPLC was performed on a Hewlett-Packard 1090 Series II instrument equipped with a photodiode array detector and a reverse-phase C18 column (Waters, 3.5 μm particle size, 4.6 mm i.d. × 150 mm, Symmetry) employing two buffers: A, H2O + 0.1% trifluoroacetic acid (TFA); and B, CH3CN + 0.1% TFA. The solvent flow rate was 1 mL/min and the eluent was spectroscopically monitored between 200 and 500 nm. The column was eluted from 0–25 min with a linear gradient from 5 to 30% buffer B, and from 25–30 min isocratically with 30% buffer B. The enzymatically produced metabolites were quantitated by comparing the integrated HPLC peak areas from the enzymatic reactions with the corresponding peak areas from standard curves prepared in the same manner using synthetic standards.
LC-MS Analyses
Liquid chromatography-mass spectrometry (LC-MS) was performed on a Waters Micromass ZQ coupled to a Waters Alliance HPLC system equipped with a 2695 separations module, a Waters 2487 Dual λ Absorbance detector, and a reverse-phase C18 column (Waters, 3.5 μm, 2.1 mm i.d. × 150 mm, Symmetry). The column was eluted with the following protocol at a flow rate 0.1 mL/min (buffer A, H2O + 0.1% formic acid (FA); buffer B: CH3CN + 0.1% FA): 0–5 min, 5% B (isocratic); 5–10 min, 5–20% B (linear gradient); 10–20 min, 20–30% B (linear gradient); and 20–30 min, 30% B (isocratic). The eluent was monitored at 280 and 320 nm. The mass spectrometer settings were as follows: mode, ES+; capillary voltage, 3.5 kv; cone voltage, 25 v; desolvation temperature, 250 °C.
Synthesis of TAZ-Sulfinic Acid
TAZ (100 mg) was slowly added to a mixture of H2O2 (1 mL, 20 eq) and acetic acid (4 mL) at 0 °C. The mixture was stirred at 0 °C for 5~10 min until the solution became clear. The solution was then poured into cold water (20 mL) and the mixture was allowed to stand at 4 °C for 0.5 h until a white precipitate appeared. After filtration, the filter cake was washed with cold water and dissolved in 5% aqueous KHCO3 (7 mL). After filtration, acidification of the filtrate with concentrated HCl to pH = 3 yielded 28 mg of a solid white product, which was recrystallized from EtOH and was subsequently dried on a lyophilizer: 1H NMR (d6-Me2SO, 400 MHz) δ 2.072 (s, 3H), 7.678 (d, J = 8.8 Hz, 2H), 7.833 (d, J = 8.8 Hz, 2H), 8.502 (s, 1H), 8.889 (s, 1H), 9.366 (s, 1H), 10.194 (s, 1H), and ppm 12.464 (br s, 1H); MS (ESI) m/z 269 (M++H).
Synthesis of TAZ-Carbodiimide
NaClO2 (30 mg/mL in H2O, 150 μL) and CuCl (saturated in H2O, 150 μL) were added to TAZ (100 mM, 150 μL) in 100 mM tricine buffer (pH 9) at room temperature. The mixture was stirred at 37 °C for 2 h. After filtration, the product was purified by preparative HPLC (Alltech, RP-C18, 5 μm, 22 mm i.d. × 250 mm, Alltima) using a linear gradient from 0 to 30% CH3CN (0.1% TFA) in 20 min (flow rate 20 mL/min, detection 280 nm). The product eluted at 16.4 min and fractions (1 mL) were collected, concentrated (lyophilizer), and characterized by NMR and MS: 1H NMR (d6-Me2SO, 400 MHz) δ 2.008 (s, 3H), 7.539 (d, J = 8.8 Hz, 2H), 7.591 (d, J = 8.8 Hz, 2H), 7.938 (s, 1H), 10.068 (s, 1H), and 11.154 ppm (s, 1H); MS (ESI) m/z 203 (M++H).
Reactions of TAZ with Chemical Oxidants
H2O2 (3 μL, 100 mM) or NaClO2 (10 μL, 30 mM in H2O) was added to TAZ (1 mM) in buffer (100 mM phosphate pH 6.5, pH 7.5, or 100 mM tricine pH 9). CuCl (10 μL, saturated in H2O) or NaClO2 (10 μL, 30 mM in H2O) was added to TAZ-sulfinic acid (1 mM) in tricine (pH 9). The reactions were stirred at 37 °C for 2 h or overnight. The crude reaction mixtures were filtered and the filtrates were analyzed by HPLC. The products were identified by comparing their elution times and UV spectra with those of synthetic standards. The reaction of [18O]H2O2 and TAZ was run under conditions identical to those described for H2O2 and TAZ.
Synthesis of TAZ-Carbodiimide-GSH Adduct
Synthetic TAZ-carbodiimide (1.5 mg, 7 mmol) in 300 μL of DMSO was added to GSH (11.5 mg, 37 mmol) in 700 μL of tricine buffer (pH 9) at room temperature. The mixture was stirred at room temperature for 1 h. After filtration, the filtrate was directly subjected to preparative HPLC (Alltech, reverse phase C18 column, 5 μm, 22 mm i.d. × 250 mm, Alltima) using a 20 min linear gradient from 0 to 30% CH3CN (0.1% TFA) at a flow rate 20 mL/min with the detector set at 280 nm. The product eluted at 13.5 min and fractions (1 mL) were collected, concentrated, and analyzed by NMR and MS: 1H NMR (d6-Me2SO with 5% D2O, 400 MHz) δ 1.965 (m, 2H), 2.008 (s, 3H), 2.324 (q, J = 7.4, 8.0 Hz, 2H), 2.457 (s, 2H), 3.28 (t, J = 10.8 Hz, 1H), 3.553 (m, 2H), 4.574 (t, J = 7.6 Hz, 1H), 7.601 (d, J = 8.4 Hz, 2H), 7.774 (d, J = 8.4 Hz, 2H), and 8.200 ppm (s, 1H); MS (ESI) m/z 510 (M++H).
MS/MS Analysis of the TAZ-Carbodiimide-GSH Adduct
MS/MS experiments were performed on a hybrid mass spectrometer (Thermo Finnigan) consisting of an LTQ (linear ion trap) front end and an FTICR (Fourier Transform Ion Cyclotron Resonance) back end. The molecular ion 510 was fragmented by CID (collision assisted dissociation) using helium as the collision gas. The collision energy was manually adjusted. The ions formed were analyzed in the FTICR at high mass resolution 50000 and mass accuracy of 5.0 ppm.
Results
Catalytic Oxidation of TAZ by EtaA
Incubation of EtaA and TAZ in the presence of NADPH resulted in the formation of two major metabolites with reverse phase HPLC retention times of 9.7 min (M1) and 17.5 min (M2) (Figure 2). The absorption spectrum of M1 exhibited a peak at 325 nm and a shoulder at approximately 230 nm. M2 had a similar UV spectrum except that the peak and the shoulder were shifted to 295 nm and 220 nm, respectively (Figure 3). No products were observed in incubations from which either EtaA or NADPH was omitted, or in incubations carried out under anaerobic conditions. NADH could not be substituted for NADPH in the incubation (data not shown). The requirement for both NADPH and O2 is consistent with involvement of the flavoprotein monooxygenase EtaA.
Figure 2.
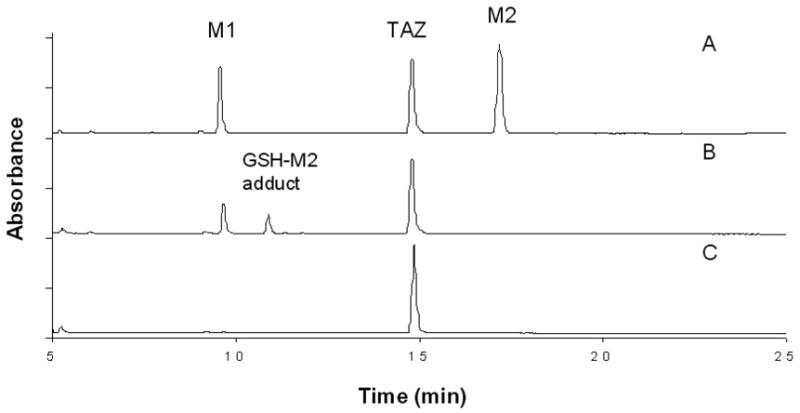
UV-HPLC chromatograms (280 nm) of the products from incubations of TAZ, NADPH and EtaA at pH 9.0 and 37 °C for 1h: (A) No GSH added; (B) GSH (2.5 mM) added after 30 min of the incubation; and C) GSH (5 mM) added at the beginning of the incubation.
Figure 3.
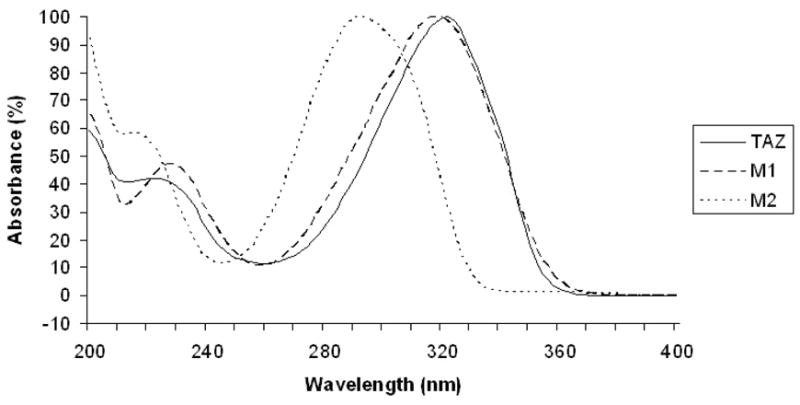
UV absorption of TAZ and its two metabolites, M 1 and M2, from the incubation of TAZ with EtaA and NADPH.
Analysis of the incubation mixtures by LC-MS identified the two metabolites, M1 and M2, formed by the action of EtaA on TAZ. The mass spectrum of M1 had a molecular ion [M+H]+ at m/z 269.09, with fragment ions at m/z 205.18 and 163.18, in accord with a structure in which TAZ has incorporated two oxygen atoms (M+H, 269 = 237+32) (Figure 4). This molecular ion supports identification of the metabolite as the TAZ-sulfinic acid. The molecular ion peak [M+H]+ of metabolite M2 at m/z 203.17 suggests that it is the carbodiimide generated by elimination of the oxidized sulfur atom (Figure 4).
Figure 4.
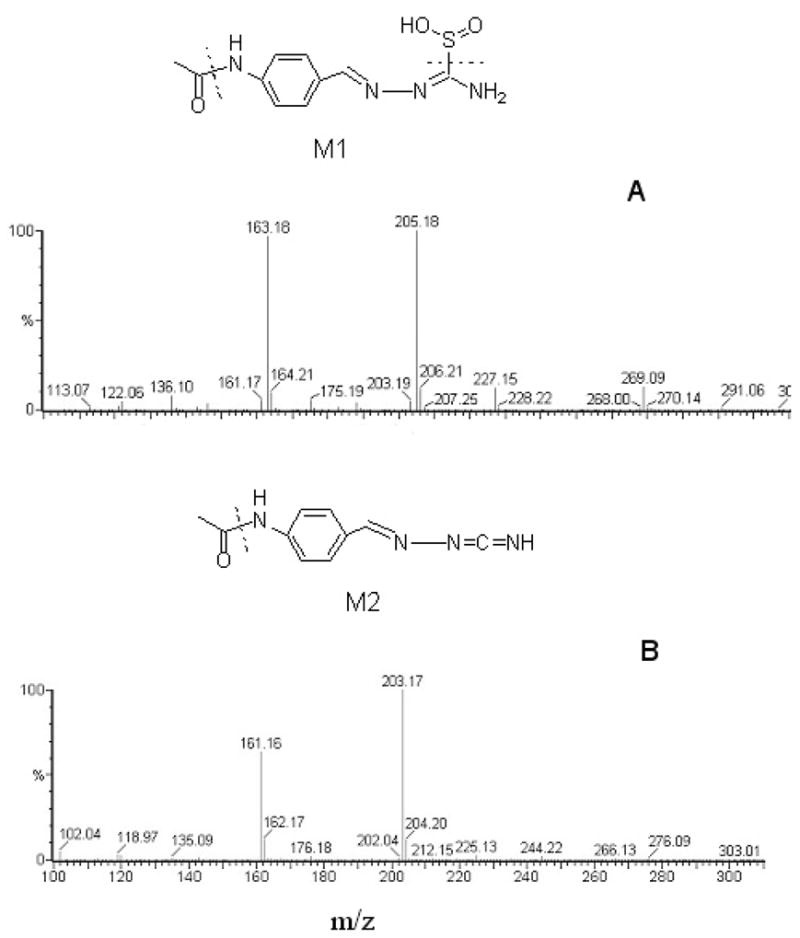
Mass spectra and structures of M1 and M2, the two products from incubations of TAZ, NADPH and EtaA in 100 mM tricine buffer, pH 9.0, at 37 °C for 1 h: (a) M1 has a molecular ion [M+H]+ = m/z 269.09, with fragment ions at m/z 205.18 and 163.18; (b) M2 has a molecular ion [M+H]+ = m/z 203.17 with a fragment ion at m/z 161.16.
To confirm the structures of the two metabolites, authentic standards were synthesized. The TAZ-sulfinic acid was prepared by the reaction of TAZ with peracetic acid (21), and the TAZ-carbodiimide by the treatment of TAZ with sodium chlorite in the presence of CuCl (22). Both of these compounds were purified by preparative HPLC and were fully characterized by MS and 1H-NMR spectroscopy. By comparing their elution times, UV spectra, and MS spectra with those of the synthetic standards, the metabolites M1 and M 2 were unambiguously identified as the TAZ-sulfinic acid and TAZ-carbodiimide, respectively.
Kinetics of TAZ oxidation by EtaA
The kinetics of the catalytic oxidation of TAZ by purified EtaA were evaluated by measuring the initial rates of TAZ metabolite formation as a function of the concentration of TAZ. The steady state kinetic parameters were calculated from Michaelis-Menten plots. The Km, Vmax, and kcat values for the formation of TAZ-sulfinic acid were found to be 131 ± 29 μM, 5.07 ± 0.4 μM min−1, and 5.07 ± 0.4 min−1, respectively. For the formation of TAZ-carbodiimide, the Km, Vmax, and kcat values were 147 ± 25 μM, 2.89 ± 0.2 μM min−1, and 2.89 ± 0.2 min−1, respectively.
TAZ Reactions with Chemical Oxidants
To determine if M2 was formed by desulfuration of M1, TAZ-sulfinic acid was incubated with the desulfurating agent CuCl at 37°C. After overnight incubation, no conversion to TAZ-carbodiimide was observed (Figure 5, A). Incubation of TAZ and NaClO2 at 37°C for 2 h led to the production of both M1 and M2. Under the same conditions, a small amount of TAZ-sulfinic acid was converted to TAZ-carbodiimide by NaClO2 oxidation (Figure 5, B and C). To further determine whether M2 was formed by oxidation of M1 or by an alternative enzymatic oxidation of TAZ, the synthetic standard TAZ-sulfinic acid was incubated with EtaA and NADPH as described earlier. No further oxidative transformation was detected by HPLC analysis (data not shown). All the metabolites were identified by comparison with the synthetic standards.
Figure 5.
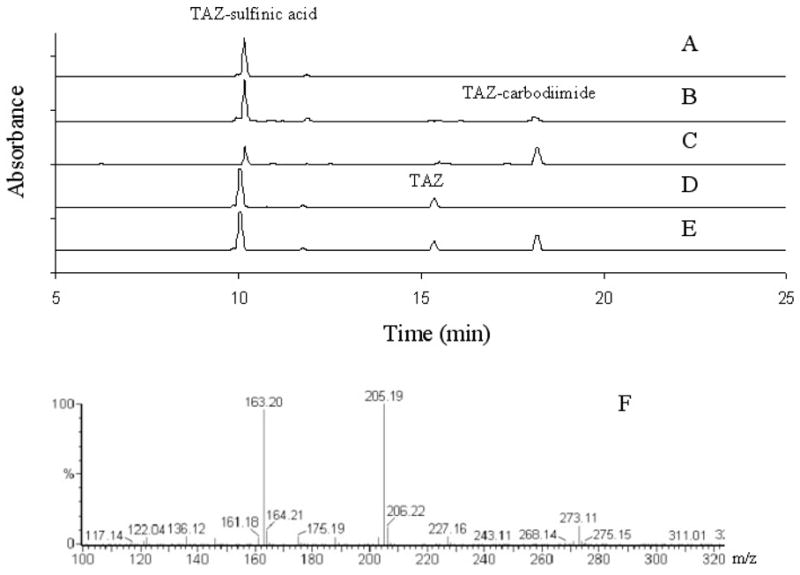
HPLC chromatograms of the products from chemical reactions of (A) TAZ-sulfinic acid and CuCl; (B) TAZ-sulfinic acid and NaClO2; (C) TAZ and NaClO2. (D) TAZ and H2O2 at pH 6.5 or pH 7.5; (E) TAZ and H2O2 at pH 9. The HPLC UV detector was set at 280 nm. In (F) is shown the mass spectrum of the main product from the reaction of TAZ with [18O]H2O2. Reactions (A) and (E) were performed at 37 °C for 2 h, and reactions (B), (C) and (D) were performed at 37 °C for overnight.
LC-MS analysis showed that the main product of the reaction of TAZ with H2O2 at either pH 6.5 or 7.5 had the same retention time on LC and the same MS spectrum as M1 and was therefore identical to it (Figure 5, D). The mass spectrum of the main product from the reaction of TAZ with [18O]H2O2 at pH 7.5 showed an M+H ion at m/z 273, confirming the incorporation of two atoms of oxygen from the peroxide into M1 (273 = 237 + 18 + 18) (Figure 5, F). At pH 9.0, both M1 and M2 were observed after 2 h incubation with H2O2 (Figure 5, E).
pH-Dependence of the Oxidation of TAZ by EtaA, FMO1, and FMO3
The effect of pH on the oxidation of TAZ was determined by HPLC analysis after incubation of TAZ with EtaA, FMO1, or FMO3 at 37 °C for 1 h in buffers that ranged from pH 5 to 10. The yield of each metabolite was determined by linear regression analysis from correlations of the integrated experimental HPLC peaks with standard curves of HPLC peaks generated with authentic metabolites. The results show that the same metabolites are generated by EtaA, FMO1, and FMO3 (Figure 6). Furthermore, the pH optimum of all three enzymes for the oxidation of TAZ was ~pH 9. For EtaA and FMO1, the formation of metabolite M1 was optimal at pH 7.5, whereas the formation of M2 was favored at pH 9. Higher pH is expected to favor the formation of M2 because basic conditions facilitate intramolecular elimination of the oxidized sulfur moiety to generate the carbodiimide function. This difference in product profile with pH was not observed with human FMO3, for which the generation of both M1 and M2 is optimal at pH 9.
Figure 6.
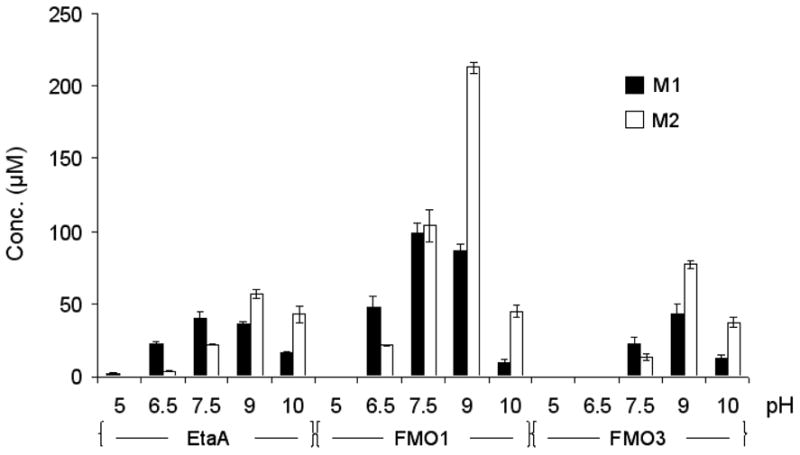
Effect of pH on the S-oxygenation of TAZ by EtaA, human FMO1, and FMO3. The results shown are the means of the normalized yields of metabolites M1 and M2 determined by integrating the corresponding HPLC peak areas and using standard curves with linear regression to calculate the metabolite concentrations.
Glutathione Adduct
GSH capture experiments were performed during the TAZ, EtaA and NADPH incubations (Figure 2). Addition of increasing amounts of GSH at the beginning of incubations of TAZ with EtaA and NADPH resulted in a concentration-dependent reduction in the yields of both M1 and M2 by GSH (Figure 7, A). Both of the TAZ metabolites were completely suppressed when 5 mM GSH was added at the beginning of the incubation. When GSH was added 30 min after the start of the incubation, i.e., halfway through the normal 60 min incubation period, the formation of M2 was suppressed while a new peak was observed in the HPLC (Figure 2, B). LC-MS analysis suggested the formation of a GSH-M2 adduct (M+H, 510 = 202 + 308). When GSH was added at the end of the normal 60 min incubation period to incubations of TAZ with EtaA and NADPH, only M2 reacted with GSH to form a GSH-M2 adduct (Figure 7, B). The concentration of M1 was not significantly altered, in agreement with the independent observation that no GSH adduct was formed when excess GSH (10 eq) was incubated with authentic TAZ sulfinic acid at 37 °C for 1 h.
Figure 7.
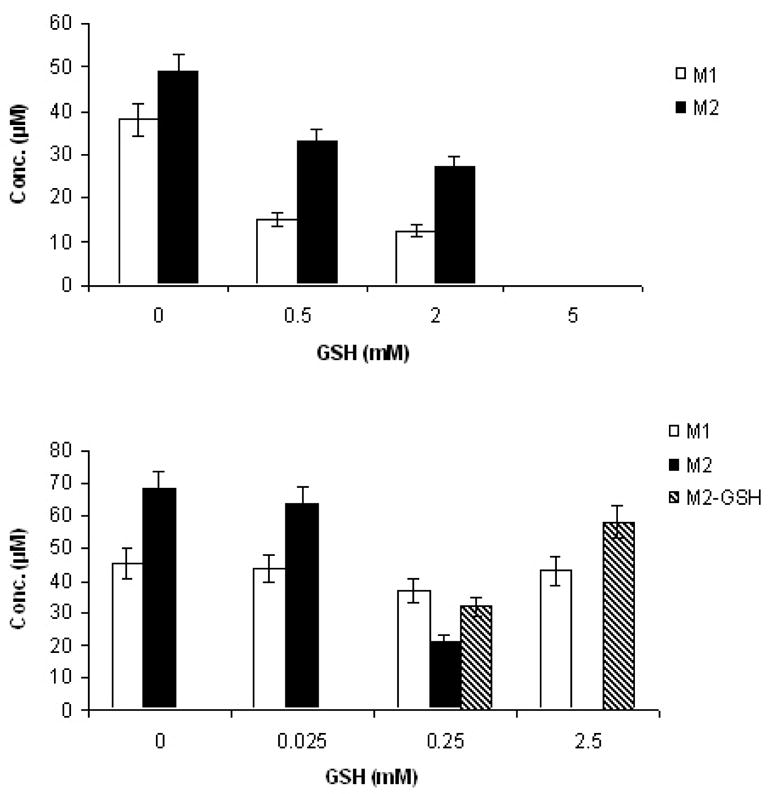
Effects of GSH addition on the yields of the sulfinic acid (M1) and carbodiimide (M2) metabolites of TAZ: (Top) Addition of increasing amounts of GSH to incubations of TAZ, NADPH and EtaA resulted in a concentration-dependent reduction in the yields of both M1 and M2; (Bottom) When GSH was added after the incubations were complete, only M2 reacted with GSH to form an adduct. GSH-M2 adduct formation was observed after adding 0.25 mM or more GSH.
The GSH-M2 adduct was isolated and its structure was determined. LC-MS/MS analysis displayed a molecular ion [M+H]+ at m/z 510.18. High resolution FTICR analysis predicted its elemental composition to be C20H28N7O7S. Subsequent collision-induced dissociation of the ion at m/z 510 produced fragment ions at m/z 364, 261 and 204, consistent with the loss of protonated ammoniated pyroglutamic acid (M = 146), glycine and acetamide, respectively. The proposed structure of the GSH adduct is shown in Figure 8. To confirm the structure of the adduct, the GSH-M2 adduct was prepared by addition of GSH to the synthetic TAZ-carbodiimide and the purified product was analyzed by 1H-NMR spectrometry. The MS/MS fragmentation patterns, UV spectra, and HPLC retention times of the synthetic adduct were identical to those of the product derived from the enzyme incubation.
Figure 8.
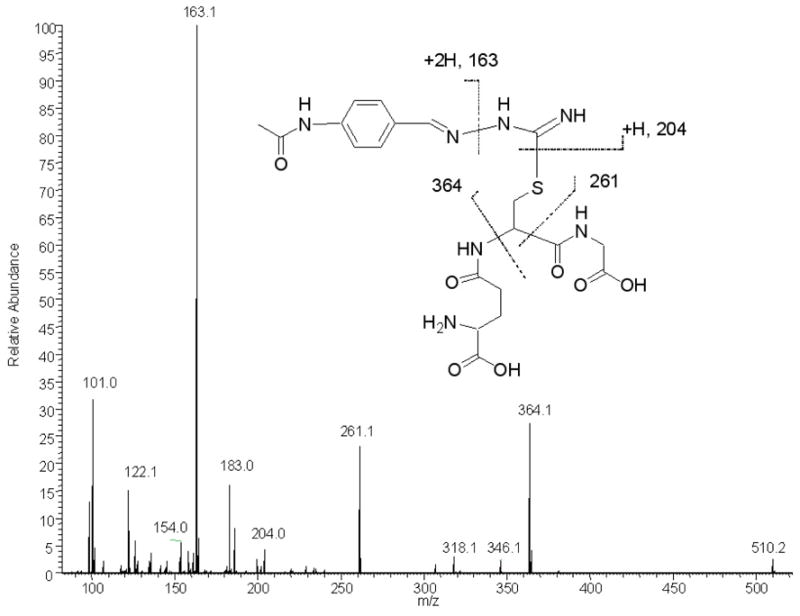
Collision-induced dissociation spectrum of the GSH-M2 adduct ion m/z 510 on an LTQ-FT hybrid mass spectrometer.
Discussion
It is generally accepted that the toxicity of thioamide and thiourea compounds stems, in most or all cases, from their biotransformation to reactive metabolites rather than from the parent compounds (23). The rapid enzymatic oxidation of thiourea derivatives is known to be catalyzed by the mammalian FMO enzymes (24, 25). However, both the structure/function properties of EtaA, the FMO from M. tuberculosis, and the physiological functions of this enzyme are still largely unknown. Some thiocarbonyl compounds in addition to ETA, notably thiobenzamide and isothionicotinamide, have been examined as substrates for EtaA and only sulfoxide-like products have been identified (16). We have shown here in vitro that oxidation of TAZ by EtaA in the presence of NADPH yields a sulfinic acid and a carbodiimide as the final metabolites. The Km, Vmax, and kcat values for the EtaA-catalyzed oxidation indicate that TAZ, like ETA (16), is a reasonable substrate for the enzyme.
The FMOs can catalyze two (or even three) thioamide or thiourea oxygenation steps, the first forming the sulfenic acid (RSOH) and the second the sulfinic acid (RSO2H) (25–27). In this study, only the sulfinic acid was observed, but we propose that it arises by further EtaA-catalyzed oxidation of the initially formed sulfenic acid. Sulfenic acids usually exhibit electrophilic reactivity and can react with GSH, whereas the sulfinic acid functionality is negatively charged and does not react with GSH (23, 28). Since formation of a carbodiimide through oxidative desulfuration of a sulfenic or sulfinic acid is feasible, as evidenced by the chemical reactions with NaClO2, we have explored the pathway for formation of the carbodiimide metabolite. To test the intermediacy of a sulfenic acid species, GSH capture experiments were performed. Addition of a large excess of GSH to the incubation mixtures at the beginning of the reaction completely eliminated both of the metabolites without the formation of a GSH adduct. Furthermore, EtaA and GSH did not react with the synthetic sulfinic acid and no carbodiimide-GSH adduct was again formed. This implicates an intermediate, presumably the sulfenic acid, as the direct precursor of both the sulfinic acid and the carbodiimide. Interestingly, the sulfenic but not sulfinic acid was readily isolated in the oxidation of ETA (16), whereas the converse is true in the oxidation of TAZ. The putative ETA sulfinic acid is rapidly hydrolyzed to the amide, but the TAZ sulfinic acid only slowly hydrolyzes to the urea if it is allowed to stand in water. Addition of water to the sulfur-bearing carbon should be more difficult in the TAZ than ETA sulfinic acid because it requires disruption of a more extensively conjugated system.
Mycobacteria do not produce GSH but make mycothiol (MSH; acetyl-L-cysteine-1-D-myo-inosityl-2-deoxy-D-glucopyranoside) (29, 30). MSH, like GSH, serves to protect the cell against oxidative damage and electrophilic toxins and is maintained in the reduced state by a reductase (31, 32). Mycothiol is the principal thiol in mycobacteria and is essential for the growth of M. tuberculosis (33). Both the sulfenic acid intermediate and the carbodiimide metabolite generated by EtaA from TAZ can react with GSH and should similarly react with mycothiol, which suggests that their formation within the mycobacterium may lower the intracellular concentration of MSH and thus sensitize the M. tuberculosis cell to oxidative damage.
Like other FMOs, EtaA is likely to generate a C4a peroxyflavin (FADOOH) intermediate, although this species, in contrast to that in the human FMOs, has not been spectroscopically detected with EtaA (14). Nucleophilic attack by the substrate on the terminal hydroperoxy oxygen of the FADOOH intermediate results in substrate oxidation. The products of FMO-mediated oxygenations therefore often coincide with the products formed by reaction of the substrates with peroxides or peracids (34). We examined the reaction of TAZ with hydrogen peroxide and found the sulfinic acid product as the only observable metabolite at both pH 6.5 and pH 7.5. At pH 9.0, a small amount of the carbodiimide was observed after overnight incubation. This is consistent with the pH effects observed on the oxidation of TAZ by EtaA. Generation of the carbodiimide metabolite is accelerated at higher pH values, where intramolecular elimination of the oxidized sulfur atom is favored by deprotonation of the adjacent nitrogen, whereas the sulfinic acid metabolite is favored by acidic conditions. In the pH 6.5 to 10 range, both human FMO1 and FMO3 formed the same metabolites as EtaA and showed a similar reactivity pattern towards TAZ. This suggests that TAZ oxidation by the FMO enzymes may not only contribute to the antitubercular action of the drug in M. tuberculosis but also to its human toxicity.
In conclusion, we have demonstrated TAZ can be oxidized by EtaA, human FMO1 and human FMO3, and have established that S-oxygenation of TAZ leads to the formation of a reactive sulfenic acid species that is further converted to two metabolites, a sulfinic acid and a carbodiimide (Scheme 1). The reaction between the TAZ sulfenic acid and GSH (or MSH in M. tuberculosis) regenerates the parent TAZ molecule with concomitant conversion of GSH to oxidized GSH (GSSG), although GSSG formation was not independently evaluated in this study. In the presence of GSH and GSH reductase, or of MSH and its reductase in M. tuberculosis cells, a redox cycle may be established that depletes the protective thiol agent and thus potentiates oxidative stress and cellular injury (23). Clearly, the TAZ carbodiimide metabolite has the potential to bind covalently to cysteine or other nucleophilic residues in proteins and, through covalent adduct formation, to exert a deleterious effect on the function of the cell. Future studies should clarify the roles of the sulfenic acid intermediate and carbodiimide metabolite in both the antimycobacterial mechanism of TAZ action and TAZ toxicity.
Scheme 1.
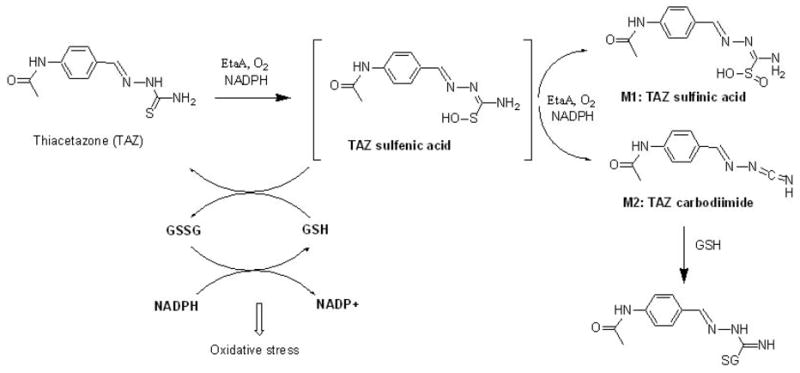
Diagram depicting the proposed EtaA-mediated metabolism of TAZ and the sites of interaction of GSH. GSSG represents oxidized GSH.
Acknowledgments
This research was supported by National Institutes of Health grants GM25515 and GM56531
Footnotes
Address editorial correspondence to: Paul R. Ortiz de Montellano, University of California San Francisco, 600 16th Street, San Francisco, CA 94143-2280, Tel: (415) 476-2903, FAX: (415) 502-4728, email: ortiz@cgl.ucsf.edu
Abbreviations: TAZ, thiacetazone; ETA, ethionamide; FMO, flavin-containing monooxygenase; TB, tuberculosis; MDR, multidrug-resistant; GSH, glutathione; MSH, mycothiol.
References
- 1.Murray JF. A century of tuberculosis. Am J Respir Crit Care Med. 2004;169:1181–1186. doi: 10.1164/rccm.200402-140OE. [DOI] [PubMed] [Google Scholar]
- 2.Ghiladi RA, Medzihradszky KF, Rusnak FM, Ortiz de Montellano PR. Correlation between isoniazid resistance and superoxide reactivity in Mycobacterium tuberculosis KatG. J Am Chem Soc. 2005;127:13428–13442. doi: 10.1021/ja054366t. [DOI] [PubMed] [Google Scholar]
- 3.Cardoso EM. Multiple drug resistance: a threat for tuberculosis control. Rev Panam Salud Publica. 2004;16:68–73. doi: 10.1590/s1020-49892004000700013. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention, N. C. f. H., STD and TB Prevention, Division of Tuberculosis Elimination. Surveillance Report: Reported Tuberculosis in the United States. 2003. [Google Scholar]
- 5.Moore M, Onorato IM, McCray E, Castro KG. Trends in drug-resistant tuberculosis in the United States, 1993–1996. J Am Med Assoc. 1997;278:833–837. [PubMed] [Google Scholar]
- 6.Rattan A, Kalia A, Ahmad N. Multidrug-resistant Mycobacterium tuberculosis: molecular perspectives. Emerg Infect Dis. 1998;4:195–209. doi: 10.3201/eid0402.980207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peloquin CA. Pharmacology of the antimycobacterial drugs. Med Clin North Am. 1993;77:1253–1262. doi: 10.1016/s0025-7125(16)30191-2. [DOI] [PubMed] [Google Scholar]
- 8.Espinal MA. The global situation of MDR-TB. Tuberculosis (Edinb) 2003;83:44–51. doi: 10.1016/s1472-9792(02)00058-6. [DOI] [PubMed] [Google Scholar]
- 9.Oliva B, Comanducci A, Chopra I. Antibacterial spectra of drugs used for chemotherapy of mycobacterial infections. Tuber Lung Dis. 1998;79:107–109. doi: 10.1054/tuld.1998.0012. [DOI] [PubMed] [Google Scholar]
- 10.Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs. 2002;62:2169–2183. doi: 10.2165/00003495-200262150-00001. [DOI] [PubMed] [Google Scholar]
- 11.Newton RW. Side effects of drugs used to treat tuberculosis. Scott Med J. 1975;20:47–49. doi: 10.1177/003693307502000204. [DOI] [PubMed] [Google Scholar]
- 12.Holdiness MR. Neurological manifestations and toxicities of the antituberculosis drugs. Med Toxicol. 1987;2:33–51. doi: 10.1007/BF03259859. [DOI] [PubMed] [Google Scholar]
- 13.Onyebujoh P, Zumla A, Ribeiro I, Rustomjee R, Mwaba P, Gomes M, Grange JM. Treatment of tuberculosis: present status and future prospects. Bull World Health Org. 2005;83:857–865. [PMC free article] [PubMed] [Google Scholar]
- 14.Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, Locht C, Besra GS. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem. 2000;275:28326–28331. doi: 10.1074/jbc.M003744200. [DOI] [PubMed] [Google Scholar]
- 15.DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE., 3rd Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2000;97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vannelli TA, Dykman A, Ortiz de Montellano PR. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem. 2002;277:12824–12829. doi: 10.1074/jbc.M110751200. [DOI] [PubMed] [Google Scholar]
- 17.Pattyn SR, Colston MJ. Cross-resistance amongst thiambutosine, thiacetazone, ethionamide and prothionamide with Mycobacterium leprae. Lepr Rev. 1978;49:324–326. [PubMed] [Google Scholar]
- 18.Osato T, Tsukagoshi K, Shimizu H. Studies on thiacetazone resistance of tubercle bacilli. 3 Cross resistance between thiacetazone and ethionamide. Kekkaku. 1971;46:89–92. [PubMed] [Google Scholar]
- 19.Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- 20.Grosset JH, Cole ST. Genomics and the chemotherapy of leprosy. Lepr Rev. 2001;72:429–440. doi: 10.5935/0305-7518.20010051. [DOI] [PubMed] [Google Scholar]
- 21.Hoggarth E. Compounds related to thiosemicarbazide. VIII The oxidation of thiosemicarbazones. J Am Chem Soc. 1951:2202–2204. [Google Scholar]
- 22.Koketsu M, Suzuki N, Ishihara H. Preparation of isoselenocyanate and synthesis of carbodiimide by oxidation of selenourea. J Org Chem. 1999;64:6473–6475. [Google Scholar]
- 23.Onderwater RC, Commandeur JN, Vermeulen NP. Comparative cytotoxicity of N-substituted N’-(4-imidazole-ethyl)thiourea in precision-cut rat liver slices. Toxicology. 2004;197:81–91. doi: 10.1016/j.tox.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 24.Poulsen LL, Hyslop RM, Ziegler DM. S-Oxygenation of N-substituted thioureas catalyzed by the pig liver microsomal FAD-containing monooxygenase. Arch Biochem Biophys. 1979;198:78–88. doi: 10.1016/0003-9861(79)90397-7. [DOI] [PubMed] [Google Scholar]
- 25.Smith PB, Crespi C. Thiourea toxicity in mouse C3H/10T1/2 cells expressing human flavin-dependent monooxygenase 3. Biochem Pharmacol. 2002;63:1941–1948. doi: 10.1016/s0006-2952(02)00978-4. [DOI] [PubMed] [Google Scholar]
- 26.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106:357–387. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onderwater RC, Commandeur JN, Menge WM, Vermeulen NP. Activation of microsomal glutathione S-transferase and inhibition of cytochrome P450 1A1 activity as a model system for detecting protein alkylation by thiourea-containing compounds in rat liver microsomes. Chem Res Toxicol. 1999;12:396–402. doi: 10.1021/tx980198p. [DOI] [PubMed] [Google Scholar]
- 28.Henderson MC, Krueger SK, Stevens JF, Williams DE. Human flavin-containing monooxygenase form 2 S-oxygenation: sulfenic acid formation from thioureas and oxidation of glutathione. Chem Res Toxicol. 2004;17:633–640. doi: 10.1021/tx034253s. [DOI] [PubMed] [Google Scholar]
- 29.Newton GL, Bewley CA, Dwyer TJ, Horn R, Aharonowitz Y, Cohen G, Davies J, Faulkner DJ, Fahey RC. The structure of U17 isolated from Streptomyces clavuligerus and its properties as an antioxidant thiol. Eur J Biochem. 1995;230:821–825. doi: 10.1111/j.1432-1033.1995.0821h.x. [DOI] [PubMed] [Google Scholar]
- 30.Newton GL, Av-Gay Y, Fahey RC. A novel mycothiol-dependent detoxification pathway in mycobacteria involving mycothiol S-conjugate amidase. Biochemistry. 2000;39:10739–10746. doi: 10.1021/bi000356n. [DOI] [PubMed] [Google Scholar]
- 31.Newton GL, Fahey RC. Mycothiol biochemistry. Arch Microbiol. 2002;178:388–394. doi: 10.1007/s00203-002-0469-4. [DOI] [PubMed] [Google Scholar]
- 32.Patel MP, Blanchard JS. Mycobacterium tuberculosis mycothione reductase: pH dependence of the kinetic parameters and kinetic isotope effects. Biochemistry. 2001;40:5119–5126. doi: 10.1021/bi0029144. [DOI] [PubMed] [Google Scholar]
- 33.Sareen D, Newton GL, Fahey RC, Buchmeier NA. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J Bacteriol. 2003;185:6736–6740. doi: 10.1128/JB.185.22.6736-6740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cashman JR. Human flavin-containing monooxygenase: substrate specificity and role in drug metabolism. Curr Drug Metab. 2000;1:181–191. doi: 10.2174/1389200003339135. [DOI] [PubMed] [Google Scholar]