

Abstract
Cytochrome c oxidase pumps protons across a membrane using energy from electron transfer and reduction of oxygen to water. It is postulated that an element of the energy transduction mechanism is the movement of protons to the vicinity of the hemes upon reduction, to favor charge neutrality. Possible sites on which protons could reside, in addition to the conserved carboxylate E286, are the propionate groups of heme a and/or heme a3. A highly conserved pair of arginines (R481/R482) interact with these propionates through ionic and hydrogen bonds. This study shows that the conservative mutant, R481K, although fully as active as wild-type under many conditions, exhibits a significant decrease in the midpoint redox potential of heme a relative to CuA of Em ≅ 40 mV, has lowered activity under conditions of high pH or in the presence of a membrane potential and has a slowed heme a3 reduction with dithionite. Another mutant, D132A, that strongly inhibits proton uptake from the internal side of the membrane, has <4 % the activity of wild-type and appears dependent on proton uptake from the outside. A double mutation, D132A/R481K, is even more strongly inhibited (~1 % wild-type). The more-than-additive effect supports the concept that R481K not only lowers the midpoint potential of heme a but also limits a supply route for protons from the outside of the membrane used by the D132 mutant. The results are consistent with an important role of R481 and heme a/a3 propionates in proton movement in a reversible exit path.
Cytochrome c oxidase (CcO) is an integral membrane protein and is the terminal electron acceptor in the respiratory chain. Electrons from cytochrome c enter CcO through a dinuclear copper site (CuA) and move to heme a, heme a3 and the closely associated CuB. Oxygen binding and reduction occur at the heme a3/CuB site (1). The electron transfer events and the chemical conversion of oxygen to water create a membrane potential by virtue of drawing protons from the inside and electrons from the outside of the membrane. In addition, CcO pumps protons through the protein to further build up the proton gradient ΔμH+ across the membrane. This gradient is then used to make ATP as a source of energy for the cell. The question still remains as to how the steps of electron transfer and the oxygen chemistry are coupled to the mechanism of proton pumping.
Intake paths for protons have been defined from the crystal structures (2-5) and by site-directed mutagenesis studies (6-10). A pathway discerned in bovine oxidase, the H path (11-13), has not been confirmed in prokaryotic CcO by mutagenesis studies (14), but two other paths, the D and K paths, appear to be highly conserved across a range of oxidases including the mammalian CcO. The K path has a highly conserved lysine (K362) that is essential for reduction of heme a3/CuB (10, 15-17) and appears to lead to the active site. The D path, so called because of an important aspartate (D132) at the entrance, appears to end at a glutamate (E286) that is ~12Å from both hemes at their lower edges (Figure 1). Substitution of D132 for anything other than a carboxyl group inhibits proton uptake from the inside of the membrane and net proton pumping is not observed. In fact, there is alkalinization on the outside of the D132A cytochrome c oxidase vesicles (COVs) during turnover, observed with the pH-sensitive dye phenol red (18).
Figure 1.

Structure of Rhodobater sphaeroides CcO showing the arginines and their interactions. From the crystal structure (5), the figure was made using Insight II. Waters are shown as red spheres. Dotted black lines show hydrogen-bonding interactions of 3Å or less. Dotted red lines show distances less than 4Å that depict electrostatic interactions.
D132A COVs exhibit the highest rates of oxygen consumption in the presence of a membrane potential (controlled state), a phenomenon that can be explained by backflow of protons from the outside, partially replacing the lack of protons from the D-path (18, 19). Consistent with this interpretation is the observation of inhibition of wild-type or D132A COVs by externally added zinc. Zinc reduces the activity of the controlled state but not the uncontrolled state (in the absence of ΔμH+) (20). This EDTA-reversible inhibition of activity by zinc is observed to correlate with inhibition of the rate of alkalinization of the exterior of the vesicles. The results strongly suggest that backflow of protons is important in the controlled state, both to support the activity of D132A, as well as the activity of wild-type when inhibited by high ΔμH+.
It has been proposed that protons to be pumped move from residue E286 to the vicinity of the propionates above the hemes (21-24) before being released to the outside bulk solvent, by an as yet undefined proton exit path (25). A possible location for a proton to reside, to accomplish charge neutralization of the incoming electron (26), is on the propionates of the hemes (or an associated water (27)). FTIR studies with 13C labeled propionates suggest that at least two of the four propionates in the Paracoccus denitrificans cytochrome c oxidase undergo changes in protonation or environment upon heme reduction (28, 29). In other heme proteins it has been shown that the protonation state of the propionates of the hemes has a role in controlling the redox potential and cooperativity (30, 31). In order to examine the possible involvement of the propionates and the region above the hemes in proton movement in CcO, mutants have been made of the highly conserved arginine pair (R481, R482) that interacts with both of the heme propionates (Figure 1) (22, 23, 29, 32). Previous reports of mutations of the arginines have pointed to a possible role in proton movement (23), while structural analysis and theoretical modeling studies also suggest a facilitated electron transfer pathway between CuA and heme a in this region (32-34).
Studies with various R482 mutations, the arginine closely associated with heme a, show changes in the electron transfer rates from the electron donor, cytochrome c, to CuA and between CuA and heme a (32). The conservative mutation of R482 to a lysine was wild-type in its steady-state activity, both in purified and reconstituted forms, but significantly altered in the midpoint potential difference between heme a and CuA (ΔEm = Em(heme a) − Em(CuA)) of +28 mV in R482K vs. +46 mV in wild-type (32)). Alteration of R482 to a hydrophobic residue caused loss of Mg, a non-redox active metal above heme a3, and a disturbed subunit I/II interface. The R482P mutant, which is relatively unstable structurally, shows very slow electron transfer from CuA to heme a. When both R482 and R481 are mutated to glutamine in the E.coli bo3 oxidase, the activity is reduced to less than 50% and there appears to be no proton pumping (23).
Arginine-481 is intimately associated with both heme a and a3 propionates. From the crystal structure, it appears to be within hydrogen-bonding distance (≤ 3Å) of the heme a D-propionate (Figure 1: black dashed lines) and to have strong electrostatic interactions (<4Å) with the D-propionate of heme a3 (Figure 1: red dashed lines). Mutation of the conserved R481 in CcO to a lysine results in normal activity in the purified enzyme and normal proton pumping efficiency, although the proton pumping is slowed, due to slowed cytochrome c oxidation under the reconstituted, pumping conditions (32). R481K has no spectrally detectable perturbation of the CuA site, even though the peptide backbone of R481 is hydrogen bonded through water to a ligand of CuA (H260II:in subunit II) (32) (Figure 1).
To examine the role of R481 in maintaining the structural and/or electrostatic properties of the propionates, further studies were carried out including time-resolved measurement of electron transfer rates from CuA to heme a in the R481K mutant CcO. A double mutant of D132A and R481K was made to test whether mutating the externally located arginine would affect the severely inhibited, internally located D132A mutant, since the latter CcO is postulated to rely on protons being supplied from the outside (backflow) for its activity.
Materials and Methods
Site-Directed Mutagenesis and Protein Purification
Site-directed mutants were constructed using PCR overlapping extension methods (35). For the double mutation of D132A/R481K the plasmid, pCH81, containing the entire Cox I with the D132A mutation (9), was used for the second R481K mutation. The primer and construction of R481K was described previously (32). Michigan State University Macromolecular Structural Facility, East Lansing, MI, synthesized all of the oligonucleotide primers. The D132A/R481K double mutation, after complete sequencing at the MSU facility, was placed in the histidine-tag II construct (36) before conjugation into Rhodobacter sphaeroides.
Cytochrome c oxidase was extracted from Rs grown in Sistrom’s media as described (37), and purified on a Ni-NTA affinity column (38). Modifications were made to this protocol to maintain binding of the his-tag II CcO. Ni-NTA (Qiagen) was suspended in high salt buffer (10 mM Tris-HCl pH 8.0, 10 mM imidazole, 200 mM KCl and 0.1% lauryl maltoside (LM)) for the binding procedure. High salt buffers were used for the Ni-NTA wash and elution of CcO. The protein, after Ni-NTA affinity chromatography, was further washed with 10 mM HEPES-KOH pH 7.4, 0.1% lauryl maltoside, and 40 mM KCl using a Centricon-50 concentrator (Amicon) to remove Ni-histidine. Prior to reconstitution into vesicles, a second ion exchange chromatography purification step was used with two DEAE columns in tandem (Tosohaas DEAE-5PW 10μ particle size, 8 mm × 7.5 cm) using a Pharmacia ÃKTA FPLC as previously described (36).
Reconstitution of Cytochrome c Oxidase
Cytochrome c oxidase vesicles were prepared as described (9, 37) by the sonication of 40 mg/mL asolectin (re-crystallized soybean phospholipids from Associated Concentrates) in 2% Na-cholate (Anatrace) and 75 mM HEPES-KOH pH 7.4. The lipids were mixed 1:1 with 4 μM CcO in 75 mM HEPES-KOH pH 7.4 with 4% cholate to give a final concentration of 2 μM oxidase, 20 mg/ml asolectin and 3% Na-cholate. The vesicles were dialyzed against buffer as before (20). Oxygen consumption assays were performed for the reconstituted enzymes in order to determine the respiratory control ratio (RCR), which is a test of whether the cytochrome c oxidase vesicles (COVs) are able to produce a membrane potential and pH gradient and the enzymes are inserted correctly (32, 36). The RCR for unreconstituted enzyme is expected to be 1.
Measurement of pH Dependence of CcO Activity
Steady-state measurements of the activity of cytochrome c oxidase were made in a Gilson oxygraph with purified CcO, horse heart cytochrome c, ascorbate and TMPD in 50 mM of the appropriate buffer (HEPES-KOH, MES-KOH, or glycylglycine-KOH) with 0.05% LM and KCl added to maintain a constant ionic strength. Cytochrome c oxidation measurements with COVs at different pH values were made in an OLIS-rsm stopped-flow at 0.1 to 0.2 μM aa3 in 50 μM buffer rapidly mixed with pre-reduced horse-heart cytochrome c (in 0.5 mM HEPES-KOH pH 7.4 + 44 mM KCl) diluted into 40 mM buffer of appropriate pH (MES-KOH, HEPES-KOH or glycylglycine-KOH) to give a final concentration after stopped-flow mixing of 20 mM buffer with 45 mM K+ and 22 mM sucrose. Measurements were made, 1) in the absence of ionophores for the controlled state, 2) with 2 μM valinomycin to remove the ΔΨ and, 3) with 2 μM valinomycin and 5 μM FCCP as an uncoupler to allow maximal activity. Ionophores were added to the COVs prior to mixing with Cc. The kinetic traces were fit exponentially and the kobs s-1 converted to molecular activity (catalytic turnover in electrons/sec/aa3) by multiplying kobs s-1 by [cyt. c2+] μM / [aa3] μM. The RCR was obtained by dividing the uncontrolled rate by the controlled rate.
Fast Electron Transfer of Oxidized CcO
Ru-55-Cc (ruthenium trisbipyridine labeled horse-heart cytochrome c) was used to deliver electrons to the oxidized CcO using a laser flash (39, 40). The photoexcited state was formed (Ru(II) to Ru(II*)) which rapidly transferred an electron to heme c. At low ionic strength, 5 mM Tris-HCl pH 8.0, Ru-55-Cc forms a 1:1 complex with CcO so that rapid electron transfer from heme c to CuA and heme a in CcO can be measured spectrophotometrically at 830 and 605 nm respectively. The reaction of cytochrome c was monitored at 550 nm using an extinction coefficient of Δε550 = 18.5 mM-1cm-1. The reaction of CuA was monitored at 830 nm using Δε830 = 2.0 mM-1cm-1 and the reduction of heme a was measured at 605 nm using Δε605 = 16 mM-1cm-1. The reduction of heme a was also monitored in some experiments with a high sensitivity detector with a 600 nm bandpass filter with a bandpass of 10 nm. The effective extinction coefficient for heme a in this system was 9.6 mM-1 (41). Reaction solutions typically contained 3 - 10 μM Ru-Cc, 5 - 20 μM CcO, 10 mM aniline, 1 mM 3CP in 5 mM Tris-HCl, pH 8.0, at 22 °C. The aniline and 3CP functioned as sacrificial electron donors to reduce Ru(III) and prevent the back reaction with heme Fe2+. Ionic strength was increased, up to 300 mM NaCl, in order to measure the effect of the mutation on the interaction of Ru-55-Cc with CcO. The transients were fitted to appropriate theoretical equations for Scheme 1 as described by Geren et al (39). Additionally, pH was altered, pH 6.5 and pH 8.0 were used and the effect on the rates measured.
The reduction of CcO by the photoexcited ruthenium dimer, Ru2C was studied in order to resolve the rapid electron transfer reaction between CuA and heme a. Reaction mixtures consisted of 12 μM CcO, 25 μM Ru2C, 10 mM aniline, 1 mM 3CP in 5 mM Tris Cl, pH 8.0. The absorbance transient was recorded at 605 nm using a monochromator detector with a time resolution of 20 nsec, as described in Zaslavsky et al.(42).
Dithionite Reduction of CcO
Measurements of the rates of heme a and heme a3 reduction of CcO with sodium dithionite were performed in a stopped-flow rapid scanning spectrophotometer (OLIS rsm-16) under anaerobic conditions. Sodium dithionite in argon-sparged buffer was rapidly mixed with CcO in the same anaerobic buffer to give a final concentration of 15 mM Na-dithionite and 2 μM wild-type or mutant CcO. 1,000 scans/sec were collected from 390 nm to 630 nm. At least three data sets were collected and averaged for each measurement. These measurements were then repeated with newly prepared samples. Reduced minus oxidized difference spectra were analyzed using the OLIS Global fitting software provided to determine the kinetically independent species (a and a3). This was compared to the first-order rate constants for single wavelengths at 605 nm and 428 nm for heme a and 434 nm for heme a plus a3 to ensure that a true fit was obtained. The measurements were made with 2 different buffers: A) 200 mM HEPES-KOH pH 8.0, 0.1% LM; B) 200 mM Bis-Tris-Propane pH 6.5, 0.1% LM (43).
Other Spectral Assays
Examination of CO binding to CcO was performed as before (37) on a Perkin-Elmer Lambda 40P UV/visible spectrophotometer at room temperature.
Results
Effect of Reconstitution on Activity
The maximal steady-state activity of the isolated or reconstituted R481K-mutated oxidase is like wild-type CcO at 25° C and pH 6.5 (32) but is more inhibited by increasing pH than wild-type CcO. When R481K is reconstituted into vesicles (COVs), with a highly buffered interior at pH 7.4 and in the presence of a pH gradient and membrane potential (ΔΨ), it exhibits low activity relative to wild-type CcO (Table 1). It is important to note that these measurements are not made with saturating levels of cytochrome c, but with limiting amounts under stopped-flow conditions comparable to those used for measuring proton pumping (32).
Table 1.
Cytochrome c reduction rates of wild-type and mutant CcO reconstituted into COVs. Measurements were made in an OLIS-rsm stopped-flow spectrophotometer at pH 7.4 under the controlled state (with a membrane potential and pH gradient), with valinomycin (no membrane potential) and in the uncontrolled state (with FCCP and valinomycin added) as in Figure 2. Errors shown are the standard deviation from the averaging of at least 3 data sets.
| Activity (e-/sec/aa3) | |||||
|---|---|---|---|---|---|
| COVs | Controlled | +Valinomycin | Uncontrolled | *RCR ΔΨ | ^RCR |
| Wild type | 51 ±1 | 180 ±23 | 460 ±120 | 3.5 | 9 |
| R481K | 13 ±1 | 45 ±25 | 210 ±36 | 3.5 | 16 |
| D132A | 13 ±3 | 5.6 ±3 | 14 ±1 | 0.4 | 1.1 |
| D132A/R481K | 7.9 ±1.4 | 2.6 ±5.7 | 3.7 ±3.5 | 0.3 | 0.5 |
RCRΔΨ = ΔΨ respiratory control ratio which is obtained from the +Valinomycin rate/Controlled rate.
RCR = respiratory control ratio which is obtained from the Uncontrolled rate/Controlled rate.
Measurement of activity versus external pH of wild-type COVs typically shows an apparent pKa at ~ pH 7.3 in the controlled state. In the R481K COVs there is no apparent pKa within the range of measurement; instead, activity gradually decreases with increasing pH, similar to the D path mutant, D132A (Figure 2A). However, in the D132A COVs, the controlled state has higher activity (~15 s-1) than that observed with no membrane potential (~6 s-1) (Figure 2B) likely due to the stimulation of proton backflow as indicated by an increased rate of external alkalinization (19,18). In contrast, the double mutant D132A/R481K is more inhibited in all states (Table 1), supportive of the concept that R481K may further limit proton access.
Figure 2.
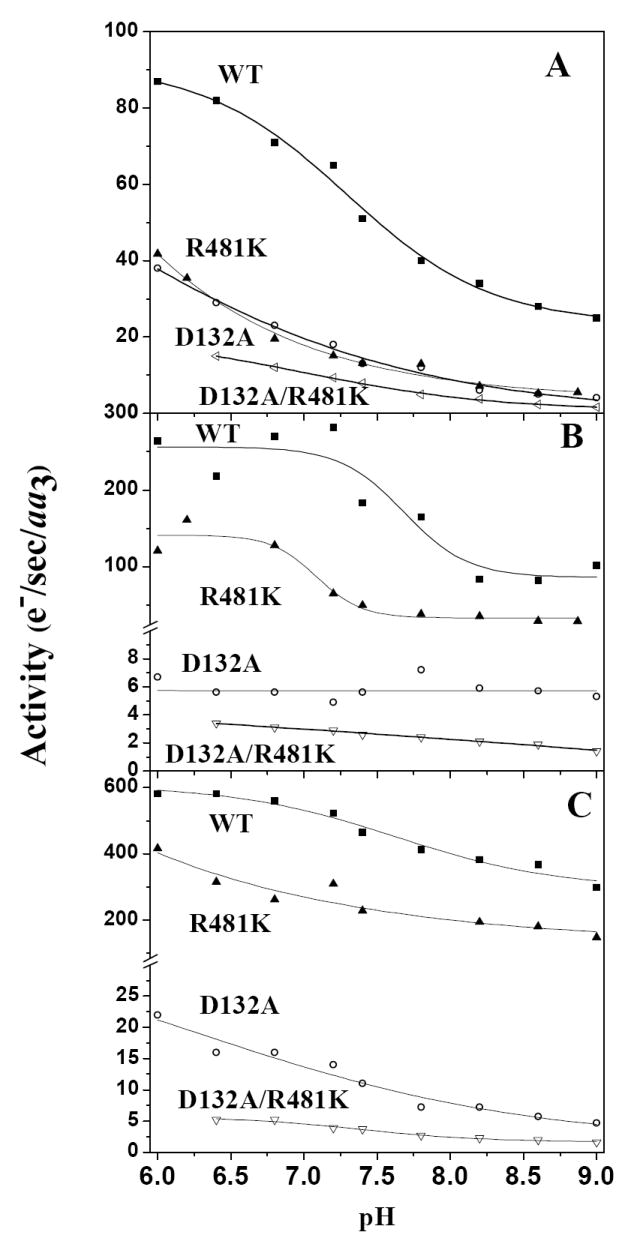
The activity of R481K CcO, reconstituted into lipid vesicles, is inhibited in the presence of a ΔΨ in the controlled state, and its activity is compared to wild type, D132A and D132A/R481K COVs (see Methods) across the pH range from pH 6.0 to pH 9.0: A. Controlled state activity, B. Activity in the absence of a membrane potential after the addition of 1 μM valinomycin, but with a pH gradient, C. Uncontrolled activity, with both valinomycin and 5 μM FCCP.
Under the conditions used to study proton pumping by stopped-flow, when the ΔΨ is removed by addition of valinomycin, activity is increased for wild-type CcO and R481K over the whole pH range, but R481K was significantly less active than wild-type. This may relate to the observed lower redox potential of heme a (Table 2) which becomes more limiting to activity when cytochrome c levels are low and CuA is not kept fully reduced. At saturating cytochrome c (Vmax) the mutant and wild-type activities are comparable, as in the free enzyme (32). In the case of D132A and the double mutant, strong inhibition, rather than stimulation, is observed across the whole pH range when the membrane potential is removed (Figure 2B). This “reverse respiratory control” has been previously observed (19). In the case of R481K, a lower than wild-type controlled rate, but stimulated uncontrolled rate results in an increase in the respiratory control ratio (RCR). The RCR for R481K is well above that seen for wild-type in stopped-flow measurements of cytochrome c oxidation (Table 1 and (44)) and more like that obtained with the addition of zinc to the outside of wild-type COVs in the controlled state (20). Both D132A and D132A/R481K show inhibition of activity when the membrane potential is removed (by the addition of valinomycin) unlike wild-type and R481K, resulting in a RCRΔΨ <1 (Table 1). This phenomenon can be explained by the removal of the driving force for proton backflow to support activity.
Table 2.
Electron transfer reaction between CuA and heme a in CcO Mutants. The equilibrium constant, K = kb/kc, is from the ratios of reduction of CuA and heme a by Ru-55-Cc, as described in Figure 4. The Em between heme a and CuA was calculated from K. The rate constant kobs for electron transfer from CuA to heme a was measured using Ru2C to photoreduce CuA, as described in Methods. The values of kb and kc were calculated from kobs and K as described in the text.
| CcO | K (kb/kc) | Δ Em (mV) (Heme a - CuA) | ka (104 s-1) | kb (104 s-1) | kc(104 s-1) |
|---|---|---|---|---|---|
| WT | 5.4 ± 1.0 | +43 ± 5 | 4.0 ± 0.5 | 9.3 ± 1.5 | 1.7 ± 3.0 |
| R481K | 1.4 ± 0.6 | +8 ± 12 | 4.0 ± 0.5 | 5.1 ± 1.0 | 3.7 ± 0.7 |
| D132A | 6.3 ± 1.0 | +47 ± 4 | 3.4 ± 0.5 | - | - |
| D132A/R481K | 1.5 ± 0.6 | +10 ± 12 | 2.5 ± 0.5 | 7.2 ± 1.5 | 4.8 ± 1.0 |
In the uncontrolled state, when both membrane potential and pH gradient are removed, the activities of wild-type CcO and R481K CcO are stimulated (Figure 2C). The D132A and the double mutant, D132A/R481K, are slightly stimulated, but these activities remain much lower than wild-type, consistent with blockage of backflow and uptake of protons in the double mutant.
When the ΔΨ is removed, both wild-type and R481K are able to pump protons with a net H+/e- stoichiometry of ~1 (32), but D132A shows no net proton pumping and has very low activity across the pH range, which correlates with rates of external alkalinization in the controlled state (18, 19)(Figure 2B). The double mutant, D132A/R481K, also shows no net proton pumping and significant but slow activity, correlated with alkalinization in the controlled state (data not shown).
Fast Electron Transfer Measurements of Oxidized CcO
A horse cytochrome c derivative with a ruthenium trisbipyridine complex covalently attached to lysine 55, Ru-55-Cc, is used as an electron donor for oxidized CcO. At low ionic strength, Ru-55-Cc forms a 1:1 complex with CcO. Photoexcitation of the Ru (II) group to a metal-to-ligand charge transfer state, Ru (II*), results in rapid reduction of heme c, followed by intracomplex electron transfer from heme c to CuA with rate constant ka (Scheme 1) (41). The intracomplex rate constant ka at low ionic strength was found to be 4.0 × 104 s-1 for wild-type CcO, compared with 4.0 × 104 s-1, 3.4 × 104 s-1, and 2.5 × 104 s-1 for the R481K, D132A, and D132A/R481D mutants, respectively (Table 2, Figure 3). Following reduction of CuA, an electron is transferred from CuA to heme a with a rate constant kb (Scheme 1). The value of kb is larger than ka and so cannot be measured using Ru-55-Cc; however this rate constant can be measured using a ruthenium dimer for photoreduction (see below; (42)). The kinetics and the ka values for wild-type CcO and the other mutants do not change significantly up to 40 mM ionic strength. At higher ionic strengths the amplitude of the fast intracomplex phase decreased due to increased dissociation of the 1:1 complex, and a new slow phase appears due to the reaction of Ru-55-Cc from solution with CcO (Figure 3). The rate constant of the slow bimolecular phase increased to a maximum at 70 mM ionic strength, and then decreased with further increases in ionic strength. The slow phase rate constants of the D132A and R481K mutants were nearly the same as that of wild-type CcO at all ionic strengths, while the values for the D132A/R481K mutant were somewhat smaller (Figure 3). The slow phase rate constant is controlled by dissociation of the Cc:CcO complex at ionic strengths up to 70 mM, and by formation of the 1:1 complex at high ionic strength (41). The present results indicate that the mutations examined in the present study had relatively small effects on the binding affinity with Ru-55-Cc.
Figure 3.
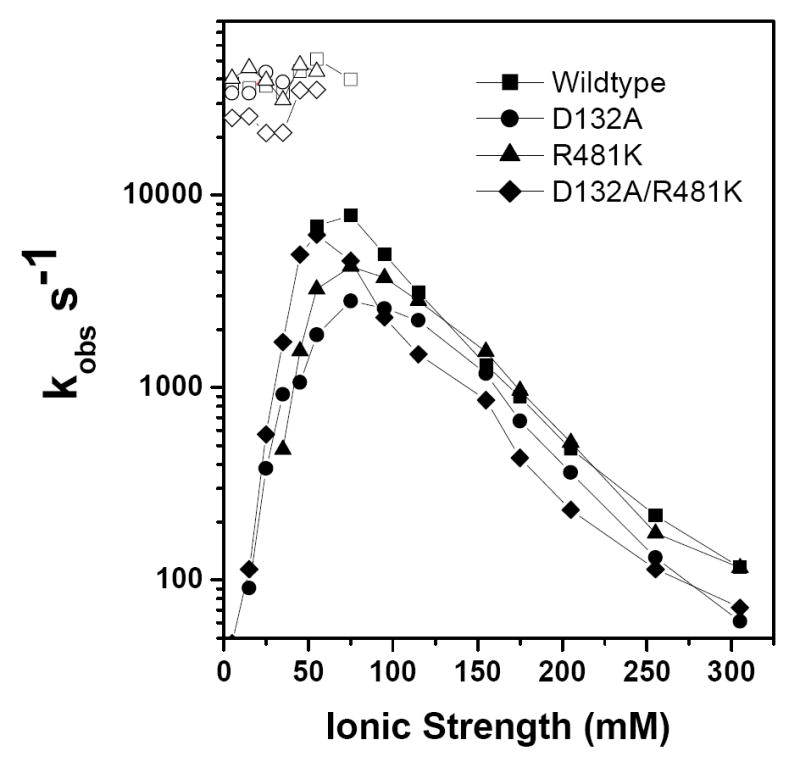
Ionic strength dependence of the fast intracomplex phase, Ka, (open symbols) and slow phase (closed symbols) of the reactions between Ru-55-Cc and CuA in CcO mutants. The solutions contained 10 μM Ru-55-Cc, and 15 μM CcO in 5 mM Tris-Cl, pH 8.0, 0 to 300 mM NaCl, 10 mM aniline, and 1 mM 3CP.
The ratio of reduced heme a to CuA, providing a measure of the equilibrium constant K, obtained from the relative amplitudes of the 600 nm and 830 nm transients after equilibrium is reached, as shown in Figure 4. These experiments were carried out at high ionic strength where only the slow bimolecular phase was present. The value of K (= kb/kc) was found to be 6.3 ± 1.0 for the D132A mutant (Figure 4A), which corresponds to a difference in midpoint potential between heme a and CuA (ΔEm = Em(heme a) − Em(CuA)) of 47 ± 4 mV) (Table 2). This is the same as previously determined for wild-type CcO, K = 5.4 ± 1.0, Em = 43 ± 5 mV (41). However, the values of K for the D132A/R481K and R481K mutants are much lower than that of wild-type CcO (Figure 4B; Table 2). The K value for D132A/R481K is 1.5 ± 0.6 (Figure 4), corresponding to Em = 10 ± 12 mV. The values for the R481K mutant are K = 1.4 ± 0.6 and Em = 8 ± 12 mV. This demonstrates that the electron has a greater tendency to remain on CuA in the R481K and D132A/R481K CcO mutants, suggesting an alteration of the Em of either the heme a (decreased) or CuA (increased). A change in the heme a site seems most probable, due to a likely change in proximity of the K481 to the propionic acid groups in heme a (see Seibold et al, accompanying paper).
Figure 4.

Photoinduced electron transfer from Ru-55-Cc to R. sphaeroides CcO mutants. A: A sample containing 10.5 μM D132A CcO and 12.7 μM Ru-55-Cc in 5mM TrisCl, pH 8 with 10 mM aniline, 1 mM 3CP and 170 mM NaCl was photoexcited with a 200 ns laser flash at 480 nm. The rate constants for reduction of heme a and CuA, measured at 600 nm and 830 nm, were 880 ± 200 s-1 and 1000 ± 200 s-1, respectively. B: The sample contained 13.4 μM D132A/ R481K CcO and 9.9 μM Ru-55-Cc in 5mM Tris-Cl, pH 8, with 150 mM NaCl.
In order to study the rapid electron transfer between CuA and heme a (kb), CcO was reduced with the photoexcited ruthenium dimer, Ru2C as described by Zaslavsky et al. (42). Ru2C has a charge of +4 which allows it to bind to the cytochrome c binding domain on subunit II near CuA. The photoexcited state of Ru2C reduces CuA within 1 μsec, allowing the electron transfer reaction between CuA and heme a to be resolved (42). The rate constants for wild-type CcO were previously found to be kb = 9.3 × 104 s-1 and kc = 1.7 × 104 s-1 from a kobs of 11 × 104 s-1 (42). For the R481K mutant CcO heme a is reduced with a rate constant of kobs = 8.8 × 104 s-1 (under the reaction conditions of 25 μM Ru2C with 12 μM R481K CcO in 5 mM TrisCl, pH 8.0 containing 10 mM aniline and 1 mM 3CP). Using the relations K = kb/kc = 1.4 and kobs = kb + kc, the values of kb and kc are calculated to be: kb = 5.1 × 104 s-1 and kc = 3.7, × 104 s-1. For the D132A/R481K double mutant, heme a is reduced with a rate constant of kobs = 12 × 104, so using K = 1.5 the rate constants are calculated to be kb = 7.2 × 104 s-1 and kc = 4.8 × 104 s-1.
Reduction Kinetics of Heme a and a3
CcO has a characteristic absorption spectrum in the Soret region with a broad peak when oxidized at 420 nm and 446 nm when fully reduced. These Soret peaks contain contributions from both the low spin six-coordinate heme a, and heme a3, the active site where oxygen binds (45). Additionally, there are absorption peaks at higher wavelengths, particularly a peak at 606 nm where ~80% of the absorption is contributed by reduced heme a (45). In order to observe the effects of R481K on the kinetic behavior of individual hemes, measurements were made using Na-dithionite as a reducing agent under anaerobic conditions with rapid mixing in a stopped-flow rapid scanning spectrophotometer.
The spectral scans of wild-type (Figure 5: left panel) and R481K CcO (Figure 5:middle panel) are shown for 2.5 sec of the reaction with Na-dithionite. The reduction of heme a is complete for both wild-type and R481K within this time, as seen by the peak absorbance at 606 nm. However, the Soret band is still split after 2.5 secs, with peaks at 420 nm and 442 nm, because heme a3 is still not fully reduced: R481K CcO shows lower absorbance at 442 nm and higher absorbance at 420 nm while the wild-type CcO shows the opposite. Heme a3 in R481K remains more oxidized than the heme a3 of wild-type CcO. This difference in the rate of reduction of heme a3 is quantified by Global fitting of the spectra to obtain the rates of heme a and heme a3 reduction, which are monophasic and complete in all cases (Figure 5: right panel). (Table 3). Both heme a and heme a3 were faster to reduce at low pH, 3.5-fold for heme a but less than 2-fold for heme a3, reflecting the complex dependence on the level of heme a reduction. In the mutant R481K, heme a3 reduction is considerably slowed at both pH 6.5 and pH 8 compared to wild-type, while the D-path mutant, D132A, shows wild-type rates of both heme a and heme a3 at both pH values (Table 3) as seen in (16) but compare (46). The double mutant D132A/R481K has lowered rates of reduction for both heme a and heme a3 at both pH values.
Figure 5.
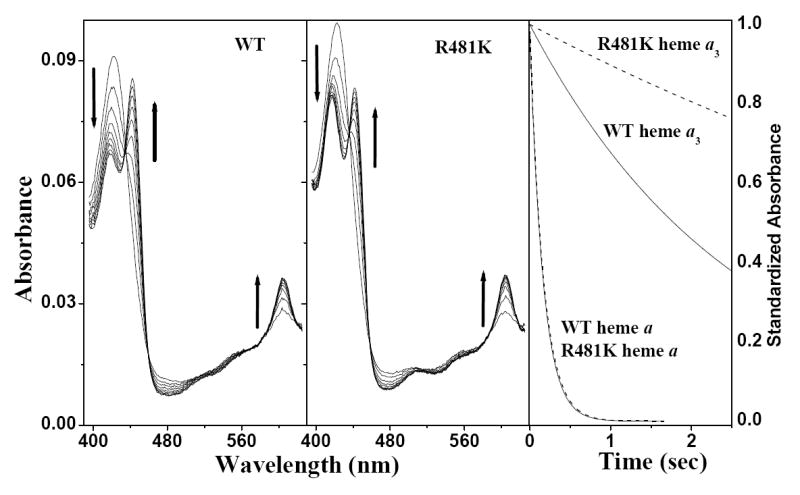
Heme a3 of R481K CcO is slow to reduce with Na-dithionite. Panels on the left show the spectral scans from OLIS-rsm stopped-flow measurements, up to 2.5 sec in steps of 310 ms, after mixing 2 μM wild type or R481K CcO with 15 mM Na-dithionite @ pH 6.5 as described in the Methods section. Arrows depict the direction of the changes associated with reduction. The panel on the right shows the kinetic traces of heme a and heme a3 reduction for wild type (solid line) and R481K (dashed line) from Global fitting of the spectral scans.
Table 3.
Reduction rates of wild type and R481K CcO from Global analysis. Na-Dithionite was rapidly mixed with wild type or R481K CcO in a stopped-flow rapid scanning spectrophotometer (OLIS rsm-16) (see Figure 5). Global fitting of the resulting spectral scans from the averaging of at least 2 separate measurements of 3 data sets gave kinetic rates (kobs s-1) of reduction for each of the hemes. Standard deviations given are from the averaging of data sets.
| Rates of Reduction (kobs s-1) | ||||
|---|---|---|---|---|
| pH6.5 | pH8 | |||
| CcO | Heme a | Heme a3 | Heme a | Heme a3 |
| Wild-type | 6.42 ±.78 | 0.49 ±.09 | 2.09 ±.54 | 0.22 ±.04 |
| R481K | 6.10 ±.30 | 0.11 ±.04 | 1.52 ±.20 | 0.05 ±.01 |
| D132A | 6.19 ± .79 | 0.48 ± .07 | 2.24 ± .16 | 0.23 ±.06 |
| D132A/R481K | 2.51 ± .18 | 0.18 ± 00 | 1.34 ± .02 | 0.09 ± .01 |
Discussion
Minor Structural Effects of the R481K Mutation
In order to interpret the results of these studies it is important to note that the conservative change of R481 to a lysine has a very minor effect on the overall structure as indicated by native visible spectra for the hemes. The CuA site was also not disturbed, as determined by visible and EPR spectroscopy (32). The non-redox active Mg/Mn above the hemes is minimally altered as seen in the hyperfine splittings of the EPR spectrum with possibly slight changes in the orientation of the bound ligands (32). There is a shift in the EPR spectrum of heme a from g = 2.83 to g = 2.85 in R481K as well as several mutants of R482. Curiously, in spite of no change in the visible spectrum of the oxidized or reduced R481K CcO, upon binding CO to heme a3 of R481K a more broadened Soret peak than for the wild-type CO-bound form was observed, with a 4nm shift to a longer wavelength. Besides these minor effects, the R481K mutation is not structurally disruptive.
Arginine 481and 482 are important for maintaining the redox potential of heme a
The control of redox potential changes in heme proteins is an important issue, particularly in CcO where the movement of protons is tightly coupled to electron movement (43, 46). Arginine 482 was shown to be important for maintaining Em of heme a in CcO, with various mutations lowering the Em between CuA and heme a, to as low as 18 mV as compared to 46 mV for wild-type.
The fast electron transfer measurements in this study show that the rate constants ka for intracomplex electron transfer from Ru-55-Cc to CuA are 4.0 × 104 s-1, 3.4 × 104 s-1, and 2.5 × 104 s-1 for the R481K, D132A, and D132A/R481D mutants, respectively, compared with 4.0 × 104 s-1 for wild-type CcO (Table 2, Figure 3,). The rate constant kb for electron transfer from CuA to heme a is 9.3 × 104 s-1 for wild-type CcO, compared with 5.1 × 104 s-1 for the R481K mutant and 7.2 × 104 s-1 for the D132A/R481K mutant (Table 2). In the case of the mutant R481K the electron transfer rates from cyt. c to CuA (ka) are similar to wild-type CcO and to D132A CcO, while the rates between CuA and heme a are altered in the R481K and the double mutant. The intrinsic electron transfer rates are all sufficiently fast so as not to be rate limiting in steady-state measurements of activity under conditions of excess reduced cytochrome c (32). However, the lower equilibrium constant between CuA and heme a in R481K and the double mutant, reflected in single electron input experiments, show that the electron has a tendency to remain on CuA in the mutant compared to wild-type. This change is calculated to be a Em of CuA minus heme a of approximately 40 mV. It is interpreted to be due to a less positive midpoint potential of heme a, since CuA is less intimately associated with the mutation and shows no change in spectral characteristics or in the electron transfer rate from Cc to CuA. This change in Em could be due to blockage of proton access from the outside via a backflow path that is needed for protonation of a group (or a bound water molecule) involved in determining the redox potential of heme a (47). Alternatively, the structural and charge distribution changes close to the heme propionates caused by the R481K mutation could be the cause of the potential shift. In a Molecular Dynamics simulation (Seibold et al: accompanying paper) the lysine substituted at position 481 is observed to interact more closely with the heme a3 propionates than the native CcO arginine, which could affect the protonation state of the propionates and/or the Em of the heme.
Heme a3 reduction is slowed in R481K CcO
Using sodium-dithionite under anaerobic conditions heme a3 reduction is slowed in R481K CcO 4-fold relative to wild-type CcO at both pH 6.5 and pH 8.0 but the mutation has no effect on the apparent heme a reduction rate, despite the fact that the Em of heme a is lowered by ≅ 40 mV. The mutant behaves as if it were caught in a “resting” state similar to that seen in bovine CcO, in which the electron transfer between heme a and heme a3 is observed to be inhibited (46, 48). In the bovine CcO there is evidence that in the fully oxidized (resting) enzyme the midpoint redox potential of heme a3 is low and a proton uptake is required to raise the Em (46, 49). It is possible that the R481K mutation affects an internal proton movement or binding site occupancy important for controlling electron transfer from heme a to heme a3 and that at high pH this proton movement/occupancy becomes even less favorable. One such proton binding site is the D-propionate of heme a3, as suggested by Brändén et al. to account for the pKa change for the F→O change in R481K (accompanying paper). The molecular dynamics studies of Seibold et al. (accompanying paper) also show major changes in structure close to this propionate. Another protonation site associated with heme a (22) may also be changed in its proton affinity, making protons less available to support electron transfer to heme a3, and causing the observed lower redox potential of heme a reported in this paper and in Brändén et al. (accompanying paper). In the reduced enzyme used in the rapid kinetic measurements, protons may be already loaded so that electron transfer is not necessarily rate limited.
In contrast, D132A CcO, which is known to be inhibited in proton uptake into the D-channel, is wild-type in its reduction with Na-dithionite for both hemes, in electron transfer from CuA to heme a and in the relative redox potentials of heme a and CuA in the oxidized enzyme (but see Ruitenberg et al.(50)), consistent with no disturbance in the structure of the region above the hemes.
Mutant R481K CcO is inhibited in the presence of a membrane potential
The measurement of activity in COVs with a stopped-flow spectrophotometer allows rapid mixing of the COVs at pH 7.4 with almost no external buffering (50 μM) with cytochrome c in high concentration buffers at varying pH. This technique allows the rapid introduction of a different pH on the outside of the COVs. Under the conditions used, there is only enough pre-reduced cytochrome c to give 10 turnovers on average. This means that substrate cytochrome c is not saturating as in steady-state conditions. These stopped-flow conditions are observed to increase the stimulatory effect of removal of the membrane potential by the addition of valinomycin, while in steady-state measurements much less stimulation is observed. This observation can be explained by the fact that electron transfer from CuA to heme a is working against the membrane potential. When this step is limiting, due to low levels of reduction of CuA, the overall CcO rate becomes very sensitive to the membrane potential (51). In the steady-state, when cytochrome c is saturating, the results with R481K show that the heme a midpoint potential can be substantially altered without causing a change in the steady-state activity of the enzyme. However, in the stopped-flow, under the constraints of a membrane potential (controlled conditions), the activity of R481K CcO is considerably slower than wild-type CcO, causing an increased RCR. This inhibition is likely due to the more negative heme a potential adding to the more negative internal membrane potential that inhibits the CuA to heme a equilibrium electron transfer.
In the D132A mutant, proton uptake from the inside becomes very slow but appears to be driven from the outside in the reverse direction by a membrane potential. Interestingly, this mutant does not affect electron transfer to heme a, or the heme a midpoint potential. These data suggest that a proton may be supplied from the outside in D132A for slow heme a/a3 reduction in the controlled state, but the kinetics of access of that proton is inhibited by the R481K mutation, causing the further inhibition of the double mutant R481K/D132A. When CcO is deprived of D-path protons (D132A), suicide inactivation is induced, particularly when subunit III is removed. It is interesting that the double mutant, D132A/R481K, suicide inactivates very rapidly even in the presence of subunit III, supporting the idea that R481K restricts proton access (52).
Role of R481 in CcO
The proposal that there is a site(s) for proton(s) in a region above the hemes involved in the reduction of heme a/a3 led to the idea that arginine 481and/or the propionates are candidates for this site (22, 23). The above results are consistent with a direct role of R481 in controlling protonation during pumping, through its influence on the protonation state of the propionates. Indeed, the role of arginine 82 in bacteriorhodopsin could serve as a model for R481 in CcO. Both arginine and lysine could perform a similar function (albeit with different facility (53)), of moving in response to a change in the protonation state (e.g. the heme a3 D-propionate) and influencing the protonation at a nearby site (e.g. the heme a D-propionate). This is consistent with the MD calculations in wild-type and mutant CcO (Seibold et al; accompanying paper). The propionates themselves, therefore, remain strong candidates for sites of protonation.
Electrostatic effects from the alteration of an arginine to a lysine could be the source of the altered Em of the hemes and of altered pKs of one or more propionates. These effects can provide an explanation of the observed activity changes reported in this paper. However, the molecular dynamic simulations reported in the accompanying paper (Seibold et al.) suggest that even the very conservative change in R481 to lysine causes significant repercussions in terms of loop movement and amino acid rotamer positions which affect water chain formation. The idea of an arginine/propionate interaction being involved in the control of water was inferred in cytochrome P450 heme enzymes (54) where an arginine that interacts with a heme propionate is proposed to act as a mediator in the formation of water chains. Further computational and kinetic studies to examine the effect of R481K on the structure and function of CcO are reported in accompanying papers (Seibold et al.; Brändén et al.) while crystallographic and other structural studies are being pursued.
Acknowledgments
We thank Dr Denis Proshlyakov for providing the CO for the binding studies and Kori Henry for help with construction of the double mutant, R481K/D132A.
Abbreviations
- CcO
cytochrome c oxidase
- COV
cytochrome c oxidase vesicles
- 3CP
3-carboxyl-2,2,5,5-tetramethyl-1-pyrolidinyloxy free radical
- DEAE
diethylaminoethyl
- EPR
electron paramagnetic resonance
- LM
lauryl maltoside
- PCR
polymerase chain reaction
- Rs
Rhodobacter sphaeroides
- RCR
respiratory control ratio
- Ru2C
ruthenium dimer
- RuCc
ruthenium complex labeled cytochrome c
- TMPD
N,N,N’,N’tetramethyl-p-phenylendiamine
References
- 1.Ferguson-Miller S, Babcock G. Heme/copper terminal oxidases. Chemical Reviews. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 3.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 4.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 Å resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody Fv fragment. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Svensson-Ek M, Abramson J, Larsson G, Tornoth S, Brzezinski P, Iwata S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 6.Hosler JP, Ferguson-Miller S, Calhoun MW, Thomas JW, Hill J, Lemieux L, Ma J, Georgiou C, Fetter J, Shapleigh JP, Tecklenburg MMJ, Babcock GT, Gennis RB. Insight into the active-site structure and function of cytochrome c oxidase by analysis of site-directed mutants of bacterial cytochrome aa3 and cytochrome bo. J Bioenerg Biomembr. 1993;25:121–136. doi: 10.1007/BF00762854. [DOI] [PubMed] [Google Scholar]
- 7.Thomas JW, Lemieux LJ, Alben JO, Gennis RB. Site-directed mutagenesis of highly conserved residues in helix VIII of subunit I of the cytochrome bo ubiquinol oxidase from Escherichia coli: an amphipathic transmembrane helix that may be important in conveying protons to the binuclear center. Biochemistry. 1993;32:11173–80. doi: 10.1021/bi00092a029. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Horsman JA, Puustinen A, Gennis RB, Wikstrom M. Proton transfer in cytochrome bo3 ubiquinol oxidase of Escherichia coli: second-site mutations in subunit I that restore proton pumping in the mutant Asp135-->Asn. Biochemistry. 1995;34:4428–33. doi: 10.1021/bi00013a035. [DOI] [PubMed] [Google Scholar]
- 9.Fetter JR, Qian J, Shapleigh J, Thomas JW, García-Horsman JA, Schmidt E, Hosler J, Babcock GT, Gennis RB, Ferguson-Miller S. Possible proton relay pathways in cytochrome c oxidase. Proc Nat Acad Sci U S A. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosler JP, Shapleigh JP, Mitchell DM, Kim Y, Pressler M, Georgiou C, Babcock GT, Alben JO, Ferguson-Miller S, Gennis RB. Polar residues in helix VIII of subunit I of cytochrome c oxidase influence the activity and the structure of the active site. Biochemistry. 1996;35:10776–10783. doi: 10.1021/bi9606511. [DOI] [PubMed] [Google Scholar]
- 11.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-itoh K, Nakashima R, Yaono R, Yoshikawa S. Structure of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Å. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 12.Yoshikawa S, Shinzawa-Itoh K, Tsukihara T. X-ray structure and the reaction mechanism of bovine heart cytochrome c oxidase. Journal of Inorganic Biochemistry. 2000;82:1–7. doi: 10.1016/s0162-0134(00)00137-9. [DOI] [PubMed] [Google Scholar]
- 13.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. The low-spin heme of cytochrome c oxidase as the driving element of the proton pumping process. Proc Nat Acad Sci (U S A) 2003;100:15304–9. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H-M, Das T, Rousseau D, Mills D, Ferguson-Miller S, Gennis R. Mutations in the putative H-channel in the cytochrome c oxidase from Rhodobacter sphaeroides show that this channel is not important for proton conduction but reveal modulation of the properties of heme a. Biochemistry. 2000;39:2989–2996. doi: 10.1021/bi9924821. [DOI] [PubMed] [Google Scholar]
- 15.Junemann S, Meunier B, Gennis RB, Rich P. Effects of mutation of the conserved lysine-362 in cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry. 1997;36:14456–14464. doi: 10.1021/bi971458p. [DOI] [PubMed] [Google Scholar]
- 16.Vygodina TV, Pecoraro C, Mitchell D, Gennis R, Konstantinov AA. Mechanism of inhibition of electron transfer by amino acid replacement K362M in a proton channel of Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 1998;37:3053–61. doi: 10.1021/bi971876u. [DOI] [PubMed] [Google Scholar]
- 17.Zaslavsky D, Gennis R. Substitution of lysine-362 in a putative proton-conducting channel in the cytochrome c oxidase from Rhodobacter sphaeroides blocks turnover with O2 but not with H2O2. Biochemistry. 1998;37:3062–3067. doi: 10.1021/bi971877m. [DOI] [PubMed] [Google Scholar]
- 18.Zhen Y, Mills D, Hoganson CW, Lucas RL, Shi W, Babcock G, Ferguson-Miller S. In: Frontiers of Cellular Bioenergetics: Molecular Biology, Biochemistry and Physiopathology. Papa S, Guerrieri F, Tager JM, editors. Kluwer Academic/Plenum Press; New York: 1999. pp. 157–178. [Google Scholar]
- 19.Fetter JR, Sharpe M, Qian J, Mills D, Ferguson-Miller S, Nicholls P. Fatty acids stimulate activity and restore respiratory control in a proton channel mutant of cytochrome c oxidase. FEBS Lett. 1996;393:155–160. doi: 10.1016/0014-5793(96)00874-5. [DOI] [PubMed] [Google Scholar]
- 20.Mills DA, Schmidt B, Hiser C, Westley E, Ferguson-Miller S. Membrane potential-controlled inhibition of cytochrome c oxidase by zinc. J Biol Chem. 2002;277:14894–14901. doi: 10.1074/jbc.M111922200. [DOI] [PubMed] [Google Scholar]
- 21.Hofacker I, Schulten K. Oxygen and proton pathways in cytochrome c oxidase. Proteins. 1998;30:100–107. [PubMed] [Google Scholar]
- 22.Michel H. Cytochrome c oxidase: Catalytic cycle and mechanisms of proton pumping--A discussion. Biochemistry. 1999;38:15129–15140. doi: 10.1021/bi9910934. [DOI] [PubMed] [Google Scholar]
- 23.Puustinen A, Wikstrom M. Proton exit from the heme-copper oxidase of Escherichia coli. Proc Natl Acad Sci U S A. 1999;96:35–7. doi: 10.1073/pnas.96.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wikström M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 25.Mills DA, Florens L, Hiser C, Qian J, Ferguson-Miller S. Where is “outside” in cytochrome c oxidase and how and when do protons get there? Biochim Biophys Acta. 2000;1458:180–187. doi: 10.1016/s0005-2728(00)00067-0. [DOI] [PubMed] [Google Scholar]
- 26.Rich P, Meunier B, Mitchell R, Moody R. Coupling of charge and proton movement in cytochrome c oxidase. Biochim Biophys Acta. 1996;1275:91–95. [Google Scholar]
- 27.Sharpe MA, Qin L, Ferguson-Miller S. In: Biophysical and Structural Aspects of Bioenergetics. Wikstrom MKF, editor. Royal Society of Biochemistry Books; London: 2005. [Google Scholar]
- 28.Behr J, Hellwig P, Mantele W, Michel H. Redox dependent changes at the heme propionates in cytochrome c oxidase from Paracoccus denitrificans: direct evidence from FTIR difference spectroscopy in combination with heme propionate 13C labeling. Biochemistry. 1998;37:7400–6. doi: 10.1021/bi9731697. [DOI] [PubMed] [Google Scholar]
- 29.Behr J, Michel H, Mantele W, Hellwig P. Functional properties of the heme propionates in cytochrome c oxidase from Paracoccus denitrificans. Evidence from FTIR difference spectroscopy and site-directed mutagenesis. Biochemistry. 2000;39:1356–1363. doi: 10.1021/bi991504g. [DOI] [PubMed] [Google Scholar]
- 30.Louro RO, Catarino T, LeGall J, Turner DL, Xavier AV. Cooperativity between electrons and proton in monomeric cytochrome c3: The importance of mechano-chemical coupling for energy transduction. Chembiochem. 2001;2:831–837. doi: 10.1002/1439-7633(20011105)2:11<831::AID-CBIC831>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 31.Mills DA, Ferguson-Miller S. Understanding the mechanism of proton movement linked to oxygen reduction in cytochrome c oxidase: lessons from other proteins. Febs Letters. 2003;545:47–51. doi: 10.1016/s0014-5793(03)00392-2. [DOI] [PubMed] [Google Scholar]
- 32.Qian J, Mills DA, Geren L, Wang K, Hoganson CW, Schmidt B, Hiser C, Babcock GT, Durham B, Millett F, Ferguson-Miller S. Role of the conserved arginine pair in proton and electron transfer in cytochrome c oxidase. Biochemistry. 2004;43:5748–5756. doi: 10.1021/bi036279o. [DOI] [PubMed] [Google Scholar]
- 33.Onuchic JN, Beratan DN, Winkler JR, Gray HB. Pathway analysis of protein electron-transfer reactions. Annu Rev Biophys Biomol Struct. 1992;21:349–77. doi: 10.1146/annurev.bb.21.060192.002025. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez BE, Malmstrom BG, Winkler JR, Gray HB. The currents of life: The terminal electron-transfer complex of respiration. Proc Natl Acad Sci USA. 1995;92:11949–11951. doi: 10.1073/pnas.92.26.11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: Gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 36.Hiser C, Mills DA, Schall M, Ferguson-Miller S. C-terminal truncation and histidine-tagging of cytochrome c oxidase subunit II reveals the native processing site, shows involvement of the C-terminus in cytochrome c binding, and improves the assay for proton pumping. Biochemistry. 2001;40:1606–1615. doi: 10.1021/bi0018988. [DOI] [PubMed] [Google Scholar]
- 37.Hosler JP, Fetter J, Tecklenburg MMJ, Espe M, Lerma C, Ferguson-Miller S. Cytochrome aa3 of Rhodobacter sphaeroides as a model for mitochondrial cytochrome c oxidase. J Biol Chem. 1992;267:24264–24272. [PubMed] [Google Scholar]
- 38.Zhen Y, Qian J, Follmann K, Hosler J, Hayward T, Nilsson T, Ferguson-Miller S. Overexpression and purification of cytochrome c oxidase from Rhodobacter sphaeroides. Protein Expression and Purification. 1998;13:326–336. doi: 10.1006/prep.1998.0903. [DOI] [PubMed] [Google Scholar]
- 39.Geren LM, Beasley JR, Fine BR, Saunders AJ, Hibdon S, Pielak GJ, Durham B, Millett F. Design of a ruthenium-cytochrome c derivative to measure electron transfer to the initial acceptor in cytochrome c oxidase. J Biol Chem. 1995;270:2466–2662. doi: 10.1074/jbc.270.6.2466. [DOI] [PubMed] [Google Scholar]
- 40.Wang K, Mei H, Geren L, Miller MA, Saunders A, Wang X, Waldner JL, Pielak GJ, Durham B, Millett F. Design of a ruthenium-cytochrome c derivative to measure electron transfer to the radical cation and oxyferryl heme in cytochrome c peroxidase. Biochemistry. 1996;35:15107–19. doi: 10.1021/bi9611117. [DOI] [PubMed] [Google Scholar]
- 41.Wang K, Zhen Y, Sadoski R, Grinnell S, Geren L, Ferguson-Miller S, Durham B, Millett F. Definition of interaction domain for the reaction of cytochrome c with cytochrome c oxidase: II. Rapid kinetics analysis of electron transfer from cytochrome c to Rhodobacter sphaeroides cytochrome oxidase surface mutants. J Biol Chem. 1999;274:38042–38050. doi: 10.1074/jbc.274.53.38042. [DOI] [PubMed] [Google Scholar]
- 42.Zaslavsky D, Sadoski RC, Wang K, Durham B, Gennis RB, Millett F. Single electron reduction of cytochrome c oxidase compound F: resolution of partial steps by transient spectroscopy. Biochemistry. 1998;37:14910–6. doi: 10.1021/bi981490z. [DOI] [PubMed] [Google Scholar]
- 43.Zhen Y, Schmidt B, Kang UG, Antholine W, Ferguson-Miller S. Mutants of the CuA site in cytochrome c oxidase of Rhodobacter sphaeroides: I. Spectral and functional properties. Biochemistry. 2002;41:2288–97. doi: 10.1021/bi0114628. [DOI] [PubMed] [Google Scholar]
- 44.Mills DA, Ferguson-Miller S. Influence of structure, pH and membrane potential on proton movement in cytochrome c oxidase. Biochimica et Biophysica Acta. 2002;1555:96–100. doi: 10.1016/s0005-2728(02)00261-x. [DOI] [PubMed] [Google Scholar]
- 45.Vanneste WH. The stoichiometry and absorption spectra of components a and a3 in cytochrome c oxidase. Biochemistry. 1966;5:838–848. doi: 10.1021/bi00867a005. [DOI] [PubMed] [Google Scholar]
- 46.Verkhovsky MI, Morgan JE, Wikstrom M. Control of electron delivery to the oxygen reduction site of cytochrome c oxidase: a role for protons. Biochemistry. 1995;34:7483–91. doi: 10.1021/bi00022a023. [DOI] [PubMed] [Google Scholar]
- 47.Forte E, Scandurra F, Richter O-MH, D’Itri E, Sarti P, Brunori M, Ludwig B, Giuffre A. Proton uptake upon anaerobic reduction of Paracoccus denitrificans cytochrome c oxidase: A kinetic investigation of the K354M and D124N mutants. Biochemistry. 2004;43:2957–63. doi: 10.1021/bi035863u. [DOI] [PubMed] [Google Scholar]
- 48.Brunori M, Giuffre A, D’Itri E, Sarti P. Internal electron transfer in Cuheme oxidases. Thermodynamic or kinetic control? J Biol Chem. 1997;272:19870–4. doi: 10.1074/jbc.272.32.19870. [DOI] [PubMed] [Google Scholar]
- 49.Gregory L, Ferguson-Miller S. Independent control of respiration in cytochrome c oxidase vesicles by pH and electrical gradients. Biochemistry. 1989;28:2655–2662. doi: 10.1021/bi00432a044. [DOI] [PubMed] [Google Scholar]
- 50.Ruitenberg M, Kaant A, Bamberg E, Fendler K, Michel H. Reduction of cytochrome c oxidase by a second electron leads to proton translocation. Nature. 2002;417:99–102. doi: 10.1038/417099a. [DOI] [PubMed] [Google Scholar]
- 51.Brunori M, Sarti P, Colosimo A, Antonini G, Malatesta F, Jones M, Wilson M. Mechanism of control of cytochrome c oxidase activity by the electrochemical-proton gradient. EMBO J. 1985;4:2365–2368. doi: 10.1002/j.1460-2075.1985.tb03940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills DA, Hosler JP. Slow proton transfer through the pathways for pumped protons in cytochrome c oxidase induces suicide inactivation of the enzyme. Biochemistry. 2005;44:4656–4666. doi: 10.1021/bi0475774. [DOI] [PubMed] [Google Scholar]
- 53.Hatanaka M, Sasaki J, Kandori H, Ebrey T, Needleman R, Lanyi JK, Maeda A. Effects of arginine-82 on the interactions of internal water molecules in bacteriorhodopsin. Biochemistry. 1996;35:6308–6212. doi: 10.1021/bi952973v. [DOI] [PubMed] [Google Scholar]
- 54.Oprea TI, Hummer G, Garcia AE. Identification of a functional water channel in cytochrome P450 enzymes. Proc Natl Acad Sci U S A. 1997;94:2133–8. doi: 10.1073/pnas.94.6.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]