

(−)-Geosmin (1) is a degraded sesquiterpene that is responsible for the characteristic odor of moist soil and is associated with unpleasant off-flavors in water, wine and fish.1 Geosmin is produced by a number of microorganisms, including most Streptomyces and several species of cyanobacteria, myxobacteria and fungi.2
A single 726-amino acid protein in Streptomyces coelicolor A3(2)catalyzes the Mg2+-dependent cyclization of conversion of farnesyl diphosphate (2, FPP) to a mixture of germacradienol (3), germacrene D (4), and geosmin (1),3,4 accompanied by small amounts of octalin 5.5 The closely related 725-amino acid GeoA protein of S. avermitilis with 78% identity and 85% similarity to the S. coelicolor enzyme catalyzes the identical reaction.6 The S. coelicolor germacradienol/geosmin synthase is a bifunctional enzyme in which the N-terminal domain of the protein converts FPP (2) to germacradienol (3) and 4, while the C-terminal domain catalyzes the transformation of germacradienol (3) to geosmin (1).7 Both the N-terminal and C-terminal halves have significant sequence similarity to the well-characterized sesquiterpene synthase, pentalenene synthase.3a,7,8
The mechanism and stereochemistry of the conversion of FPP to 3 and 4, which is thought to involve the partitioning of a common germacradienyl cation intermediate 6, has been investigated in detail (Scheme 1a).3,4,6,7 Formation of germacrene D (4) results from a 1,3-hydride shift of the original H-1si of FPP.3b The alternative formation of germacradienol (3), which involves competing loss of the H-1si proton of FPP (2), can occur by cyclization of 6 to an enzyme-bound, trans-fused bicyclic intermediate, isolepidozene (7), a compound that has been isolated from incubation of FPP with the S233A mutant of S. coelicolor germacradienol/geosmin synthase.7 Isolepidozene (7) would be converted to germacradienol (3) by proton-initiated ring opening and capture of the resulting homoallyl cation by water.4,7
Scheme 1.
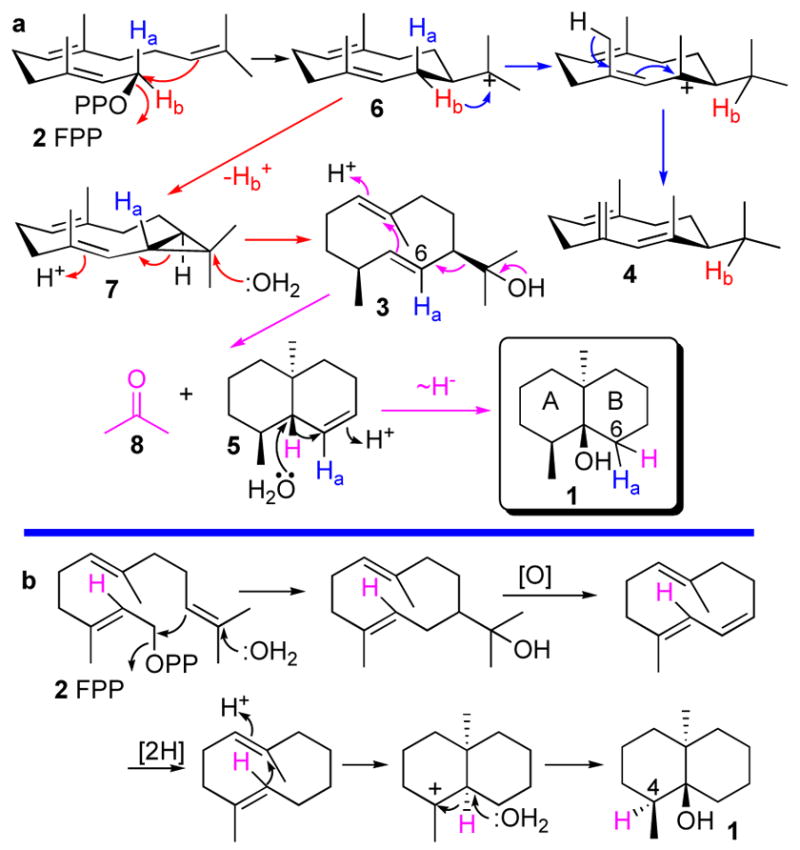
Cyclization of farnesyl diphosphate to geosmin
By contrast, the mechanistic details of the subsequent conversion of germacradienol (3) to geosmin (1) are still incomplete. Independent incorporation experiments with labeled mevalonates using Myxococcus xanthus and Stigmatella aurantiaca support the mechanism of Scheme 1a in which proton-initiated cyclization of germacradienol and retro-Prins fragmentation result in formation of octalin 5 and release of the 2-propanol side chain as acetone (8).9 Reprotonation of 5 followed by 1,2-hydride shift of the bridgehead proton into ring B and quenching of the resulting cation by water will generate geosmin (1).9 This model is supported by the isolation of octalin 5 as a coproduct of incubations of FPP with germacradienol/geosmin synthase.5–7 By contrast, an alternative 1,2-hydride shift of the same bridgehead hydrogen into ring A of geosmin during biosynthesis in the liverwort Fossombronia pusilla has also been proposed, based on incorporations of labeled mevalonate.10 It has been suggested that this mechanism is also operative in Streptomyces sp. JP95 (Scheme 1b).10 We now report evidence that conversion of germacradienol (3) to geosmin (1) by S. coelicolor germacradienol/geosmin synthase results in the release of the three-carbon side chain as acetone and involves a 1,2-hydride shift of the bridgehead hydrogen exclusively into ring B of geosmin.
To detect acetone generated in the formation of geosmin, the product mixture from incubation of FPP with recombinant S. coelicolor germacradienol/geosmin synthase was reacted with cysteamine (Scheme 2a).11 GC-MS analysis confirmed the formation of 2,2-dimethylthiazolidine (9) which displayed a parent peak at m/z 117 and a prominent [M-CH3]+ at m/z 102.. Control experiments established that neither geosmin nor acetone was formed when the protein was first inactivated by boiling. To confirm the origin of the enzymatically generated acetone, [13,13,13-2H3]FPP (2a)12 was incubated with germacradienol/geosmin synthase. The [2H3-Me]-2,2-dimethylthiazolidine (9a) derived from the resulting deuterated acetone showed a molecular ion at m/z = 120 [M+3]+ with fragment ions at m/z = 102 and 105 resulting from loss of the CD3- and CH3- groups, respectively (Figure 1). The presence of the trideuterated 2-hydroxypropyl moiety in the intermediate [12,12,12-2H3]-germacradienol (3a) was indicated by a shift of the molecular ion [d3-M]+ from m/z = 222 to 225 and a corresponding shift in the base peak from m/z = 59 to 62 [CH3(CD3)C=OH]+, while the [M-acetone]+ fragment at m/z 164 was unchanged. The mass spectrum of the [12,12,12-2H3]germacrene D (4a) co-product also displayed all the predicted changes. The mass spectra of the derived geosmin (1, m/z 182) and octalin (5, m/z 164) confirmed the complete absence of deuterium label in either of these C12 products.
Scheme 2.
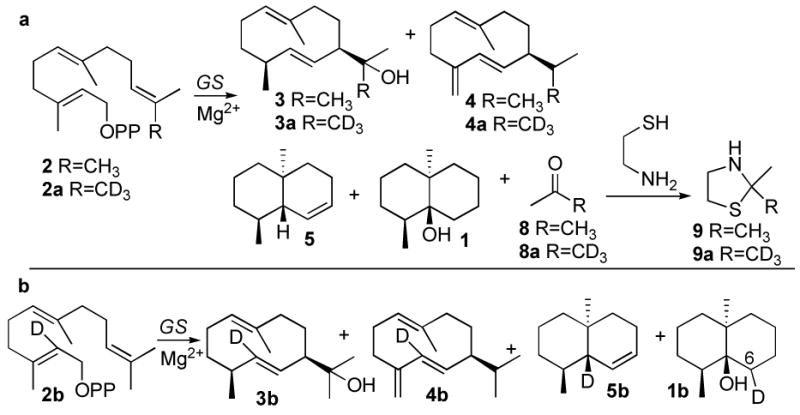
Cyclization/fragmentation of deuterated FPPs to geosmin
Figure 1.
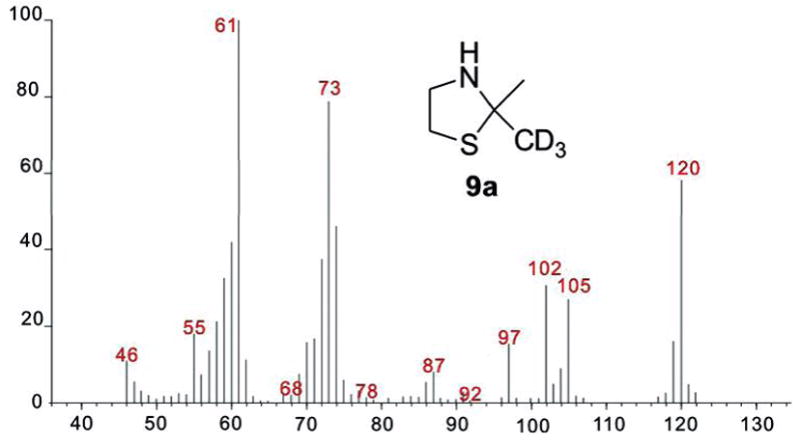
Mass spectrum of [2H3-Me]-2,2-dimethylthiazolidine (9a).
To explore the fate of the H-2 proton of FPP, the requisite [2-2H]FPP (2b) (>99 atom% deuterium) was synthesized from trideuteroacetic acid by way of [2,2-2H2]trimethylsilylacetic acid using a modified Peterson olefination procedure that avoids exchange of the deuterium label.13 GC-MS analysis of the products resulting from cyclization of [2-2H]FPP (2b) showed the predicted germacradienol-d1 (3b), germacrene D-d1 (4b), octalin-d1 (5b) and geosmin-d1 (1b) (Scheme 2b). In the mass spectrum of unlabeled geosmin, besides the weak molecular ion (m/z = 182), two other well-defined fragments at m/z = 112 and m/z = 126 correspond to the parent rings A and B (Figure 2).9.10 Cyclization of [2-2H]FPP (2b) is predicted to generate [6-2H]geosmin (1b). The observed site of deuterium labeling in 1b is consistent with the observed shift from m/z 112 to 113 of the characteristic ring B fragment ion; while the corresponding ring A-derived fragment ion from 1b, m/z 126, was devoid of deuterium (Figure 2a). Most importantly, the mass spectrum of 1b was indistinguishable from that of [6-2H]geosmin derived from (1R)-[1-2H]FPP, which should differ from 1b only in the configuration of the C-6 deuterium (Figure 2b).4
Figure 2.
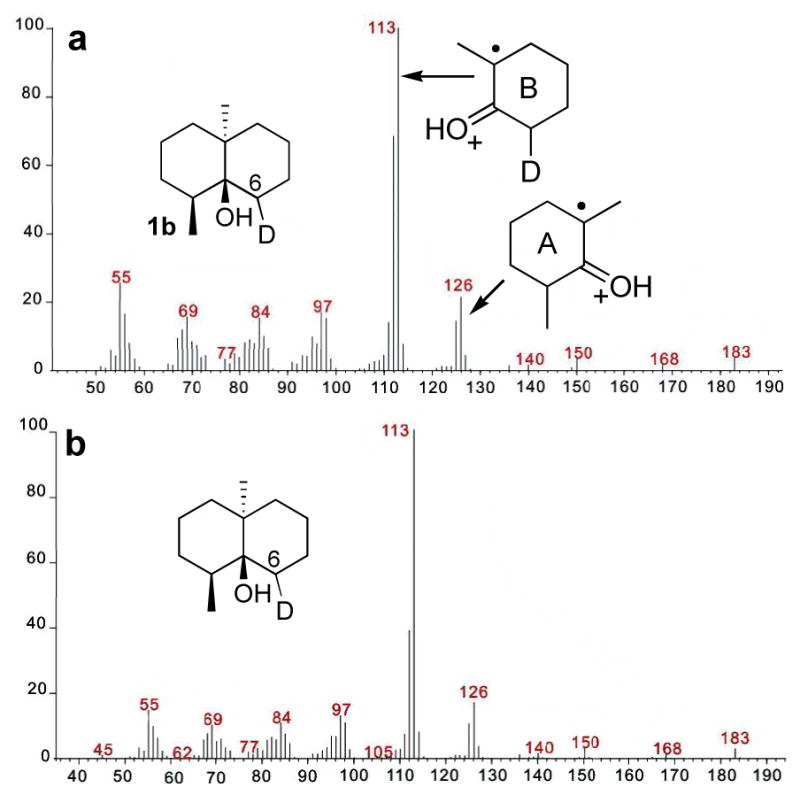
Mass spectra of [6-2H]geosmin (1b) derived from a) [2-2H]FPP and b) (1R)-[1-2H]FPP.
The results of conversion of both [13,13,13-2H3]FPP (2a) and [2-2H]FPP (2b) to geosmins 1 and 1b are fully consistent with the proposed mechanism of cyclization and fragmentation of germacradienol (3) (Scheme 1a)4,9 while firmly excluding the mechanism of Scheme 1b10 as well as alternative, mechanistically less likely proposals.2b The Retro-Prins fragmentation that results in the loss of the germacradienol side chain as acetone has no biochemical precedent. There is an exceptionally high level of amino acid sequence conservation (45–78% identity, 57–85% similarity) among more than a dozen known or presumed microbial geosmin synthases.7 The existence of two independent geosmin biosynthetic pathways, at least among microorganisms, is therefore highly unlikely.
Supplementary Material
Experimental methods, incubation conditions, and GC-MS data. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by National Institutes of Health Grant GM30301 to D.E.C.
References and Notes
- 1.a) Gerber NN. CRC Crit Rev Microbiol. 1979;7:191–214. doi: 10.3109/10408417909082014. [DOI] [PubMed] [Google Scholar]; b) Buttery RG, Garibaldi JA. J Agric Food Chem. 1976;24:1246–1247. [Google Scholar]
- 2.a) Gerber NN, Lechevalier HA. Appl Microbiol. 1965:935–938. doi: 10.1128/am.13.6.935-938.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gerber NN. Tetrahedron Lett. 1968:2971–2974. doi: 10.1016/s0040-4039(01)99008-2. [DOI] [PubMed] [Google Scholar]; b) Pollak FC, Berger RG. Appl Environ Microbiol. 1996;6:1295–1299. doi: 10.1128/aem.62.4.1295-1299.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dickschat JS, Wenzel SC, Bode HB, Muller R, Schulz S. Chembiochem. 2004;5:778–787. doi: 10.1002/cbic.200300813. [DOI] [PubMed] [Google Scholar]; Dickschat JS, Bode HB, Wenzel SC, Muller R, Schulz S. Chembiochem. 2005;6:2023–2033. doi: 10.1002/cbic.200500174. [DOI] [PubMed] [Google Scholar]; d) Scholler CE, Gurtler H, Pedersen R, Molin S, Wilkins K. J Agric Food Chem. 2002;50:2615–2621. doi: 10.1021/jf0116754. [DOI] [PubMed] [Google Scholar]; e) La Guerche S, Chamont S, Blancard D, Dubourdieu D, Darriet P. Antonie Van Leeuwenhoek. 2005;88:131–139. doi: 10.1007/s10482-005-3872-4. [DOI] [PubMed] [Google Scholar]
- 3.a) Cane DE, Watt RM. Proc Natl Acad Sci U S A. 2003;100:1547–1551. doi: 10.1073/pnas.0337625100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) He X, Cane DE. J Am Chem Soc. 2004;126:2678–2679. doi: 10.1021/ja039929k. [DOI] [PubMed] [Google Scholar]
- 4.Jiang J, He X, Cane DE. J Am Chem Soc. 2006;128:8128–8129. doi: 10.1021/ja062669x. [DOI] [PubMed] [Google Scholar]
- 5.Nawrath T, Dickschat JS, Müller R, Jiang J, Cane DE, Schulz S. J Am Chem Soc. doi: 10.1021/ja077790y. submitted (accompanying paper) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cane DE, He X, Kobayashi S, Omura S, Ikeda H. J Antibiot (Tokyo) 2006;59:471–479. doi: 10.1038/ja.2006.66. [DOI] [PubMed] [Google Scholar]
- 7.Jiang J, He X, Cane DE. Nat Chem Biol. 2007;3:711–715. doi: 10.1038/nchembio.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickschat JS, Bode HB, Mahmud T, Muller R, Schulz S. J Org Chem. 2005;70:5174–5182. doi: 10.1021/jo050449g. [DOI] [PubMed] [Google Scholar]
- 10.Spiteller D, Jux A, Piel J, Boland W. Phytochemistry. 2002;61:827–834. doi: 10.1016/s0031-9422(02)00282-0.A similar mechanism has also been proposed for the formation of (+)-dehydrogeosmin in flower heads of the cactus Rebutia marsoneri. Cf. Feng Z, Huber U, Boland W. Helv Chim Acta. 1993;76:2547–2552.
- 11.Shibamoto T. J Pharm Biomed Anal. 2006;41:12–25. doi: 10.1016/j.jpba.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 12.The [13,13,13-2H3]FPP was synthesized by Prabhakaran PC, CfCane DE, Tandon M, Prabhakaran PC. J Am Chem Soc. 1993;115:8103–8106.
- 13.Arigoni D, Cane DE, Shim JH, Croteau R, Wagschal K. Phytochemistry. 1993;32:623–631. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental methods, incubation conditions, and GC-MS data. This material is available free of charge via the Internet at http://pubs.acs.org.