

The role of fast protein structural fluctuations in enzyme-catalyzed reactions is a hotly debated topic in enzymology. Both theoretical1–4 and experimental5–8 studies suggest that collective heavy-atom motions at the femtosecond to picosecond time scale couple to the reaction coordinate. Although such motions have been identified in molecular dynamics simulations and kinetic measurements, there are few experimental observations of protein dynamics at this time scale.9–14 Specifically, no experimental data are available for systems in which fast dynamics are expected to couple to the catalyzed chemical transformation. We report infrared photon echo measurements of formate dehydrogenase (FDH) in complex with azide (N3−, a nM inhibitor and a transition state analog) and nicotinamide (NAD+). We observe only femtosecond to picosecond active-site dynamics for this ternary complex. Our results demonstrate that the active site of the reactive enzyme complex near the catalytic transition state may indeed exhibit the fast dynamics that have been invoked to explain the kinetic properties of several enzymes.
Three-pulse infrared photon echo spectroscopy is a vibrational analog of the NMR spin echo that probes the frequency fluctuations of an ensemble of oscillators. In as much as the structural dynamics of the protein environment control the frequency fluctuations of the azide antisymmetric stretch, infrared photon echo spectroscopy characterizes the spectral density of protein motions. The two-point frequency-frequency time correlation function (FFCF) is a quantitative representation of the dynamics measured by infrared echo spectroscopy. The FFCF is often expressed in the form, , where the Δ’s quantify the magnitude of the frequency fluctuations and the τ’s identify their time scales.9–14
Among the earliest measurements on proteins are studies of the FFCF of azide bound to carbonic anhydrase and hemoglobin and of carbon monoxide bound to hemoglobin by Lim et. al.9 The FFCF’s decay on time scales ranging from hundreds of femtoseconds to tens of picoseconds and longer. A more extensive series of studies by the Fayer group reports similar dynamics for CO bound myoglobin and its mutants.10–13 Finkelstein et al.14 recently measured the FFCF of CO-bound horseradish peroxidase in free form and with different substrates. They observed that the binding of the substrate slows the active-site fluctuations locking certain residues into a heterogeneous distribution of structures. These studies all report a static component in the FFCF indicating significant static heterogeneity and supporting the general conclusion that proteins exhibit both long time scale dynamics (ns-ms) and fast dynamics (fs-ps).
The model system in the current study is FDH from Candida boidinii. This enzyme catalyzes the NAD+-dependent oxidation of formate to carbon dioxide. Kinetic isotope effect (KIE) experiments with this system indicate a contribution from quantum mechanical tunneling and coupling of the dynamics of the donor, the acceptor, and their environment.15,16 Since kinetic experiments suggest that fast protein-ligand interaction dynamics may be functionally important, it is of great interest to measure such motions and their coupling to ligands in the active site. FDH serves as a good model system because azide, which is an excellent vibrational chromophore, is a tight binding, competitive inhibitor for this enzyme with an inhibition constant Ki = 7 nM. Because azide is isoelectronic with the carbon dioxide product and negatively charged like the formate anion reactant, it is a potent analog of the transition-state structure of the catalyzed reaction.
Figure 1 schematically illustrates the active-site structure of the ternary complex of FDH with azide and NAD+ based on a crystal structure of the ternary complex of FDH from Pseudomonas sp. 101, which has high sequence homology to the C. biodinii enzyme.17 The active-site structure is compact and is located between two similar structural motifs each a sandwich of α-helix, parallel β-sheet, and α-helix. In the ternary complex, the azide is “anchored” by H-bonds to two residues on one side (Arg-284 and His-332), and by two residues on the other side (Ile-122 and Asn-146). Significant to this study, these four residues connect to both rigid structural motifs.
Figure 1.
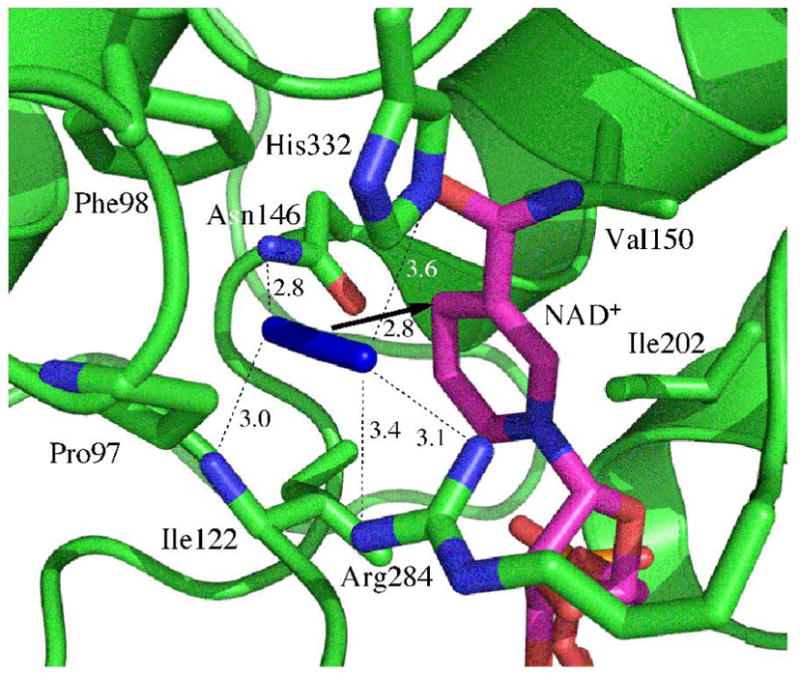
Active site structure of FDH-azide-NAD+ complex (PDB# 2NAD). The arrow indicates the reaction path from the H- donor to acceptor and the dashed lines represent the hydrogen bonds discussed in the text. All distances are in Å.
Figure 2 shows representative three-pulse infrared photon echo data for the azide-NAD-FDH ternary complex in D2O. There are two time delays between the three pulses in a photon echo experiment, T and τ. τ corresponds to the time delay between the first two pulses in the experiment, and T is the delay between the second and third pulses. We measure the echo response as a function of τ for many values of T. Although the vibrational relaxation lifetime, 2.2 ps for azide bound to FDH, limits the time scales over which we can measure dynamics, it affects the amplitude and our measurement depends on the shape of the echo response. Thus, we are able to quantify contributions to the FFCF that decay on time scales longer than the population lifetime. The dots are the measured data and the solid lines are calculated echo responses using the parameters from a global fit to both the echo data and the infrared absorption spectrum assuming a FFCF of the form, . The parameters for the FFCF from the fit are, Δ1 = 1.73 ps−1, τ1 = 0.25 ps, Δ2 = 1.43 ps−1, τ2 = 3 ps. The narrow feature centered at τ = 0 in each data set is due to the response of the D2O solvent that we excite due to the overlap between the edge of the laser pulse spectrum and the tail of the OD stretch absorption. The solvent response persists because of a thermally induced spectral shift, but does not interfere with the fits because we mask out the data 250 fs on either side of time zero to do the global fitting.
Figure 2.
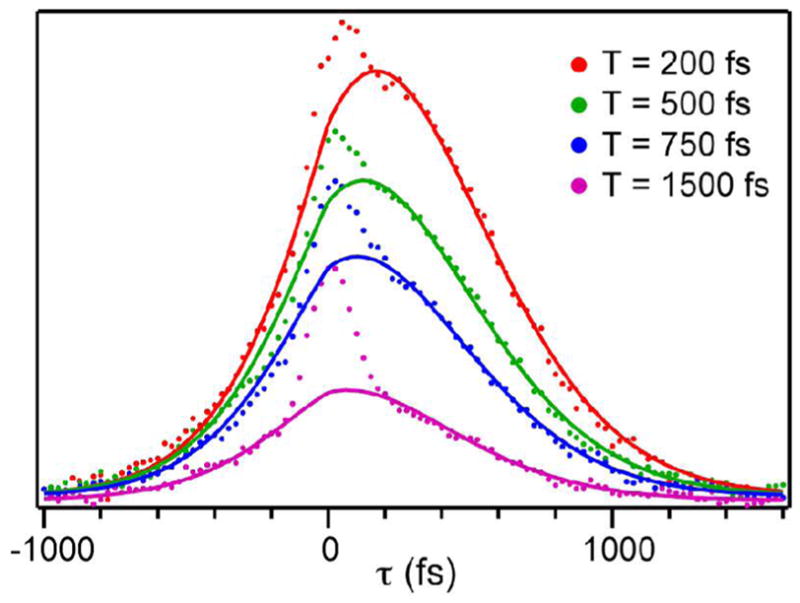
Photon echo scans of the azide-NAD-FDH ternary complex in D2O for increasing values of T (top to bottom), the time during which spectral diffusion occurs.
The data for the ternary complex with azide are distinct from the photon echo and 2D IR measurements that have been made on protein-bound ligands previously. The fit to the FFCF has two components, one with a time constant of 250 fs the other with a time constant of 3 ps. Notably, the fit does not require a static component in the FFCF decay. The maximum static contribution to the FFCF for which the global fit is still acceptable is 5% of the total amplitude. This value represents an upper limit on the amplitude of a static contribution to the FFCF. This result is unique because all the proteins that have been studied to date presented much slower FFCF components (tens of ps or more).
A key difference between previously studied protein complexes of this sort and the azide-NAD-FDH complex reported here is that no fs-ps dynamics have been implicated to have a role in the functionality of those proteins.9–14 In contrast, 2° KIE and equilibrium measurements,16 and close analogy to other nicotinamide-dependent enzymes, indicate that fast dynamics may play a critical role in the formate dehydrogenase catalyzed reaction. Although our results do not prove a role for fs-ps motions, they are consistent with the hypothesis that fs-ps dynamics are relevant to the catalyzed reaction, particularly because we observe no dynamics at longer time scales.
Another difference between FDH and the previously studied enzymes is that, in the earlier cases, the ligands are ground-state analogs. In FDH, however, azide is a potent inhibitor and an unusually close analog of the transition state.17 Consequently, our measurements reveal the time scales for structural fluctuations near the transition state where such motions exert the greatest influence over kinetic properties.
In spite of the differences between this and previous studies, such rapid spectral diffusion is still surprising. Azide spectral diffusion is most sensitive to fluctuations of hydrogen bonding interactions at the terminal nitrogens.18,19 Our results indicate that azide samples the entire distribution of hydrogen bond lengths within a few picoseconds. This result is unexpected because long-range structural motions of the secondary structural elements to which these residues are attached should modulate the hydrogen bonding interactions to the active-site-bound azide, and those motions can span many decades in time. Our measurements indicate that the bound azide does not experience protein structural fluctuations on time scales longer than a few picoseconds.
One interpretation of this observation is that there are no fluctuations of the secondary structural components to which the active-site residues are attached on time scales longer than a few picoseconds. Of course, slower motions might be important for other kinetic steps along the mechanistic cascade (e.g., binding and release of ligands, protein rearrangements, etc.), but are not present as the structure nears that of the chemical transition state. An alternative explanation is that the structural fluctuations are precisely choreographed so that the motions of the protein modulate the hydrogen bond lengths of the residues in an anticorrelated fashion. Then, the lengthening of one hydrogen bond would offset the shortening of another hydrogen bond resulting in no change in frequency. Both possibilities are interesting and should be further investigated by a variety of experimental and theoretical methods attempting to explore structural and dynamic properties of enzyme catalyzed C-H bond activation.
Acknowledgments
This work was supported by the Roy J. Carver Charitable Trust and NSF CHE-0644410 (CMC); NIH R01 GM65368 and NSF CHE-0715448 (AK); and a fellowship from the Center for Bioprocessing and Biocatalysis at the University of Iowa (JB).
References
- 1.Benkovic SJ, Hammes-Schiffer S. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 2.Truhlar DG. Variational transition state theory and multidimensional tunneling for simple and complex reacrions in the gas phase, solids, Liquids, and enzymes. In: Kohen A, Limbach HH, editors. Isotope effects in chemistry and biology. Ch. 22. Taylor & Francis, CRC Press; Boca Raton, FL: 2006. pp. 579–620. [Google Scholar]
- 3.Hammes-Schiffer S. Acc Chem Res. 2006;39:93–100. doi: 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- 4.Warshel A, Olsson MHM, Villá-Freixa J. Computer simulations of isotope effects in enzyme catalysis. In: Kohen A, Limbach HH, editors. Isotope effects in chemistry and biology. Ch. 23. Taylor & Francis, CRC Press; Boca Raton, FL: 2006. pp. 621–644. [Google Scholar]
- 5.Nagel ZD, Klinman JP. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Goodey Nm, Benkovic SJ, Kohen A. Proc Natl Acad Sci USA. 2006;103:15753–15758. doi: 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohen A. Kinetic isotope effects as probes for hydrogen tunneling in enzyme catalysis. In: Kohen A, Limbach HH, editors. Isotope effects in chemistry and biology. Ch. 28. Taylor & Francis, CRC Press; Boca Raton, FL: 2006. pp. 743–764. [Google Scholar]
- 8.Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, Ranaghan KE, Mulholland AJ, Sutcliffe MJ, Scrutton NS, Leys D. Science. 2006;312:237–241. doi: 10.1126/science.1126002. [DOI] [PubMed] [Google Scholar]
- 9.Lim MH, Hamm P, Hochstrasser RM. Proc Natl Acad Sci USA. 1998;95:15315–15320. doi: 10.1073/pnas.95.26.15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fayer MD. Ann Rev Phys Chem. 2001;52:315–356. doi: 10.1146/annurev.physchem.52.1.315. [DOI] [PubMed] [Google Scholar]
- 11.Merchant KA, Noid WG, Akiyama R, Finkelstein IJ, Goun A, McClain BL, Loring RF, Fayer MD. J Am Chem Soc. 2003;125:13804–13818. doi: 10.1021/ja035654x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merchant KA, Noid WG, Thompson DE, Akiyama R, Loring RF, Fayer MD. J Phys Chem B. 2003;107:4–7. [Google Scholar]
- 13.Finkelstein IJ, Goj A, McClain BL, Massari AM, Merchant KA, Loring RF, Fayer MD. J Phys Chem B. 2005;109:16959–16966. doi: 10.1021/jp0517201. [DOI] [PubMed] [Google Scholar]
- 14.Finkelstein IJ, Ishikawa H, Kim S, Massari AM, Fayer MD. Proc Natl Acad Sci USA. 2007;104:2637–2642. doi: 10.1073/pnas.0610027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanchard JS, Cleland WW. Biochemistry. 1980;19:3543–3550. doi: 10.1021/bi00556a020. [DOI] [PubMed] [Google Scholar]
- 16.Hermes JD, Morrical SW, OLeary MH, Cleland WW. Biochemistry. 1984;23:5479–5488. doi: 10.1021/bi00318a016. [DOI] [PubMed] [Google Scholar]
- 17.Popov VO, Lamzin VS. Biochem J. 1994;301:625–643. doi: 10.1042/bj3010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li SZ, Schmidt JR, Corcelli SA, Lawrence CP, Skinner JL. J Chem Phys. 2006;124:204110. doi: 10.1063/1.2200690. [DOI] [PubMed] [Google Scholar]
- 19.Li SZ, Schmidt JR, Piryatinski A, Lawrence CP, Skinner JL. J Phys Chem B. 2006;110:18933–18938. doi: 10.1021/jp057568k. [DOI] [PubMed] [Google Scholar]