

Abstract
The activation of phosphatidylinositol 3-kinase (PI 3-K) and subsequent production of PtdIns(3,4,5)P3 launches a signal transduction cascade that impinges on a plethora of downstream effects on cell physiology. Control of PI 3-K and PtdIns(3,4,5)P3 levels are important therapeutic targets in treatments for allergy, inflammation, cardiovascular, and malignant human diseases. We designed metabolically-stabilized, i.e., phosphatase resistant, analogues of PtdIns(3,4,5)P3 as probes for long-lived potential agonists or potentially antagonists for cellular events mediated by of PtdIns(3,4,5)P3. In particular, two types of analogues were prepared containing phosphomimetics that would be selectively resistant to the lipid 3-phosphatase PTEN. The total asymmetric synthesis of the 3-phosphorothioate-PtdIns(3,4,5)P3 and 3-methylenephosphonate-PtdIns(3,4,5)P3 analogues is described. These two analogues showed differential binding to PtdIns(3,4,5)P3 binding modules, and both were potential long-lived activators that mimicked insulin action in sodium transport in A6 cells.
The phosphoinositide 3-kinase (PI 3-K) signaling pathway contains important therapeutic targets in human pathophysiology.1,2 Phosphatidylinositol-3,4,5 –triphosphate (PtdIns(3,4,5)P3) is a ubiquitous signaling lipid found in higher eukaryotic cells3 and activates a plethora of downstream cellular processes.4 These signaling events include cell proliferation and transformation,5 cell shape and motility,6 and insulin action and alteration of glucose transport.7 PtdIns(3,4,5)P3-regulated signaling is modulated by the lipid 3-phosphatase PTEN8 and SH2 domain-containing inositol 5-phosphatase SHIP.9
A metabolically-stabilized (ms) analogue of PtdIns(3,4,5)P3 that resists lipid 3- and 5-phosphatases would have numerous applications in understanding the role of PtdIns(3,4,5)P3 in cell physiology. The ms-PtdIns(3,4,5)P3 analogues could separate the activation of signal transduction from the degradation of the signal by phosphatase action in cells. This chemical biology approach to dissection of the PI 3-K pathway is complementary to the use of siRNA knockdowns or genetic knockouts for PTEN and SHIP. We focused first on a 3-stabilized PtdIns(3,4,5)P3 analog, i.e., one resistant to hydrolysis by PTEN, and we selected two stabilized phosphomimetic isosteres to replace the 3-phosphate of PtdIns(3,4,5)P3.
Phosphorothioates are phosphomimetics that show reduced rates of enzyme-mediated hydrolysis.10 However, the replacement of P = O by P = S also affects the pKa of the phosphate and removes a H-bond acceptor.11,12 For example, the phosphorothioate analogue of PtdIns(3)P had reduced binding activity for cognate binding proteins, due in part to reduced H-bonding.13 We hypothesized that a 3-phosphorothioate of PtdIns(3,4,5)P3 could be either an antagonist or a long-lived agonist in the PI 3-K signaling pathway, because of reduced dephosphorylation by PTEN. Moreover, the methylenephosphonate analogue of PtdIns(3)P bound selectively to one of two cognate binding proteins.14 We now describe the first asymmetric total syntheses of two PtdIns(3,4,5)P3 analogues that are resistant to the 3-phosphatase PTEN - 3-PT-PtdIns(3,4,5)P3 and 3-MP-PtdIns(3,4,5)P3. Further, we show both selective binding to a PtdIns(3,4,5)P3-binding protein and the ability of these analogues to increase sodium transport in A6 cell monolayers.
The synthetic sequence to 3-phosphorthioate-PtdIns(3,4,5)P3 (3-PT-PtdIns(3,4,5)P3) is illustrated in Scheme 1. Treatment of TBDPS-ether 315,16 with the bulky bifunctional reagent TBDPSCl2, in the presence of imidazole selectively afforded the diol 4,5-bis-silyl ether in 88% yield as a single product; the diols were then protected to give compound 4. Next, TIPDS deprotection, bisphosphorylation with dimethyl N, N-diisopropyl-phosphoramidite and subsequent m-CPBA oxidation generated the protected 4,5-bisphosphate 5 in good yield. Since attempts to remove TBDPS in the presence of the cyanoethyl phosphate protecting groups failed to give a satisfactory result, the TBDPS was replaced with TES at this stage. Reduction of the benzoyl ester 6 with Dibal-H at −78 °C followed by thiophosphorylation with bis(2-cyanoethoxy)(diisopropylamino)phosphine in the presence of 1H-tetrazole and phenylacetyl disulfide to provide the desired TES ether.17 Deprotection of TES with the weakly acidic reagent NH4F in methanol gave the key advanced intermediate 7 in 80% yield. Condensation of 7 with each of four different freshly prepared 1,2-di-O-acyl-sn-glycero cyanoethyl (N,N -diisopropylamino) phosphoramidites 8a–8d in the presence of 1H-tetrazole, followed by t-BuOOH oxidation, gave the fully protected lipids 9a–9d.13 Removal of the cyanoethyl groups with triethylamine and bis(trimethylsilyl)trifluoro-acetamide (BSTFA), followed by removal of the MOM and methyl ester groups with TMSBr afforded the 3-PT-PtdIns(3,4,5)P3 analogues 1a–1d.
Scheme 1.
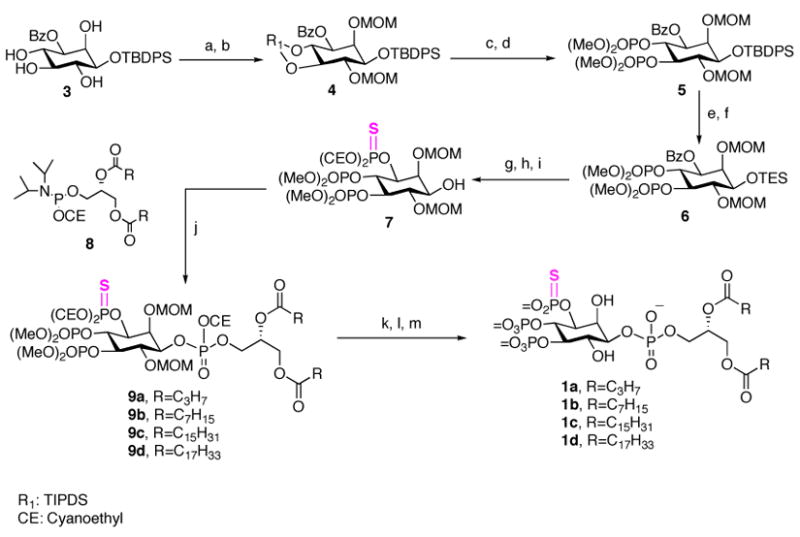
Synthesis of Phosphorothioates 1a
a Conditions: (a) TIPDSCl2, imidazole, Py, 88%; (b) MOMCl, DIPEA, DMF, 65 °C, 63%; (c) TBAF, THF, 77%; (d) N,N -dimethylphosphor-midite, 1H -tetrazole,; m-CPBA, 81%; (e) TBAF·3H2O, DMF, 91%; (f) TESCl, imidazole, CH2Cl2, 88%; (g) Dibal-H, CH2Cl2, −78 °C, 84%; (h) Bis(2-cyanoethoxy) (diisopropylamino)phosphine, 1H-tetrazole; phenylacetyl disulfide, 72%; (i) NH4F, MeOH, 85%; (j) 1H -tetrazole, CH2Cl2, rt; then t-BuOOH; (k) TEA, BSTFA, CH3CN; (l) TMSBr/CH2Cl2 (2:3), rt, 40 min; (m) MeOH.
Scheme 2 summarizes the preparation of the 3-methylenephosphonate-PtdIns(3,4,5)P3 (3-MP-PtdIns(3,4,5)P3, 2), in which reduction of 4 with Dibal-H was followed by alkylation with dimethyl phosphonomethyltriflate (n-BuLi/HMPA) to give methylenephosphonate 10 in 80% yield. Use of excess HMPA to chelate the Li+ cation and enhance the nucleophilicity of the alkoxide was the key to obtaining a high yield. Selective desilylation of 10 with 1 M TBAF in THF provided the 4,5-diol, which was bisphosphorylated to give TBDPS ether 11. Removal of the TBDPS group followed by coupling with the phosphoramidites 8a–8d to give protected lipids 12a–12d. Removal of the protective groups gave the 3-MP-PtdIns(3,4,5)P3 analogues 2a–2d.
Scheme 2.
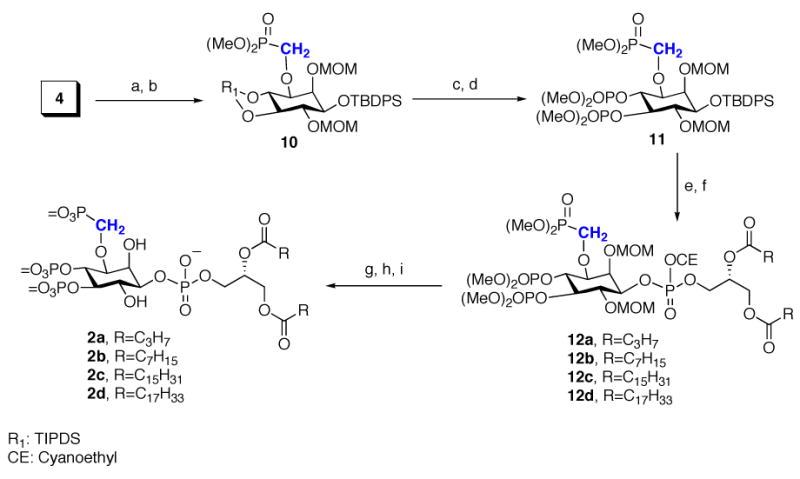
Synthesis of Methylenephosphonates 2a
a Conditions: (a) Dibal-H, CH2Cl2, −78 °C, 88%; (b) n-BuLi, HMPA, dimethyl phosphonomethyltriflate, THF, −78 °C to rt, 80%; (c) TBAF, 90%; (d) N,N-dimethylphosphoramidite, 1H -tetrazole; m-CPBA, 95%; (e) TBAF·3H2O, DMF, 75%; (f) 8a–8d, 1H -tetrazole, CH2Cl2; then t-BuOOH; (g) TEA, BSTFA, CH3CN; (h) TMSBr/CH2Cl2 (2:3); (i) MeOH.
To test the function of these analogues, we used carrier-mediated intracellular delivery18 of PtdIns(3,4,5)P3 which is known to activate GLUT4 translocation to the plasma membrane7 and sodium transport.19 The physiological function of the 3-PT-and 3-MP-PtdIns(3,4,5)P3 analogues was examined in A6 cell monolayers, a renal epithelium model that expresses epithelial sodium channels (ENaC).20 ENaC activity is the rate-limiting step of the sodium transport and is stimulated by insulin.21 DiC16-PtdIns(3,4,5)P3 is an early mediator of the insulin-stimulated sodium transport in A6 cells.19 Thus, we compared the effect of the unmodified diC16-PtdIns(3,4,5)P3 with diC16-3-PT-PtdIns(3,4,5)P3 1c and diC16-3-MP-PtdIns(3,4,5)P3 2c on sodium transport across confluent monolayers of A6 cells. As shown in Figure 1, apical addition of either 1c or 2c increased sodium transport. Moreover, the 3-MP analogue 2c was the most potent and long-lived mediator of sodium transport, and the 3-PT-analogue 1c also extended sodium transport compared to unstabilized PtdIns(3,4,5)P3. The lag time observed between PtdIns(3,4,5)P3 analogue addition and the final effect on sodium transport was due to intracellular delivery. The spatiotemporal coordination of lipid production and removal are likely required for normal physiology, and thus PtdIns(3,4,5)P3 is necessary but not sufficient to fully mimic the action of insulin.
Figure 1.
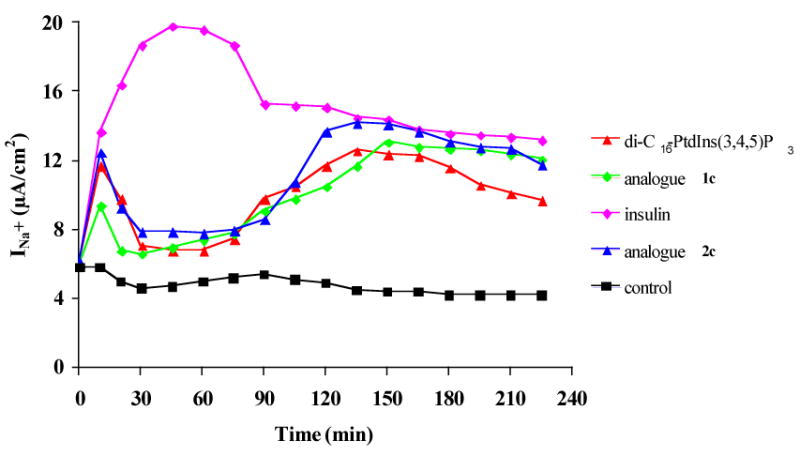
Stimulation of A6 cell monolayers. Experimental details for triplicate measurements of sodium transport (INa+, μA/cm2)19 are in the Supplementary Information. A representative result is illustrated.
We tested the binding of 3-PT and 3-MP analogues to the specific binding protein Grp1 (Supplementary Figure 2). DiCg-3-PT-PtdIns(3,4,5)P3 1b bound to Grp1 with 5-fold reduced affinity relative to diC8-PtdIns(3,4,5)P3, but the diC8-3-MP analogue 2b showed no binding at all. Moreover, while PTEN rapidly hydrolyzed diC8-PtdIns(3,4,5)P3, no hydrolysis was observed with either 1b or 2b (Supplementary Figure 3). Interestingly, diCg-3-PT analogue 1b showed > 90% inhibition of PTEN activity at 0.3 μM, while the diC8-3-MP analogue 2b required 30 μM for >90% inhibition (A. Branch, P. Neilsen, personal communication). Thus, analogues 1 and 2 have potential as protein-selective biological tools in the PI 3-K signaling pathway. Additional functional assays and interactions with PTEN will be reported in due course.
Supplementary Material
Experimental details for synthesis, characterization of new compounds, binding data, and PTEN assays. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We thank the NIH (NS 29632 to GDP) and the “Fonds de la Recherche Scientifique Médicale” for support, Dr. C. Ferguson (Echelon Biosciences, Inc.) for Grp1 binding data, and Dr. P. Neilsen and Ms. A. Branch (Echelon) for PtdIns(3,4,5)P3, histone, PTEN, and assistance with the PTEN assays.
References
- 1.Prestwich GD. Chem Biol. 2004;11:619–637. doi: 10.1016/j.chembiol.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 2.Drees BE, Mills GB, Rommel C, Prestwich GD. Expert Opin Ther Patents. 2004;14:703–732. [Google Scholar]
- 3.Traynor-Kaplan AE, Harris AL, Thompson BL, Taylor P, Sklar LA. Nature. 1988;334:353–356. doi: 10.1038/334353a0. [DOI] [PubMed] [Google Scholar]
- 4.Toker A, Cantley LC. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 5.Cantley LC, Auger KR, Carpenter C, Duckworth B, Graziani A, Kapeller R, Soltoff S. Cell. 1991;64:281–302. doi: 10.1016/0092-8674(91)90639-g. [DOI] [PubMed] [Google Scholar]
- 6.Final N, Goberdhan DC, Collinson L, Fujita Y, Cox IM, Wilson C, Pichaud F. Curr Biol. 2006;16:140–149. doi: 10.1016/j.cub.2005.11.068. [DOI] [PubMed] [Google Scholar]
- 7.Sweeney G, Garg RR, Ceddia RB, Li D, Ishiki M, Somwar R, Foster LJ, Neilsen PO, Prestwich GD, Rudich A, Klip A. J Biol Chem. 2004;279:32233–32242. doi: 10.1074/jbc.M402897200. [DOI] [PubMed] [Google Scholar]
- 8.Maehama T, Dixon JE. Trends Cell Biol. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- 9.Pesesse X, Deleu S, De Smedt F, Drayer L, Erneux C. Biochem Biophys Res Commun. 1997;239:697–700. doi: 10.1006/bbrc.1997.7538. [DOI] [PubMed] [Google Scholar]
- 10.Lampe D, Liu C, Potter BV. J Med Chem. 1994;37:907–912. doi: 10.1021/jm00033a007. [DOI] [PubMed] [Google Scholar]
- 11.Murray AW, Atkinson MR. Biochemistry. 1968;7:4023–4029. doi: 10.1021/bi00851a032. [DOI] [PubMed] [Google Scholar]
- 12.Hampton A, Brox LW, Bayer M. Biochemistry. 1969;8:2303–2311. doi: 10.1021/bi00834a011. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y, Lee SA, Kutateladze TG, Sbrissa D, Shisheva A, Prestwich GD. J Am Chem Soc. 2006;128:885–897. doi: 10.1021/ja0554716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gajewiak J, Xu Y, Lee SA, Kutateladze T, Prestwich GD. Org Lett. 2006;8:2811–2813. doi: 10.1021/ol060903i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruzik KS, Tsai MD. J Am Chem Soc. 1992;114:6361–6374. [Google Scholar]
- 16.Kubiak RJ, Bruzik KS. J Org Chem. 2003;68:960–968. doi: 10.1021/jo0206418. [DOI] [PubMed] [Google Scholar]
- 17.Dreef CE, Mayr GW, Jansze J-P, Roelen HCPF, Van deer Marel GA, van Boom JH. Bioorg Med Chem Lett. 1991;1:239–242. [Google Scholar]
- 18.Ozaki S, DeWald DB, Shope JC, Chen J, Prestwich GD. Proc Natl Acad Sci USA. 2000;97:11286–11291. doi: 10.1073/pnas.210197897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markadieu N, Blero D, Boom A, Erneux C, Beauwens R. Am J Physiol Renal Physiol. 2004;287:F319–328. doi: 10.1152/ajprenal.00314.2003. [DOI] [PubMed] [Google Scholar]
- 20.Handler J, Perkins F, Johnson J. Am J Physiol Cell Physiol. 1981;240:C103–C105. doi: 10.1152/ajpcell.1981.240.3.C103. [DOI] [PubMed] [Google Scholar]
- 21.Rossier BC, Canessa CM, Schild L, Horisberger JD. Curr Opin Nephrol Hypertens. 1994;3:487–496. doi: 10.1097/00041552-199409000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for synthesis, characterization of new compounds, binding data, and PTEN assays. This material is available free of charge via the internet at http://pubs.acs.org.