

Abstract
Batzelladine F (1) was synthesized in enantioselective and stereoselective fashion in 15 steps (longest linear sequence) and 1.7% overall yield from two readily available enantioenriched β-hydroxy esters, methyl (R)-3-hydroxydecanoate and methyl (R)-3-hydroxybutyrate. Tethered Biginelli condensations are used to assemble both tricyclic guanidine fragments, with the second tethered Biginelli condensation (14 + 16 → 17) also being employed to join the guanidine fragments. Three diastereomers of batzelladine F, 2−4, were prepared also. A combination of HPLC, optical rotation and CD spectroscopy was employed to distinguish stereoisomers 1−4, proving that 1 is the correct structure of the hexacyclic marine alkaloid batzelladine F.
Introduction
The hexacyclic diguanidine alkaloid batzelladine F was first reported in 1997 from a marine natural products isolation program directed at identifying small molecule inhibitors of protein–protein interactions.2 Isolated from a red Jamaican sponge, batzelladine F is one of the most structurally complex of the marine alkaloids of the batzelladine family.3 Batzelladine F was reported to induce the dissociation of the tyrosine kinase p56lck from the CD4 co-receptor expressed on the surface of T cells, an event that was postulated as potentially being useful in the treatment of autoimmune diseases.2
In the preceding contribution, we described the evolution of a concise strategy for the chemical synthesis of complex guanidine alkaloids that contain two linked octahydro-5,6,6a-triazaacenaphthalene moieties.4 We also disclosed the use of this chemistry to prepare four stereoisomers of the presumed structure of batzelladine F,5,6 none of which were identical with the marine isolate.4 After reexamining the natural product, we proposed a different constitution for batzelladine F in which a ten-atom chain tethers the two guanidine moieties and the right-hand tricyclic fragment contains an n-heptyl side chain. As the configurational relationship between the two octahydro-5,6,6a-triazaacenaphthalene fragments was not known, nor the relative configuration of the methyl substituent of the tether, the eight stereoisomers (four enantiomer pairs) depicted in Figure 1 all became possibilities for the structure of batzelladine F. In this article, we describe our enantioselective total synthesis of batzelladine F (1) and the studies that establish 1 to be the correct structure of the natural alkaloid.
Figure 1.
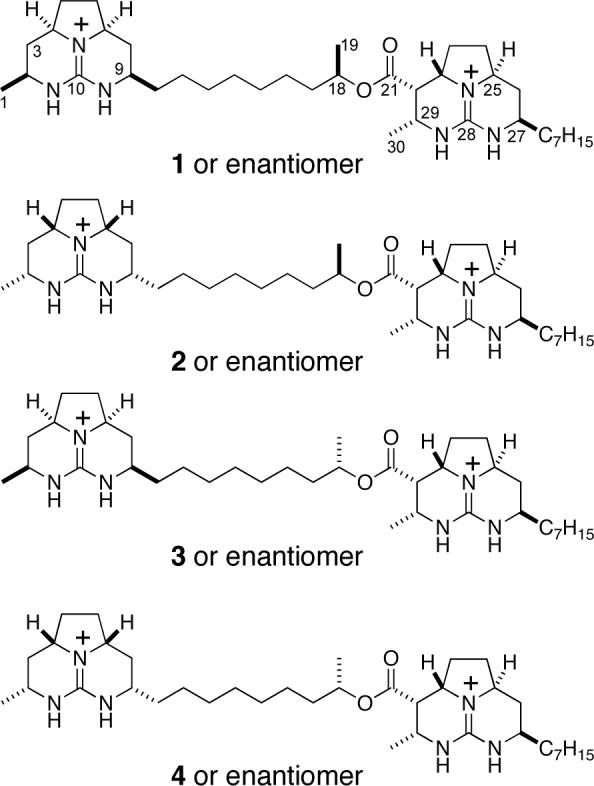
Eight possibilities for the revised structure of batzelladine F.
Results and Discussion
Total Synthesis of Batzelladine F
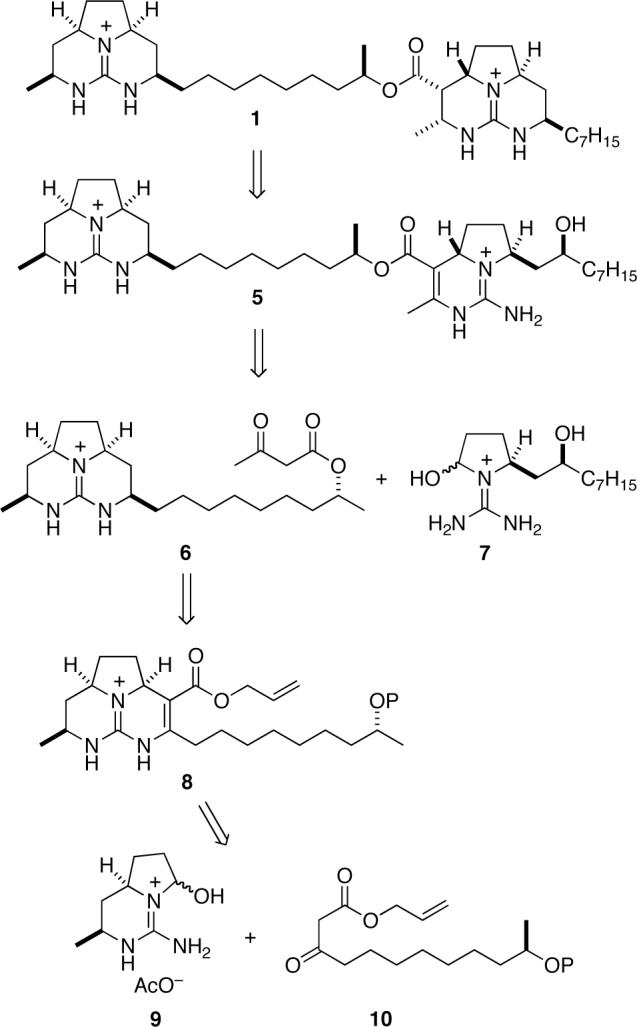
Our plan for the synthesis of batzelladine F is outlined antithetically in Scheme 1 and follows the strategy developed during the studies described in the preceding article.4 We envisaged the right-hand guanidine fragment of 1 as evolving from pentacyclic precursor 5 by ring-closure and catalytic hydrogenation.7 This intermediate would be the product of a convergent, anti-selective, tethered Biginelli condensation8 between β-keto ester 6 and guanidine hemiaminal 7. The allyl ester precursor 8 of β-keto ester 6 would be the product of a syn-selective tethered Biginelli condensation9 between β-keto ester 10 and bicyclic guanidine hemiaminal 9 whose synthesis was described in the preceding article.4
Scheme 1.
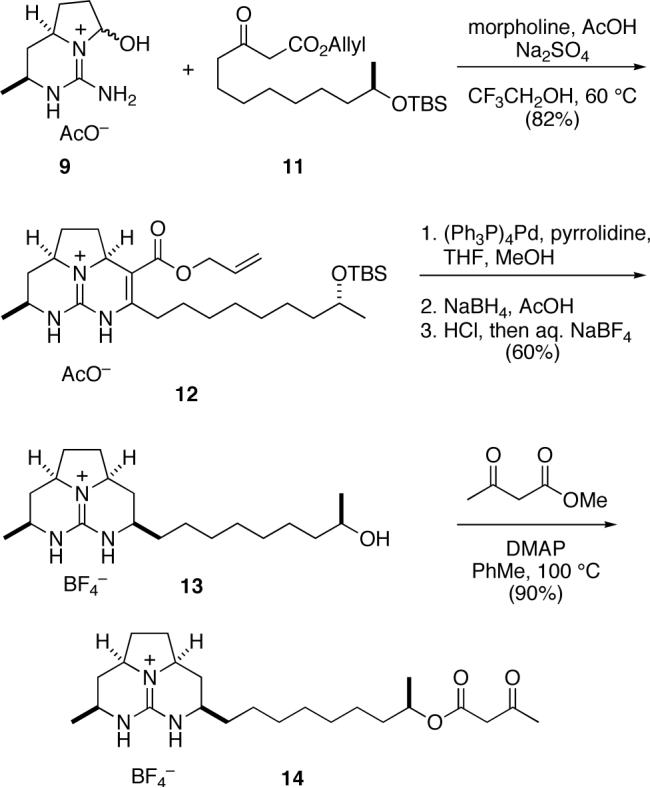
Construction of the left-hand tricyclic guanidine fragment of batzelladine F is summarized in Scheme 2. Tethered Biginelli condensation of β-keto ester 1110,11,12 with aminohexahydropyrrolopyrimidinol 94 proceeded smoothly under standard conditions to provide hexahydro-5,6,6a-triazaacenaphthalene 12 in 82% yield, as a 5:1 mixture of syn:anti stereoisomers. Using a sequence of transformations developed in our earlier studies,4 this product was deallylated with concomitant decarboxylation and reduced stereoselectively to form the octahydro-5,6,6a-triazaacenaphthalene product. The silyl-protecting group was removed from this intermediate by reaction with aqueous HCl at room temperature. After chromatography on silica gel to remove the minor anti stereoisomer, the product fractions were washed with aqueous NaBF4 to provide tricyclic guanidinium tetrafluoroborate 13. These three steps proceeded in 60% overall yield. Acylation of guanidine alcohol 13 with methyl acetoacetate13 then provided octahydro-5,6,6a-triazaacenaphthalene β-keto ester 14 in 90% yield.
Scheme 2.
As the prelude to the central Biginelli fragment coupling, the (S,S)-1-(diaminomethylene)-2-hydroxypyrrolidinium acetate 16 was assembled in eight high-yielding steps from (R)-β-hydroxy ketone 159 using the sequence we had developed earlier to prepare the analogous structure having a (S)-2-hydroxyundecyl side chain (Scheme 3).7,12 The pivotal fragment-coupling tethered Biginelli condensation was carried out by combining guanidine β-keto ester 14 with 3 equiv of guanidine hemiaminal 16 and 3 equiv of morpholinium acetate in 2,2,2-trifluoroethanol, followed by heating the resultant solution at 60 °C for 48 h. The product of this reaction, pentacyclic diguanidine 17, was isolated in 59% yield as the ditrifluoroacetate salt after HPLC separation from minor amounts (<10%) of isomer 18 and remaining guanidine hemiaminal 16.
Scheme 3.
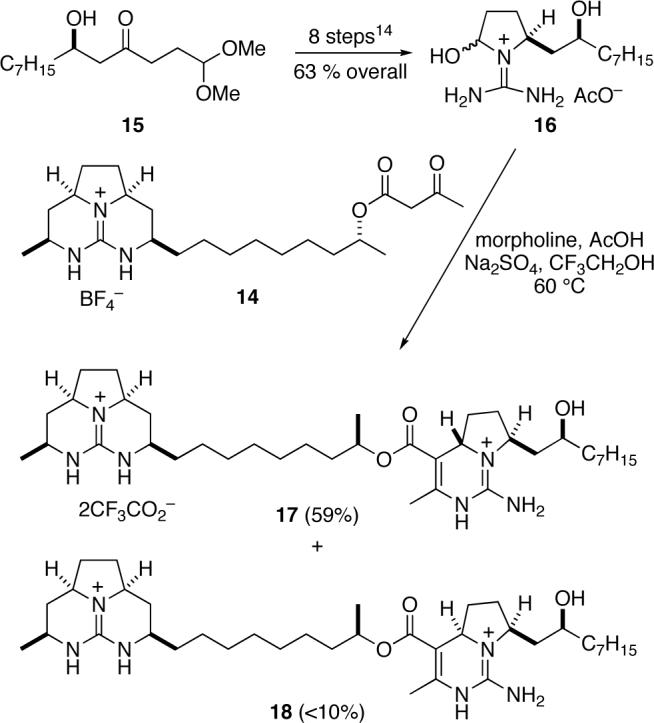
To complete the synthesis of batzelladine F, we needed to close the final ring and reduce the carbon-carbon double bond (Scheme 4). To this end, the trifluoroacetate counter ions were exchanged for tetrafluoroborate by washing a CHCl3 solution of pentacyclic diguanidine 17 with saturated aqueous NaBF4.14 The alcohol was then converted to its mesylate derivative, which cyclized smoothly in the presence of Et3N in hot CHCl3 to deliver hexacyclic diguanidine 19 in 68% yield. Finally, reduction of 19 over Rh·Al2O3 with H2 at 100 psi provided batzelladine F (1) as its ditrfluoroacetate salt, (c 0.25, MeOH), in 21% yield after HPLC separation from isomer 20. NMR spectra of synthetic 1 (1H and 13C in CD3OD) agreed well with the corresponding data reported for natural batzelladine F.2
Scheme 4.
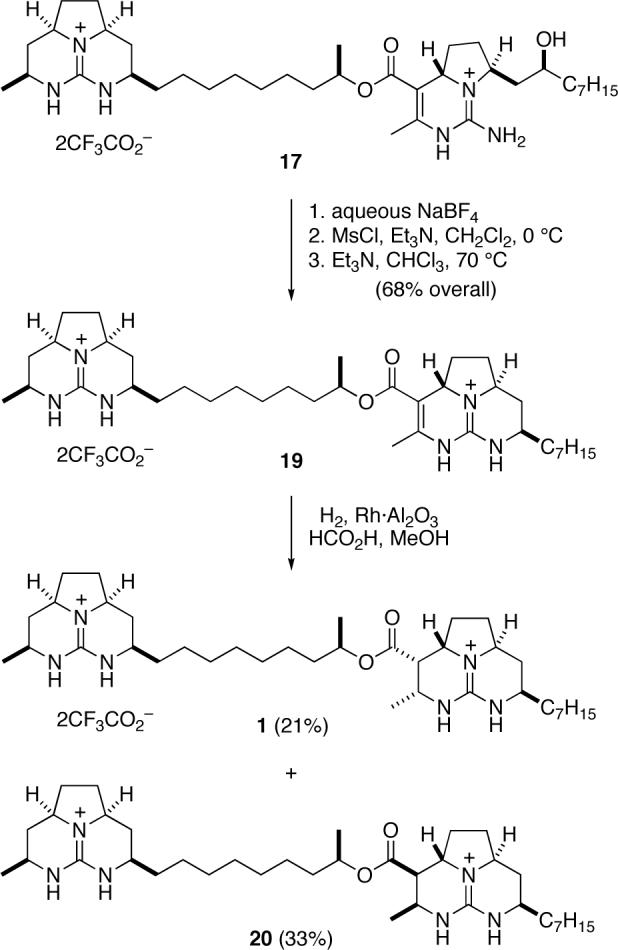
Synthesis of Three Stereoisomers of Batzelladine F and Proof that 1 is the Structure of Batzelladine F
Because of the large distance between the two tricyclic guanidine fragments and the separation between the C18 and C21 stereocenters, we anticipated that the diastereomers of 1 depicted in Figure 1 would show NMR spectra identical, or nearly identical, to those of isomer 1. Thus to establish rigorously the stereostructure of batzelladine F, we synthesized stereoisomers 2−4 depicted in Figure 1. These syntheses were accomplished using chemistry identical to that described for the synthesis of 1; full details of these syntheses are provided in the Supporting Information.
With the four synthetic isomers 1−4 in hand, each as its ditrifluoroacetate salt, we turned to investigate what criteria could distinguish them from each other. We began with HPLC co-injection. To our chagrin, all four isomers co-eluted on the C18 column that had cleanly separated the four related diastereomers we had prepared during our earlier studies that corrected the misassigned connectivity of batzelladine F.4 After screening a variety of combinations of solid and mobile phases, we found that an Altima© Phenyl column with acetonitrile–water as the eluent could separate the C18 epimers, that is separate isomer 1 from 3, and 2 from 4. Isomer 4 did not coelute with an authentic sample of batzelladine F, thus eliminating it and its enantiomer as possibilities for the structure of the natural product. As diastereomers 3 and 4 co-eluted under these conditions, isomer 3 and its enantiomer could also be eliminated. Unfortunately, isomers 1 and 2 coeluted under all conditions. Moreover, these samples showed 1H and 13C NMR spectra indistinguishable from one another and batzelladine F.
Fortunately, the ditrifluoroacetate salts of synthetic isomers 1, (c 0.25, MeOH), and 2, (c 0.36, MeOH), could be distinguished by polarimitry as they showed different optical rotations at four wavelengths. However, the natural isolate, counter ion unspecified, is reported to show (c 0.87, MeOH).2
We thought it likely that this discrepancy in optical rotation resulted from impurities in the natural marine isolate.15 Therefore, we purified the small sample of authentic batzelladine F in our possession by HPLC. The CD spectrum of this purified material closely resembled that of synthetic 1 and was distinct from that of isomer 2 (Figure 2). Therefore, we conclude that 1 is the correct structure of batzelladine F.
Figure 2.

CD spectra of the ditrifluoroacetate salts of authentic batzelladine F (0.3 mg/mL), and synthetic products 1 (3.9 mg/mL) and 2 (3.6 mg/mL) in MeOH.
Conclusion
An enantioselective and stereoselective total synthesis of batzelladine F (1) was accomplished in 15 steps (longest linear sequence) and 1.7% overall yield from two readily available enantioenriched β-hydroxy esters: methyl (R)-3-hydroxydecanoate16 (the precursor of 15)7 and commercially available methyl (R)-3-hydroxybutyrate (the precursor of 9).4 The synthesis is punctuated by the use of stereoselective tethered Biginelli condensations to assemble both tricyclic guanidine fragments,17 with the second tethered Biginelli condensation also allowing these complex fragments to be joined. This modular assembly allowed three diastereomers of 1 to be readily prepared also, permitting the structure 1 of batzelladine F to be rigorously defined. This study is one example in a growing list of recent investigations in which stereocontrolled total syntheses corrected misassigned structures of recently isolated natural products.18 Furthermore, the collection of polycyclic guanidines prepared during this and cognate studies4 has proven to be of considerable value in identifying small molecular inhibitors of pharmacologically relevant protein–protein interactions.19
Experimental Section20
(2aS,7S,8aR)-3-Allyloxycarbonyl-4-[6R-(tert-butyldimethylsiloxy)nonyl]-7-methyl-1,2,2a,5,6,7,8,8a-octahydro-5,6,8b-triazaacenaphthylenium acetate (12)
A mixture of guanidine 9 (1.70 mmol), β-keto ester 11 (1.96 g, 5.1 mmol), morpholinium acetate (250 mg, 1.7 mmol), Na2SO4 (250 mg), and trifluoroethanol (3.5 mL) was heated at 60 °C for 2 d. After cooling to room temperature, the mixture was filtered, and concentrated. The residue was chromatographed (SiO2, gradient elution with 2−5% MeOH–CHCl3 with 1% AcOH) to provide 810 mg (82%) of tricyclic guanidine 12 as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 5.99−5.86 (m, 1H), 5.34−5.23 (m, 2H), 4.66−4.58 (m, 2H), 4.42 (dd, J = 9.8, 6.0 Hz, 1H), 3.77−3.72 (m, 1H), 3.70−3.63 (m, 1H), 3.57−3.50 (m, 1H), 2.85−2.60 (m, 2H), 2.55−2.48 (m, 1H), 2.32−2.24 (m, 1H), 2.12−2.05 (m, 1H), 2.00 (s, 3H), 1.66−1.50 (m, 4H), 1.45−1.20 (m, 15H), 1.09 (d, J = 6.1 Hz, 3H), 0.87 (s, 9H), 0.03 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 177.7, 165.0, 150.5, 147.0, 132.2, 118.4, 99.0, 68.7, 64.9, 57.0, 55.6, 55.3, 54.3, 47.4, 45.4, 39.7, 35.7, 33.2, 30.7, 29.6, 29.4, 28.7, 27.0, 25.9, 25.8, 23.8, 22.9, 19.4, 18.1, −4.5, −4.7; IR (film) 2930, 2857, 1688, 1626, 1538, 1258 cm−1; (c 0.95, MeOH); HRMS (ESI) m/z 518.3781 (518.3778 calcd for C29H52N3O3Si, M).
(2aS,4R,7R,8aR)-4-[6R-Hydroxynonyl]-7-methyl-1,2,2a,3,4,5,6,7,8,8a-decahydro-5,6,8b-triazaacenaphthylenium tetrafluoroborate (13)
A solution of guanidine ester 12 (200 mg, 0.35 mmol), (PPh3)4Pd (10 mg, 0.009 mmol), pyrrolidine (140 μL, 1.7 mmol), THF (2 mL), and MeOH (2 mL) was maintained at room temperature. After 6 h, the reaction was concentrated and acetic acid (5 mL) was added. Solid NaBH4 (64 mg, 1.7 mmol) then was added in portions over 20 min, and the mixture was stirred overnight. The solvent was removed by azeotroping with heptane (2 × 5 mL) under reduced pressure, 1 N HCl (5 mL) was added, and the resulting solution was maintained at room temperature for 1 d. This solution was diluted with 1 N HCl (10 mL) and extracted with CHCl3 (6 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated. The residue was chromatographed (SiO2, gradient elution with 2−5−10% MeOH–CHCl3); the fractions containing the product were combined, concentrated, and azeotroped with heptane. The residue was dissolved in CHCl3 (20 mL), washed with saturated aqueaous NaBF4 (3 × 5 mL), dried (Na2SO4), filtered, and concentrated to provide 86 mg (60%) of tricyclic guanidine 13 as a colorless oil: 1H NMR (500 MHz, CD3OD) δ 3.77−3.66 (m, 3H), 3.57−3.50 (m, 1H), 3.45−3.38 (m, 1H), 2.26−2.17 (m, 4H), 1.72−1.64 (m, 2H), 1.61−1.53 (m, 2H), 1.46−1.33 (m, 13H), 1.30−1.20 (m, 5H), 1.13 (d, J = 6.1 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 151.1, 68.5, 57.52, 57.46, 51.6, 47.3, 40.2, 36.7, 35.8, 34.7, 31.02, 30.99, 30.7, 30.51, 30.47, 26.8, 26.2, 23.5, 20.8; IR (film) 2929, 2856, 1621, 1062 cm−1; , , , (c 0.73, MeOH); HRMS (FAB) m/z 322.2849 (322.2858 calcd for C19H36N3O).
3-Oxo-butyric acid (1R)-1-methyl-8-((2aR,4R,7S,8aS)-7-methyl-1,2,2a,3,4,5,6,7,8,8a-decahydro-5,6,8b-triazaacenaphthylenium-4-yl)octyl ester tetrafluoroborate(14)
Following the general procedure of Taber,13 a solution of alcohol 13 (65 mg, 0.16 mmol), methyl acetoacetate (170 μL, 1.2 mmol), DMAP (20 mg, 0.16 mmol), and toluene (2 mL) was heated at reflux for 18 h. The reaction was allowed to cool to room temperature, and the residue was chromatographed (SiO2, gradient elution with 2−5% MeOH–CHCl3 with 1% AcOH) to provide 71 mg (90%) of β-keto ester 14, as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 6.86 (br s, 1H), 6.75 (br s, 1H), 4.98−4.90 (m, 1H), 3.72−3.61 (m, 2H), 3.58−3.47, (m, 1H), 3.45 (s, 2H), 3.40−3.32 (m, 1H), 2.27−2.15 (m, 6H), 1.86−1.75 (m, 2H), 1.70−1.63 (m, 3H), 1.60−1.55 (m, 1H), 1.52−1.44 (m, 2H), 1.40−1.19 (m, 16H); 13C NMR (125 MHz, CDCl3) δ 200.8, 166.8, 149.2, 72.3, 56.0, 55.9, 50.5, 50.4, 46.0, 36.0, 35.8, 35.7, 34.4, 33.6, 33.0, 30.2, 30.1, 29.1, 25.1, 24.8, 20.1, 19.8; IR (film) 3356, 2933, 2858, 1713, 1622, 1326, 1060 cm−1; (c 0.62, MeOH); HRMS (ESI) m/z 406.3069 (406.3070 calcd for C23H40N3O3, M).
(4aS,7S,2′S,1′′R,2a′′′S,4′′′R,7′′′S,8a′′′R)-7-(2′-Hydroxynonyl)-1-imino-3-methyl-1,2,4a,5,6,7-hexahydropyrrolo[1,2-c]pyrimidinium-4-carboxylic acid 1′′-methyl-9′′-(7′′′-methyl-1′′′,2′′′,2a′′′,3′′′,4′′′,5′′′,6′′′,7′′′,8′′′,8a′′′-decahydro-5′′′,6′′′,8b′′′,-triazaacenaphthylenium-4′′′-yl)nonyl ester ditrifluoroacetate (17)
A mixture of β-keto ester 14 (42 mg, 0.09 mmol), guanidine 16 (0.27 mmol), morpholinium acetate (40 mg, 0.27 mmol), Na2SO4 (40 mg), and 2,2,2-triflouroethanol (1 mL) was maintained in a sealed tube at 60 °C for 2 d. After cooling to room temperature, the mixture was filtered through cotton, concentrated, and further filtered through a 0.45 μm nylon filter with MeOH. The filtrate was concentrated, and the residue was purified by HPLC (5 μm C18, 50% MeCN-H2O with 0.1% trifluoroacetic acid) to provide 46 mg (59%) of 17 as a colorless oil: 1H NMR (500 MHz, CD3OD) δ 5.04−4.98 (m, 1H), 4.50 (dd, J = 9.9, 5.3 Hz, 1H), 4.41−4.35 (m, 2H), 3.57−3.48 (m, 2H), 3.44−3.38 (m, 1H), 2.58−2.52 (m, 1H), 2.33−2.15 (m, 8H), 1.84 (ddd, J = 14.2, 11.5, 2.9 Hz, 1H), 1.72−1.63 (m, 4H), 1.62−1.55 (m, 5H), 1.51−1.45 (m, 3H), 1.45−1.20 (m, 31H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.2, 151.8, 151.1, 143.1, 104.1, 72.6, 69.0, 58.6, 57.5, 57.4, 51.1, 47.2, 42.6, 38.6, 36.9, 36.8, 35.7, 35.1, 34.8, 33.0, 31.1, 31.0, 30.7,21 30.5, 30.4, 30.3, 29.0, 26.5, 26.4, 26.1, 23.7, 20.7, 20.2, 17.5, 14.4; IR (film) 3281, 3200, 2930, 2860, 1675, 1629, 1540, 1177 cm−1; , , , (c 0.90, MeOH). HRMS (ESI) m/z 641.5118 (641.5118 calcd for C37H65N6O3 M–H+).
(2aS,7R,8aS,1′R,2a′′S,4′′R,7′Ś,8a′′R)-4-Methyl-7-heptyl-1,2,2a,5,6,7,8,8a-octahydro-5,6,8b,-triazaacenaphthylenium-3-carboxylic acid 1′-methyl-9′-(7′′-methyl-1′′,2′′,2a′′,3′′,4′′,5′′,6′′,7′′,8′′,8a′′-decahydro-5′′,6′′,8b′′-triazaacenaphthylenium-4′′-yl)nonyl ester ditriflouroacetate (19)
Diguanidine 17 (25 mg, 0.03 mmol) was dissolved in CHCl3 (20 mL), washed with saturated aqueous NaBF4 (3 × 5 mL), and the combined aqueous layers were extracted with CHCl3 (1 × 5 mL). The combined organic phases were dried (Na2SO4), filtered, concentrated, and azeotroped with C6H6 (3 × 1 mL) to provide diguanidine 17 as its BF4− salt, which was used directly.
Methanesulphonyl chloride (52 μL, 1.0 M in CH2Cl2, 0.052 mmol) was added over 10 min to a stirring 0 °C solution of this guanidinium BF4− salt (21 mg, 0.03 mmol), Et3N (105 μL, 1.0 M in CH2Cl2, 0.10 mmol) and CH2Cl2, (2 mL). After an additional 1 h at 0 °C, ESMS indicated complete consumption of the starting material. The solution then was diluted with CH2Cl2, (20 mL), washed with saturated aqueous NaBF4 (3 × 5 mL), and the combined aqueous phases were extracted with CHCl3 (1 × 2 mL). The combined organic phases were dried (Na2SO4), filtered, concentrated, to provide the corresponding mesylate as a yellow oil. The mesylate intermediate was dried azeotropically with C6H6 (3 × 1 mL), and combined with CHCl3 (3 mL, filtered through basic Al2O3) and Et3N (0.3 mL) in a heavy-walled sealable tube. This solution was sparged with N2 for 15 min, sealed, shielded from light, and heated at 70 °C for 3 d. The resultant red solution was concentrated, the residue was dissolved in MeOH, this mixture was filtered through a 0.45 μm nylon filter, and concentrated. The residue was purified by HPLC (5 μm C18, 50% MeCN-H2O with 0.1% trifluoroacetic acid) to provide 14 mg (68%) of 19 as a colorless oil: 1H NMR (500 MHz, CD3OD) δ 5.04−4.97 (m, 1H), 4.27−4.23 (m, 1H), 3.64−3.48 (m, 2H), 3.44−3.38 (m, 1H), 2.65−2.59 (m, 1H), 2.40−2.28 (m, 2H), 2.28−2.16 (m, 7H), 1.96−1.86 (m, 1H), 1.80−1.65 (m, 5H), 1.65−1.50 (m, 5H), 1.45−1.20 (m, 32H), 0.89 (t, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 165.8, 151.1, 150.0, 147.3, 106.6, 72.7, 57.5, 57.4, 56.0, 53.5, 51.5, 47.3, 36.9, 36.8, 36.0, 35.7, 34.7, 33.5, 33.1, 33.0, 32.5, 31.1, 31.0, 30.6, 30.42, 30.37, 30.31, 26.6, 26.3, 26.1, 23.7, 20.7, 20.3, 17.7, 14.4; IR (film) 2930, 2860, 1679, 1633, 1324, 1200, 1131 cm−1; , , , (c 0.7, MeOH); HRMS (ESI) m/z 623.5027 (623.5012 calcd for C37H63N6O2, M–H+).
Batzelladine F (1) and Isomer 20
A mixture of diguanidine 19 (12 mg, 0.014 mmol), 5% Rh·Al2O3 (40 mg), HCO2H (10 drops) and MeOH (2 mL) was maintained under 90 psi of H2 for 48 h. Celite was added, the mixture was filtered through Celite, and the filtrate was further filtered through a 0.45 μm nylon filter, rinsing with MeOH. This filtrate was concentrated, and the residue was purified by HPLC (5 μm C18, 40−50% MeCN-H2O with 0.1% trifluoroacetic acid) to provide 3.9 mg (33%) of 20 and 2.5 mg (21%) of batzelladine F (1) both as colorless oils. Batzelladine F (1): 1H and 13C NMR data for synthetic batzelladine F agreed well with those of a natural specimen (see Tables S1 and S2 in Supporting Information); IR (film) 3196, 3123, 2930, 2860, 1725, 1679, 1637, 1328, 1200 cm−1; , , , (c 0.25, MeOH); HRMS (ESI) m/z 625.5182 (625.51695 calcd for C37H65N6O2, M–H+). Batzelladine F stereoisomer 20: 1H NMR (500 MHz, CD3OD) δ 5.00−4.94 (m, 1H), 4.05−3.99 (m, 1H), 3.80−3.68 (m, 2H), 3.65−3.55 (m, 2H) 3.55−3.41 (m, 2H), 3.41−3.38 (m, 1H), 2.88 (dd, J = 10.8, 5.7 Hz, 1H), 2.48−2.42 (m, 1H), 2.34−2.28 (m, 1H), 2.27−2.16 (m, 4H), 1.70−1.50 (m, 8H), 1.40−1.22 (m, 25H) 1.16 (d, J = 6.6 Hz, 3H), 0.90 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 170.2, 151.1, 73.7, 57.5, 57.4, 53.2, 52.9, 51.6, 47.3, 36.8, 36.7, 34.8, 34.1, 33.0, 31.6, 31.5, 31.1, 31.0, 30.6, 30.5, 30.4, 30.3, 26.5, 26.2, 23.7, 20.7, 20.0, 19.5, 14.4;, , , (c 0.2, MeOH); HRMS (ESI) m/z 625.5165 (625.5169 calcd for C37H65N6O2, M–H+).
Supplementary Material
Acknowledgment
The National Heart, Lung, and Blood Institute (HL-25854) supported this research. F.C was supported in part by an ACS Organic Division Graduate Fellowship funded by Pharmacia and a Pharmacia Graduate Fellowship. We particularly thank Dr. J. A. Chan of SmithKline Beecham for providing an authentic sample of batzelladine F, and Dr. John Greaves for mass spectrometric analyses. NMR and mass spectra were determined at UCI with instruments purchased with the assistance of NSF and NIH.
Footnotes
Supporting Information. Reaction schemes for the preparation of compounds reported only in Supporting Information, experimental details and tabulated characterization data for new compounds not reported in the Experimental Section, 1H and 13C NMR data for natural batzelladine F and synthetic isomer 2, HPLC co-injection of authentic batzelladine F and batzelladine F stereoisomers 2−4, andcopies of 1H and 13C NMR spectra of new compounds (76 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Current Address: Department of Medicinal Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080.
- 2.Patil AD, Freyer AJ, Taylor PB, Carté B, Zuber G, Johnson RK, Faulkner DJ. J. Org. Chem. 1997;62:1814–1819. [Google Scholar]
- 3.For reviews summarizing the isolation, structure and synthesis of batzelladine alkaloids, see: Berlinck RGS, Kossuga MH. J. Nat. Prod. 2005;22:516–550. doi: 10.1039/b209227c. and earlier reviews in this series. For the recent isolation of a new batzelladine alkaloid, see: Gallimore WA, Kelly M, Scheuer PJ. J. Nat. Prod. 2005;68:1420–1423. doi: 10.1021/np050149u. For a recent synthetic study, see: Arnold MA, Duron SG, Gin DY. J. Am. Chem. Soc. 2005;127:6924–6925. doi: 10.1021/ja0519029.
- 4.Cohen F, Overman LE. preceding contribution in this issue.
- 5.As discussed in detail in the preceding article,4 the relative configuration originally proposed for the right-hand tricyclic guanidine fragment of batzelladine F2 had been revised by the synthesis of model compounds.4,6
- 6.a Snider BB, Busuyek MV. J. Nat. Prod. 1999;62:1707–1711. doi: 10.1021/np990312j. [DOI] [PubMed] [Google Scholar]; b Black GP, Murphy PJ, Thornhill AJ, Walshe NDA, Zanetti C. Tetrahedron. 1999;55:6547–6554. [Google Scholar]
- 7.Cohen F, Overman LE, Sakata SK. Org. Lett. 1999;1:2169–2172. doi: 10.1021/ol991269u. [DOI] [PubMed] [Google Scholar]
- 8.McDonald AI, Overman LE. J. Org. Chem. 1999;64:1520–1528. doi: 10.1021/jo981972g. and references therein. [DOI] [PubMed] [Google Scholar]
- 9.Franklin AF, Ly SK, Mackin GH, Overman LE, Shaka AJ. J. Org. Chem. 1999;64:1512–1519. doi: 10.1021/jo981971o. [DOI] [PubMed] [Google Scholar]
- 10.Prepared in 82% overall yield from (R)-7-tert-butyldimethylsiloxyoctanol11 using a sequence identical to the one employed to synthesize the dodecanoate congener.4,12
- 11.a Mori K, Maemoto S. Liebigs Ann. Chem. 1987;10:863–869. [Google Scholar]; b Jones GB, Huber RS, Chapman BJ. Tetrahedron: Asymmetry. 1997;8:1797–1809. [Google Scholar]
- 12.Full details are available in the Supporting Information.
- 13.Taber DF, Amedio JC, Patel YK. J. Org. Chem. 1985;50:3618–1619. [Google Scholar]
- 14.In our earlier studies, we found that this counter anion exchange was essential in order to obtain good yields of clean material in the mesylate cyclization step.4,7 Without this counter ion exchange, the product was contaminated with colored impurities that were difficult to remove.
- 15.The authentic sample we acquired in 1999 showed significant impurities by 1H NMR and HPLC analysis, some of which could have arisen during storage. However, the published 1H NMR spectrum of batzelladine F shows significant impurities as well.2
- 16.Noyori R, Ohkuma T, Kitamura H, Takaya H, Sayo N, Kumobayashi H, Akutagawa S. J. Am. Chem. Soc. 1987;109:5856–5858. [Google Scholar]
- 17.For a recent review, see: Aron ZD, Overman LE. Chem. Commun. 2004:253–265. doi: 10.1039/b309910e.
- 18.For recent reviews, see: Weinreb SM. Acc. Chem. Res. 2003;36:59–65. doi: 10.1021/ar0200403.Nicolaou KC, Snyder SA. Angew. Chem., Int. Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864.
- 19.HIV-1 envelope-mediated fusion: Bewley CA, Ray S, Cohen F, Collins SK, Overman LE. J. Nat. Prod. 2004;67:1319–1324. doi: 10.1021/np049958o. Interactions of HIV-1 Nef with p53, actin, and p56ick: Olszewski A, Sato K, Aron ZD, Cohen F, Harris A, McDougall BR, Robinson WE, Overman LE, Weiss GA. Prod. Nat. Acad. Sci. USA. 2004;39:14709–14084. doi: 10.1073/pnas.0406040101.
- 20.General experimental details have been described: MacMillan DWC, Overman LE, Pennington LD. J. Am. Chem. Soc. 2001;123:9033–9044. doi: 10.1021/ja016351a.
- 21.Three overlapping resonances.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.