

Abstract
Thrombin is a dual action serine protease in the blood clotting cascade. Similar to other clotting factors, thrombin is mainly present in the blood in a zymogen form, prothrombin. Although the two cleavage events required to activate thrombin are well known, little is known about why the thrombin precursors are inactive proteases. Although prothrombin is much larger than thrombin, prethrombin-2, which contains all of the same amino acids as thrombin, but has not yet been cleaved between Arg320 and Ile321, remains inactive. Crystal structures of both prethrombin-2 and thrombin are available and show almost no differences in the active site conformations. Slight differences were, however, seen in the loops surrounding the active site, which are larger in thrombin than in most other trypsin-like proteases, and have been shown to be important for substrate specificity. To explore whether the dynamics of the active site loops were different in the various zymogen forms of thrombin, we employed amide H/2H exchange experiments to compare the exchange rates of regions of thrombin with the same regions of prothrombin, prethrombin-2 and meizothrombin. Many of the surface loops showed less exchange in the zymogen forms, including the large loop corresponding to anion binding exosite 1. Conversely, the autolysis loop and sodium-binding site exchanged more readily in the zymogen forms. Prothrombin and prethrombin-2 gave nearly identical results while meizothrombin in some regions more closely resembled active thrombin. Thus, cleavage of the Arg320 - Ile321 peptide bond is the key to formation of the active enzyme, which involves increased dynamics of the substrate-binding loops and decreased dynamics of the catalytic site.
Keywords: Thrombin, prothrombin, prethrombin-2, meizothrombin, zymogen
Thrombin is a dual action serine protease in the blood clotting cascade. Similar to other clotting factors, thrombin is mainly present in the blood in a zymogen form, prothrombin, to maintain normal hemostasis. While the plasma concentration of prothrombin is estimated at 1.2 μM, the concentration of active thrombin is only 0.06–0.09 μM (1). Activation of thrombin occurs via two cleavages by factor Xa (Figure 1A). Cleavage between Arg271 and Thr272 separates the pro domain (37 kD) from the catalytic domain (37 kD), creating an inactive precursor called prethrombin-2. Cleavage within the catalytic domain between residues Arg320 and Ile321 (Arg15CT-Ile16CT), create the light chain and heavy chain that remain connected via a disulfide bond in the active protease (2). Although the cleavage events required to activate thrombin are well known, little is known about why the thrombin precursors are inactive proteases.
Figure 1.

A) Schematic diagram showing the cleavages that occur in the conversion of prothrombin to meizothrombin or to prethrombin-2 and then to thrombin (23)). B) Aligned sequences of human and bovine α-thrombin showing all the peptides generated by pepsin cleavage as lines below the sequence. Thick lines denote peptides from the human protein digest, and thin lines denote those from the bovine digest. Dashed lines indicate new peptides that were obtained from the digests done in the presence of TCEP. Taken together, the peptides cover 65% of the thrombin sequence. The light chain residues of thrombin are indicated by bold text, and significant residues including the catalytic triad (H57CT, D102CT, and S195CT) are in both bold text and annotated above the sequence in the chymotrypsin numbering system. The four disulfide bonds in the protease domain are drawn in vertical lines connecting the cysteines.
Serine proteases work by formation of a tetrahedral intermediate between the active site serine (Ser195CT) and the substrate. This intermediate is stabilized by His57CT which acts as a general base and the backbone amides of Gly193CT and Ser195CT which stabilize the negative charge on the carbonyl oxygen of the tetrahedral intermediate (3). The most well-studied examples of serine proteases, trypsin and chymotrypsin, are produced by a single proteolytic cleavage of their zymogen forms trypsinogen and chymotrypsinogen. Remarkably, crystal structures of the zymogens showed that the active site is almost fully formed prior to proteolysis (3, 4). The only difference between the active enzyme and the zymogen was in the conformation of the “oxyanion hole”. In the crystal structures of the zymogens, Gly193CT was too far from Ser195CT to play a role in correctly forming the “oxyanion hole,” which is required for stabilization of the intermediate in the catalytic mechanism (4).
Several crystal structures of zymogen forms of thrombin are also available, but these too reveal little difference in structure between the inactive zymogens and their active enzyme counterparts except, again, for the ill-formation of the oxyanion hole in the active site. In fact, it appears that the catalytic residues are in an active conformation even in the inactive precursors (5, 6). One such structure represents the noncovalent complex between fragment 2 from prothrombin (or its kringle 2 domain) and PPACK-thrombin. This structure shows a binding site on the catalytic domain of thrombin for fragment 2 which may play a role in blocking the active site from entering substrate (5). The other structure is of prethrombin-2 and shows minimal conformational changes in loops of thrombin around the active site of thrombin (Gly142CT-Pro152CT), near the oxyanion hole (Pro186CT-Asp194CT), and in the cleavage region of Arg15CT-Ile16CT (6, 7).
In order to assess structural and stability changes upon prothrombin activation, Stevens and Nesheim did a fluorescence study in which they measured the modification of tryptophan residues in thrombin upon activation from prethrombin-2 to thrombin. Their results suggest that the dynamics of the two proteins differ, and revealed structural changes near His57 and at the substrate binding site of thrombin (8). Differential scanning calorimetry was used to measure the stability of prothrombin and prethrombin-2. These experiments showed that despite prethrombin-2 losing half its mass when converted from prothrombin, its inherent stability did not differ from that of the full zymogen (9). This lead us to speculate that because there is no drastic overall structural change or stability change, the key to the activity of thrombin over prothrombin or prethrombin-2 may be due to subtle changes, perhaps mainly in backbone dynamics, upon activation.
In this study we used amide H/2H exchange and mass spectrometry to study the dynamic differences between prothrombin, meizothrombin (a precursor cleaved between Arg320 and Ile321), prethrombin-2 (an immediate precursor to active thrombin which lacks the pro domain), and active thrombin. Since prethrombin-2 differs from thrombin only by the cleavage of a single peptide bond, it is possible to interpret the amide H/2H exchange data in terms of dynamics. Our results show major differences in dynamics between the active enzyme and its inactive precursors which help to deduce a mechanism of activation.
EXPERIMENTAL PROCEDURES
Proteins
Bovine blood was processed to the barium citrate eluate (prepared from bovine plasma) according to previously published methods (10). The eluate powder (8 g) was redissolved overnight in 200 mL of 100 mM EDTA, 150 mM NaCl, 10 mM sodium citrate, containing 11.1 g ammonium sulfate and 0.03 g of benzamidine. Following resuspension, the concentration of ammonium sulfate was increased from 10% to 40%. After centrifugation at 10,000g, the supernatant was kept, brought to 70% ammonium sulfate, and centrifuged. The pellet was dissolved in 5 mL of 50 mM Tris, pH 7.5, 150 mM NaCl, loaded onto a G-25 sephadex gel filtration column (2.5 × 100 cm) to remove the ammonium sulfate, and the fraction containing protein was collected. The protein was further purified by HiLoad 16/60 Superdex 200 size-exclusion chromatography (Amersham/GE Healthcare) using 50 mM Tris base pH 7.4, 150 mM NaCl and buffer exchanged into 25 mM KH2PO4 pH 6.5, 50 mM NaCl and concentrated to ~6 mg/mL. This process was shown to produce bovine meizothrombin as assessed by the observations that the protein that eluted from Superdex 200 at an apparent molecular weight of 60 kD and that had no measurable clotting activity was shown by reducing SDS PAGE to be cleaved to the appropriately-sized fragments. Others have shown that clotted bovine blood yields primarily meizothrombin when prepared in this way (11–13). The bovine meizothrombin was stored as 5 μL aliquots (750 pmol/tube) at −80°C until use.
Bovine thrombin was obtained from purified bovine prothrombin (described above). The prothrombin was activated by incubating it with 2.0 mg/mL Echis carinatus venom, 10 mM CaCl2, 1 mg/mL PEG-8000 for 45 minutes at 37° C. The mixture was loaded onto a G-25 sephadex column (2.5 × 100 cm) equilibrated in 25 mM KH2PO4 pH 6.5, 100 mM NaCl, and the protein fraction was collected. Finally, the G-25 fraction containing active thrombin was loaded onto a MonoS FPLC column 16/10 (Amersham/GE Healthcare) equilibrated with buffer A (25 mM KH2PO4 pH 6.5, 100 mM NaCl). The thrombin was eluted with a linear gradient of B (25 mM KH2PO4 pH 6.5, 500 mM NaCl) over 1 hour. Alpha-thrombin was identified by fibrinogen clotting and the most concentrated fractions were pooled. The specific activity of these pooled fractions was typically 4000 U/mg (14). Bovine thrombin was buffer exchanged into 25 mM KH2PO4 pH 6.5, 50 mM NaCl, concentrated to ~3 mg/mL, and stored as 5 μL aliquots (750 pmol/tube) at −80°C until use. Control experiments were performed with lyophilized bovine thrombin to verify that there was no difference in deuterium incorporation or pepsin digestion in lyophilized samples compared to non-lyophilized samples.
Human prothrombin and prethrombin-2 were obtained from Haematalogic Technologies. The prothrombin was purified by HiLoad 16/60 Superdex 200 size-exclusion chromatography (Amersham/GE Healthcare) using 50 mM Tris base pH 7.4, 150 mM NaCl. The prethrombin-2 was purified by HiLoad 16/60 Superdex 75 size-exclusion chromatography in the same Tris buffer. Human prothrombin was buffer exchanged into 25 mM KH2PO4, pH 6.5, and 50 mM NaCl, concentrated to ~6 mg/mL, and stored as 5 μL aliquots (750 pmol/tube) at −80°C until use. Human prethrombin-2 was exchanged into 12.5 mM KH2PO4, pH 6.5, and 25 mM NaCl, and concentrated to ~1 mg/mL. Aliquots of 750 pmol protein were lyophilized and stored at −80°C until use. The final buffer upon resuspension of the lyophilized protein was 25 mM KH2PO4, pH 6.5, and 50 mM NaCl.
Human thrombin was obtained from purified prothrombin (Haematologic Technologies) as described previously (15). Optimal yields were obtained when the prothrombin concentrate was dissolved in 50 mM Tris pH 7.5, 150 mM NaCl, 10 mM CaCl2, 1 mg/mL PEG-8000 so that the final prothrombin concentration was 1.6 mg/mL. The prothrombin was then activated for two hours at 37° C with 5 mg/mL Echis carinatus venom, and purified as already described for bovine thrombin. Alpha-thrombin was identified by fibrinogen clotting assay, and protein concentration was determined by absorbance at 280 nm (ε=1.92 cm mL unit−1 mg−1). Human thrombin was buffer exchanged into 12.5 mM KH2PO4, pH 6.5, and 25 mM NaCl, and concentrated to ~1 mg/mL. Aliquots of 750 pmol protein were lyophilized and stored at −80°C until use. The final buffer upon resuspension of the lyophilized protein was 25 mM KH2PO4, pH 6.5, and 50 mM NaCl.
Mass Spectrometry
Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were acquired on a Voyager DE-STR instrument (Applied Biosystems) as previously described (16). The matrix used was 5.0 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) dissolved in a solution containing a 1:1:1 mixture of acetonitrile, ethanol, and 0.1% TFA. The pH of the matrix was adjusted to pH 2.2 using 2% TFA. The matrix solution was chilled on ice for at least 60 min prior to use and the MALDI-target plates were chilled overnight at 4 °C. Peptides produced by pepsin cleavage of human and bovine thrombin were identified previously (15, 17).
Amide H/2H Exchange Experiments
The pH conditions during various stages of the reaction were determined on a Accumet Inlab 423 pH electrode (Mettler-Toledo) using non-deuterated mock solutions (to avoid having to recalibrate the electrode for deuterons). The number of deuterons incorporated into different regions of thrombin over ten minutes (on-exchange) was measured in 2H2O. The kinetics of incorporation (on-exchange) are a good measure of the solvent accessibility of each region of thrombin. The lyophilized thrombin was rehydrated in 12 μL of 2H2O. The samples were simultaneously quenched and diluted after varying on-exchange times (0.5 to 10 min) by addition of 120 μL of H2O (0°C) with ~5 μL of 2% TFA to give a final pH of 2.2. The non-lyophilized thrombin samples were allowed to come to room temperature prior to exchange. They were then diluted with 45 μL of 2H2O to initiate exchange for times from 0.5 to 10 min. The samples were then simultaneously quenched and diluted by addition of 250 μL of H2O (0°C) with ~10 μL of 2% TFA to give a final pH of 2.2. Each sample was then incubated with a two-fold molar excess of immobilized pepsin (Pierce Chemicals) for 5 min (human proteins) or 10 min (bovine proteins) at 4°C. Pepsin beads were removed by centrifugation at 14,000 rpm for ten seconds, and the resulting thrombin peptide mixture was aliquoted into several fractions, rapidly frozen in liquid N2, and stored at −80°C.
All samples were analyzed by MALDI-TOF mass spectrometry one at a time. Samples were rapidly thawed, mixed with cold matrix, spotted on a pre-chilled MALDI target plate, and dried under vacuum (18). The mass spectra were analyzed to determine the average number of deuterons present in each peptic peptide. The number of deuterons incorporated into each peptide was quantified by subtracting the centroid of the undeuterated control from the centroid of the isotopic peak cluster for the deuterated sample. All values reported represent only the deuterons exchanged onto the backbone amide-hydrogen (NH) positions. The residual deuterium content (4.5% for the lyophilized samples and 7.5% for the non-lyophilized samples) that incorporated into rapidly exchanging side chain positions was subtracted. Finally, data were corrected for back-exchange loss (~ 45%) as described previously (15). On-exchange was measured at 0, 0.5, 1, 2, 5, and 10 min and the data were fit using a bi-exponential fit in Kaleidagraph as described previously (19).
Partial reduction with TCEP
Further H/2H experiments were performed in which the deuterium exchanged protein was exposed to TCEP for 5 min prior to pepsin digestion using a protocol developed by T. M. Sabo and M. C. Maurer (Biochemistry, in press). After this partial reduction, both groups were able to identify the same additional peptides from bovine thrombin, which included MH+1924.92 (residues -2-15, -3CT-7CT), MH+1548.82 (residues 55-68, 34CT-46CT), MH+2267.99 (residues 256-275, 208CT-227CT), MH+1845.82 (residues 259-275, 211CT-227CT), and MH+1788.80 (residues 260-275, 212CT-227CT). Additionally, the following peptides were identified from the digest of human thrombin: MH+1569.74 (residues 1-15, 1ACT-7CT), MH+1548.82 (residues 55-68, 34CT-46CT), MH+2267.99 (residues 256-275, 208CT-227CT), MH+1845.82 (residues 259-275, 211CT-227CT), MH+1788.80 (residues 260-275, 212CT-227CT), and MH+1489.62 (residues 263-275, 215CT-227CT). All new peptides were identified using MALDI-TOF-TOF mass spectrometry. A peptide covering H57CT and C58CT, which is disulfide bonded to C42CT (in MH+1548.82, residues 34CT-46CT) was not found. It is not unusual for some cysteine containing peptides not to be observed in MALDI-TOF mass spectrometry and should not be interpreted as being due to residual structure during pepsin digestion.
The H/2H exchange experiments were carried out as described above except that the 120 μL quench solution was 25 mM TCEP in 0.1% TFA, giving a final TCEP concentration of 22.7 mM. After the combination quenching-reduction step was allowed to occur for 5 min on ice, the entire mixture was incubated with a two-fold molar excess of immobilized pepsin (Pierce Chemicals) for 5 min at 4° C. Pepsin beads were then removed by centrifugation at 14,000 rpm for ten seconds, and the resulting thrombin peptide mixture was aliquoted into several fractions, rapidly frozen in liquid N2, and stored at −80°C.
Differential Scanning Calorimetry (DSC)
DSC scans were collected using a MicroCal VP DSC instrument. Human prothrombin, prethrombin-2, and thrombin were dialyzed into 25 mM PIPES pH 6.5, and 150 mM NaCl, and the final protein concentration was 5 μM for each. Buffer alone was used in the calorimeter reference cell. Protein samples and buffer were degassed prior to loading in the calorimeter cells. The temperature range scanned was 25–90°C with a scan rate of 90°C/hour. Scans were performed in duplicate using fresh sample for each scan.
RESULTS
Thrombin conformational dynamics probed by amide H/2H exchange
On-exchange experiments were performed to probe the solvent accessibility of backbone amides in human prothrombin, prethrombin-2 and active thrombin. Additionally, we have used partial reduction with TCEP to increase the sequence coverage from our pepsin digestion to approx. 65% of the thrombin sequence (Figure 1B). Regions of interest to thrombin function showed dramatic differences in deuterium incorporation depending on the activity state of the enzyme.
The side of the active site that connects to ABE1 becomes more dynamic upon thrombin activation
Amide H/2H exchange experiments revealed several regions of thrombin that had slower on-exchange when the protease was inactive either as prothrombin or as prethrombin-2. These regions included both sites where cofactors bind the enzyme as well as loops around the active site of the enzyme.
Exchange at ABE1 was markedly slower in prothrombin than in active thrombin. Exchange at ABE1 in prethrombin-2 was also slower than in active thrombin, but not as slow as in prothrombin. These changes were evident in two peptides from the digest of human thrombin: MH+2127.19, comprising residues 96-112 (65CT-80CT) and MH+2586.44, comprising residues 97-117 (66CT-85CT) (Figure 2).
Figure 2.

Amide H/2H exchange at ABE1 of thrombin. A) MALDI-TOF mass specetra of exchange into peptide MH+ 2586.44 (i) before deuteration, (ii) after 30 sec deuteration of active thrombin, (iii) after 30 sec deuteration of prethrombin-2. B) On-exchange into residues 97-117 (peptide MH+ 2586.44) of thrombin (●) and prethrombin-2 (■). C) On-exchange into residues 96-112 (peptide MH+ 2127.19) of thrombin (●) and prethrombin-2 (■). D) MALDI-TOF mass specetra of exchange into peptide MH+ 2586.44 (i) before deuteration, (ii) after 30 sec deuteration of active thrombin, (iii) after 30 sec deuteration of prothrombin. E) On-exchange into residues 97-117 (peptide MH+ 2586.44) of thrombin (●) and prothrombin (▲). F) On-exchange into residues 96-112 (peptide MH+ 2127.19) of thrombin (●) and prothrombin (▲).
More evidence for conformational change upon activation of the enzyme was seen in the loop near the P2 position of the active site. A peptide covering residues 117-132 (Leu85CT-Leu99CT) in this region (MH+2144.14) was seen to have slowed exchange in prothrombin and prethrombin-2 suggesting that the active site is in a locked conformation prior to activation (Figures 3). Thus, the 90sCT loop shows decreased dynamics in all zymogen forms of thrombin that are unable to readily cleave fibrinogen.
Figure 3.
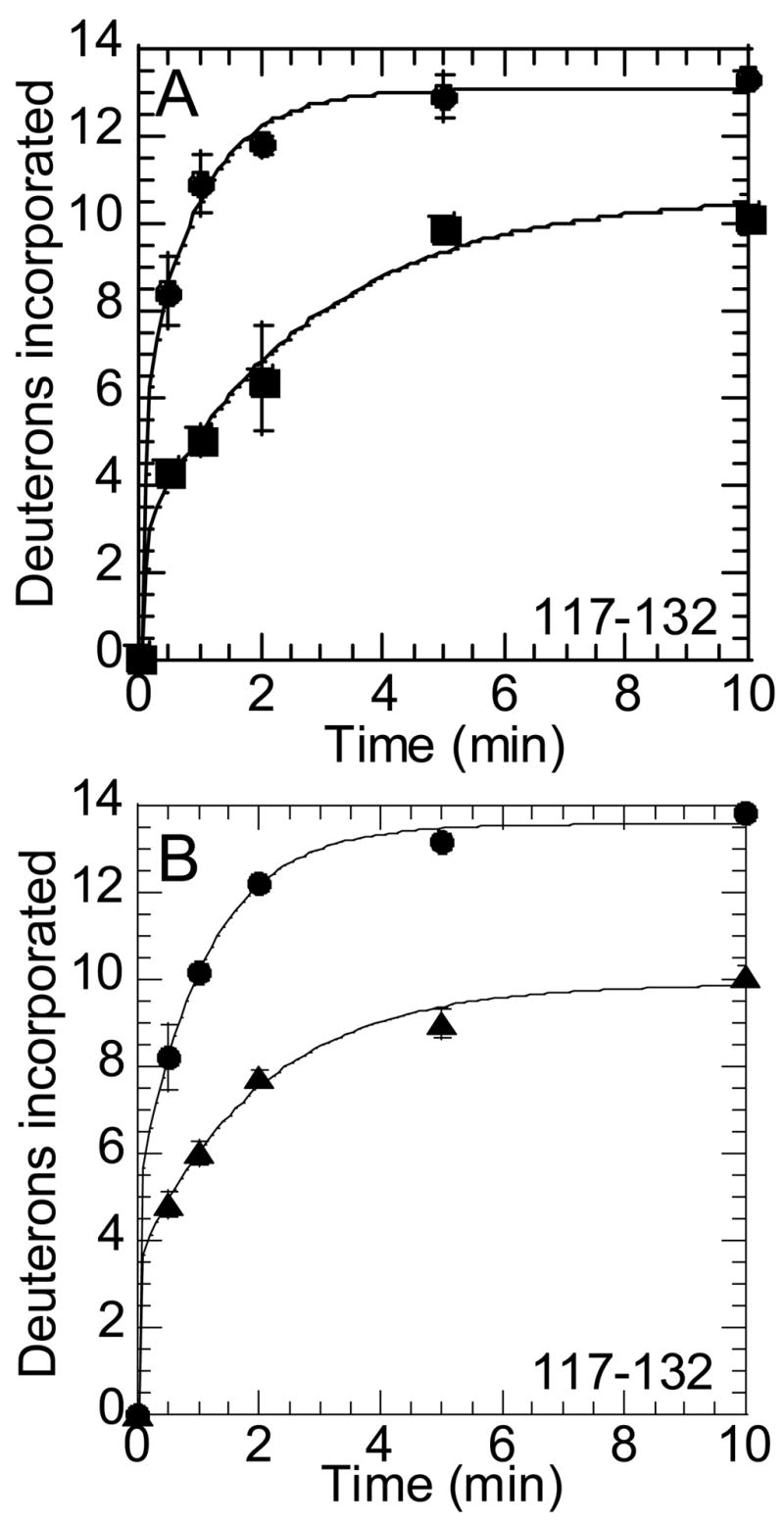
Amide H/2H exchange at the 90s loop of thrombin. A) On-exchange into residues 117-132 (peptide MH+ 2144.14) of thrombin (●) and prethrombin-2 (■). B) On-exchange into residues 117-132 (peptide MH+ 2144.14) of thrombin (●) and prothrombin (▲).
Continuing along the linear sequence of the protein is another surface strand, residues 139-149 (106CT-116CT), that forms the bottom side of ABE1 and contains two Lys residues that interact with thrombomodulin. This segment, represented by a peptide of mass MH+1263.71, was also seen to become more dynamic upon activation (Figure 4). As with the other surface segments and loops of this region of linear sequence, this region was less solvent exposed in both prethrombin-2 and prothrombin.
Figure 4.
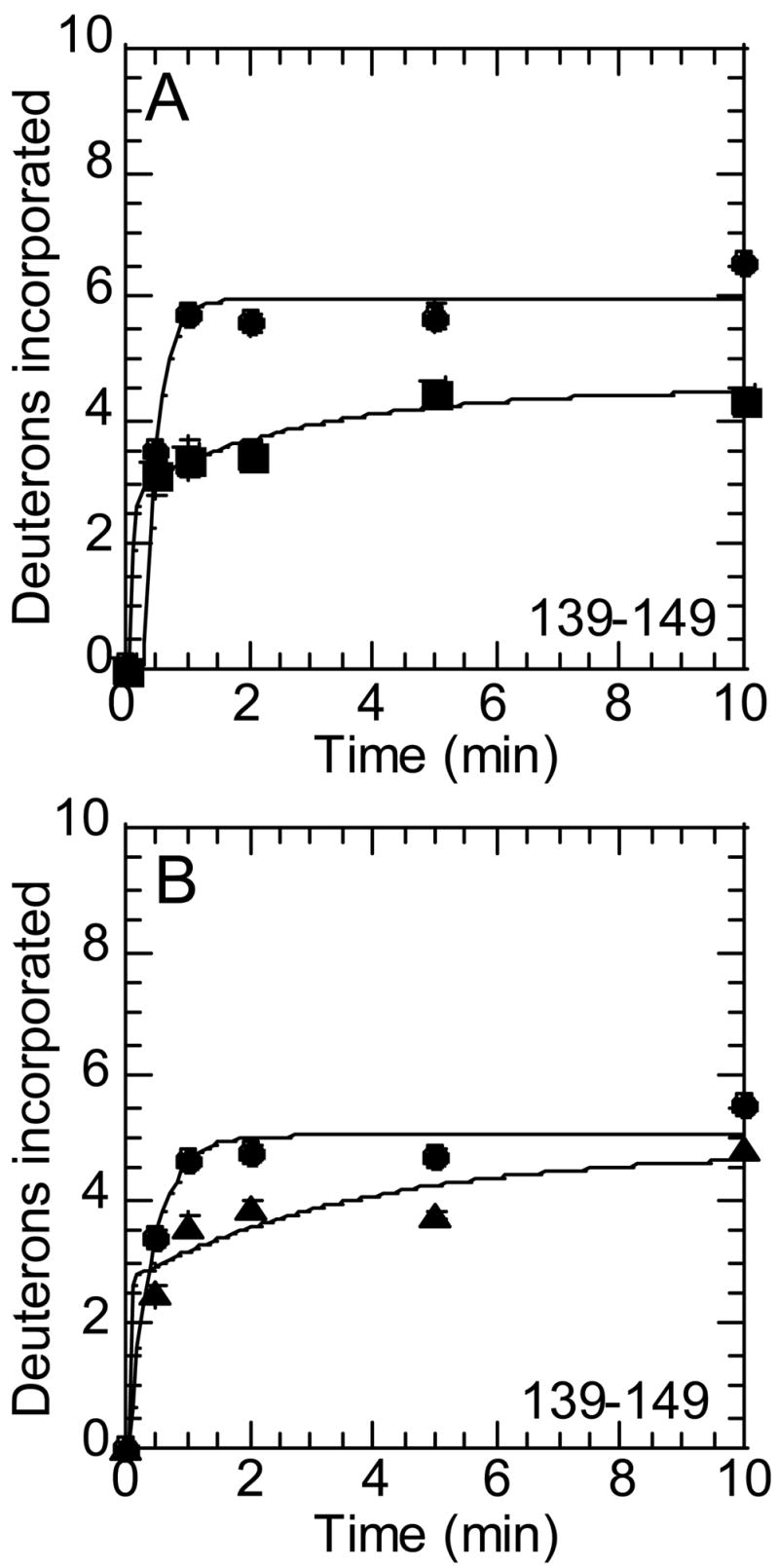
Amide H/2H exchange adjacent to ABE1 of thrombin. A) On-exchange into residues 139-149 (peptide MH+ 1263.71) of thrombin (●) and prethrombin-2 (■). B) On-exchange into residues 139-149 (peptide MH+ 1263.71) of thrombin (●) and prothrombin (▲).
The C-terminal helix of thrombin is seen in the crystal structure to be lying just below the 90sCT loop, and appears to also be protected by fragment 2 in the co-crystal structure. This region also showed differences in H/2H exchange when active thrombin was compared to prethrombin-2. Two peptides from the digest of human thrombin: MH+1702.02 comprising residues 281-293 (Arg233CT-Phe245CT), and MH+2202.26 comprising residues 276-292 (Tyr228CT-Gln244CT) showed decreased amide exchange in both prothrombin and prethrombin-2 (Figure 5).
Figure 5.

Amide H/2H exchange at the C-terminus of thrombin. A) On-exchange into residues 281-293 (peptide MH+ 1702.02) of thrombin (●) and prethrombin-2 (■). B) On-exchange into residues 276-292 (peptide MH+ 2202.25) of thrombin (●) and prethrombin-2 (■). C) On-exchange into residues 281-293 (peptide MH+ 1702.02) of thrombin (●) and prothrombin (▲). B) On-exchange into residues 276-292 (peptide MH+ 2202.25) of thrombin (●) and prothrombin (▲).
The catalytic site and autolysis loop become less dynamic upon thrombin activation
Two thrombin loops that make up the active site cleft near the catalytic serine (Ser195CT) showed faster exchange in two of the inactive forms of the enzyme (prothrombin and prethrombin-2). One of these loops is known as the autolysis loop. Several overlapping peptides from the pepsin digest covered the autolysis loop. Data for the peptide from human thrombin of mass MH+1619.87 comprising residues 166-180 (Leu130CT-Leu144CT) are shown in Figure 6 A and B. The peptide of mass MH+1506.78 comprising residues 167-180 (Gln131CT-Leu144CT) gave identical results. In all of the peptides that cover this region, we saw slowed exchange in the active thrombin.
Figure 6.
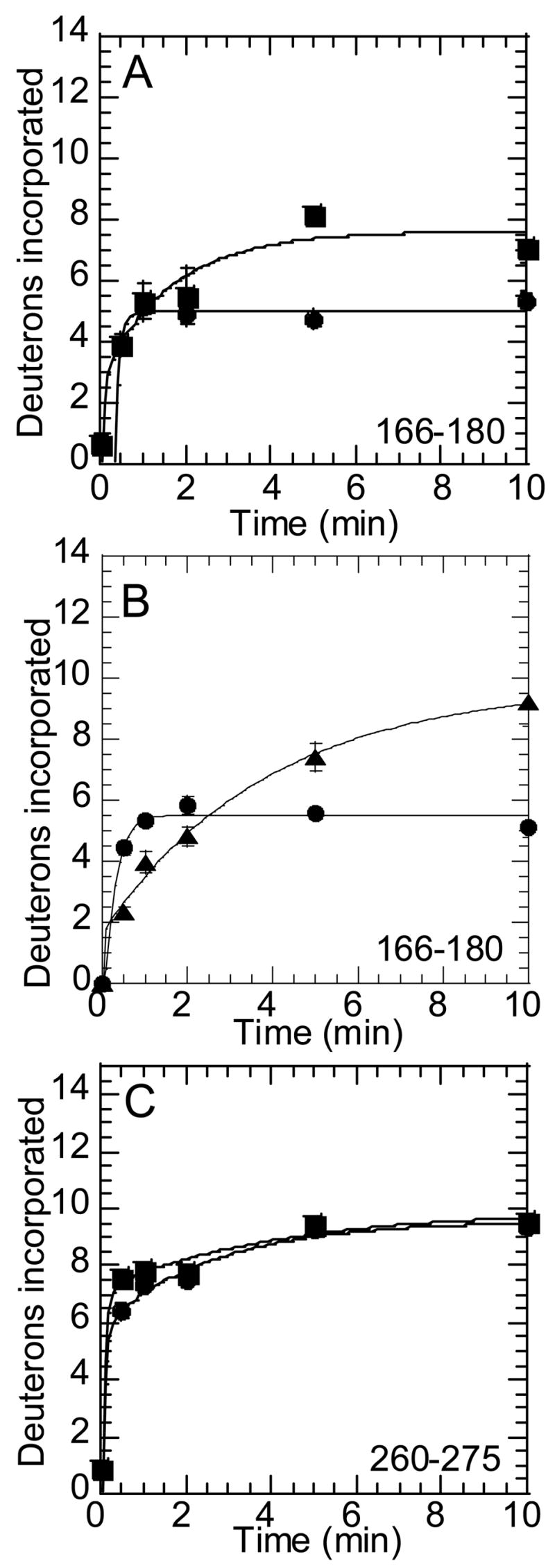
Amide H/2H exchange at the autolysis loop and Na+-binding site of thrombin. A) On-exchange into residues 166-180 (peptide MH+ 1619.87) of thrombin (●) and prethrombin-2 (■). B) On-exchange into residues 166-180 (peptide MH+ 1619.87) of thrombin (●) and prothrombin (▲). C) On-exchange into residues 260-275 (peptide MH+ 1788.80) of thrombin (●) and prethrombin-2 (■).
Another loop in this region was covered by the human peptide of mass MH+1788.80 (residues 260-275, Ile212CT-Phe227CT). This site is also known as the Na+-binding site. This peptide showed somewhat faster exchange in prethrombin-2 than in active thrombin, but this was only evident in the shortest exchange times (30 s and 1 min) (Figure 6C).
Differential scanning calorimetry showed that all states of thrombin have similar stability to unfolding
Duplicate scans of the human thrombin species revealed that the melting temperatures of human active thrombin, prethrombin-2 and prothrombin do not differ significantly. The scans revealed a non-reversible melting occurring in the range of 54–58°C. Because of the nonreversibility, we are observing an aggregation event and the temperature is really an aggregation temperature rather than a melting temperature. Regardless, neither prothrombin nor prethrombin-2 appears significantly more stable than active thrombin (Table 1, Supplementary Figure 1).
Table 1.
Differential Scanning Calorimetric Measurement of Protein Stabilities.
| protein | melting temperature (°C) |
|---|---|
| active thrombin | 56.6 |
| prothrombin | 58.2 |
| prethrombin-2 | 53.8 |
Meizothrombin is partially dynamic
Meizothrombin had dynamic behavior that partially resembled the zymogen forms and partially resembled active thrombin. Despite the fact that prothrombin and prethrombin-2 showed lower solvent accessibility of the ABE1 loop, no difference was seen in the exchange at ABE1 when bovine meizothrombin was compared to active bovine thrombin as shown by the peptide comprising residues 96-116 (65CT-84CT, MH+2586.44) (Figure 7A). Continuing along the linear sequence, the 90sCT loop did show less solvent accessibility than active thrombin (Figure 7B). The next contiguous segment, residues 139-149 (106CT-116CT) also showed less solvent accessibility than active thrombin, again similar to the zymogen forms (Figure 7C). On the other side of the active site, the autolysis loop also resembled the zymogen forms, as seen in the peptide of mass MH+2162.12 comprising residues 171-189 (Lys135CT-Ala149DCT) from bovine thrombin, which had higher solvent accessibility in meizothrombin than in active thrombin (data not shown). Finally, the Na+-binding site, which was represented by a peptide from bovine thrombin of mass MH+1367.65 (residues 252-262, Asn204BCT-Ser214CT), showed identical H/2H exchange in meizothrombin and active thrombin (Figure 7D).
Figure 7.

Amide H/2H exchange in bovine meizothrombin. A) On-exchange into residues 96-116 (peptide MH+ 2586.48) of thrombin (○) and meizothrombin (⋄). B) On-exchange into residues 117-132 (peptide MH+ 2102.12) of thrombin (○) and meizothrombin (⋄). C) On-exchange into residues 139-149 (peptide MH+ 1311.82) of thrombin (○) and meizothrombin (⋄). D) On-exchange into residues 252-262 (peptide MH+ 1367.65) of thrombin (○) and meizothrombin (⋄).
DISCUSSION
Using amide H/2H exchange experiments followed by pepsin digestion and mass spectrometry we previously probed the relative solvent accessibility of regions of thrombin when different effector molecules were bound. In earlier work, we mapped the interface between thrombin and a fragment of thrombomodulin containing the 4th and 5th EGF-like domains (TMEGF45) using deuterium off-exchange experiments. The thrombin-TMEGF45 interface, which corresponded to anion-binding exosite 1 (ABE1), was found to have solvent-inaccessible amides when TMEGF45 was bound (18). During these studies, other changes in thrombin were observed that could not be readily explained. This led to the further studies of thrombin conformational dynamics using on-exchange experiments to compare PPACK-bound thrombin to the active site open form (15) and to thrombin bound to active and inactive fragments of thrombomodulin (20).
These studies revealed how the β-sheet architecture of thrombin connects ABE1 to the active site. The large surface loop that comprises ABE1, residues 97-117 (66CT-85CT) is connected by a surface β-strand, residues 117-126 (85CT-94CT) to the 90sCT loop, residues 127-132 (95CT-99CT), near the active site. Thus, binding of effector molecules such as thrombomodulin alters the conformation of the 90sCT loop(20), and binding of substrates at the active site alters the dynamics of residues 97-117 (66CT-85CT) at ABE1 (15). The 90sCT loop further connects back to the strand that runs at the bottom of ABE1 comprised of residues 139-149 (106CT-116CT) which contains Lys 109CT and 110CT. Thus this entire side of the thrombin molecule is a network of β-strand-to-loop-to-β-strand connections between ABE1 and the active site. The C-terminal helix of thrombin runs under this β-sheet network and is likely a secondary sensor for its dynamics. Consistent with this idea, the C-terminal helix showed decreased exchange upon PPACK binding and thrombomodulin binding that reflected the decreased exchange seen in the β-sheet network (15, 20).
The connection between ABE1 and the 90sCT loop at the active site are more dynamic in active thrombin
In the studies presented here, the entire β-strand-to-loop-to-β-strand network connecting ABE1 to the active site appears to be more constrained in prothrombin and prethrombin-2. Indeed, significantly fewer amides exchanged in the segments corresponding to ABE1, (residues 97-117, 66CT-85CT), the β-sheet network (residues117-132, 85CT-99CT, and residues 139-149, 106CT-116CT) and the C-terminal helix (residues 276-292, 228CT-244CT, and residues 281-293, 233CT-245CT) in prothrombin and prethrombin-2 as compared to active thrombin (Figure 8). It is possible that the decrease in exchange observed for this β-sheet network in prothrombin may be due to solvent exclusion by fragment 2, since it was seen to bind at this site (ABE2) in the co-crystal structure (5). This can not be said, however, for prethrombin-2, which does not contain fragment 2. Thus, our results show that a change in dynamics of an entire side of the active site occurs as a result of cleavage of the Arg320-Ile321 peptide bond. Cleavage of this peptide bond causes the entire β-sheet network (residues117-132, 85CT-99CT, and residues 139-149, 106CT-116CT) and the C-terminal helix (residues 276-292, 228CT-244CT, and residues 281-293, 233CT-245CT) to become more dynamic.
Figure 8.
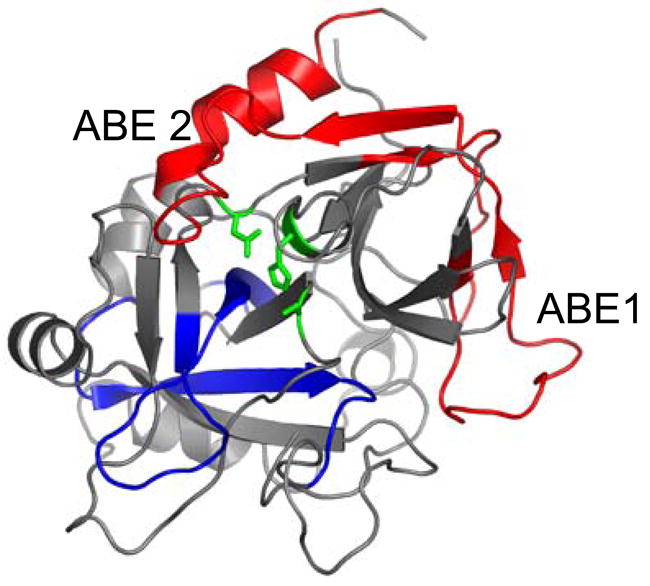
Structure of PPACK thrombin (24) with loops colored according to the change in amide H/2H exchange seen upon activation of thrombin. The catalytic triad residues (H57CT, D102CT, and S195CT) are shown as sticks and in green and these indicate where the active site is located, between the two β-barrels. The location of each anion binding exosite is indicated. Loops in red become more dynamic upon thrombin activation. Loops in blue become less dynamic upon thrombin activation.
The situation was somewhat different for meizothrombin, which still has the pro domain, but which has already been cleaved at the Arg320-Ile321 peptide bond. In meizothrombin, ABE1 was as dynamic as in active thrombin indicating that zymogen cleavage forms ABE1 to its fully dynamic state. The 90sCT loop in meizothrombin, however, still showed somewhat slower exchange than in fully active thrombin. This could be due either to incomplete dynamic changes as was seen for prethrombin-2, or to surface interactions with fragment 2, which is still present in meizothrombin and could simply be occluding solvent accessibility to this loop, or both. These results suggest that fragment 2 is also bound to ABE2 in meizothrombin as was observed for the co-crystal of thrombin with fragment 2 (5). Our results are consistent with the observation that both thrombin and meizothrombin are inhibited by antithrombin III (ATIII), but only the thrombin-ATIII interaction is accelerated by heparin. This makes sense because in meizothrombin, the heparin binding site, ABE2, is blocked by fragment 2 (12). Heparin cofactor II inhibition of meizothrombin is accelerated by heparin, because the complex of heparin cofactor II and heparin bind only to ABE1, which our results show is in its fully binding-competent dynamic state in meizothrombin (13).
It is important to note that Asp102CT is located just four residues further along this loop, and the fact that the amide H/2H exchange experiments show large differences in this region may hint at the mechanism by which the catalytic residues cannot fully attain their active conformations in the zymogen forms. It is possible that correct positioning of Asp102CT is required for correct positioning of His57CT and subsequently Ser195CT and that this positioning may require dynamic motions that accommodate induced fit movements upon substrate binding.
Regions near Ser195CT are less dynamic in active thrombin
Although we did not have a peptide that covered Ser195CT, the amide H/2H exchange experiments were able to probe segments near the catalytic site. It is interesting to note that while the region around Asp102CT appeared to lack necessary dynamic motions in the zymogen forms, the region around Ser195CT appeared to be too dynamic. The autolysis loop, corresponding to residues 166-180 (Leu130CT-Leu144CT), which runs along the backside of the strand containing Ser195CT, showed significantly more amide H/2H exchange in the zymogen forms. This region of thrombin is disordered in both the prethrombin-2 crystal structure and in the structure of thrombin bound to prothrombin fragment 2(5, 6). Interestingly, it is also more disordered in the Na+-free form of thrombin (21). It is likely that conformation of this loop affects the activation state of the enzyme due to its proximity to Gly193CT which is known to be important in active site conformation, namely the formation of the oxyanion hole.
The sodium binding site, corresponding to residues 260-275 (Ile212CT-Phe227CT) was probed by taking advantage of a new TCEP reduction protocol (T. M. Sabo and M. C. Maurer, Biochemistry, in press). At the shortest exchange times (30 s and 1 min) this peptide showed somewhat faster exchange in prethrombin-2 than in active thrombin. This again suggests that the active site may not be well-formed but instead may be too dynamic in the zymogen forms. Interestingly, no differences in H/2H exchange of the Na+-binding site were observed when we compared meizothrombin to active thrombin. This result is consistent with the observation that although meizothrombin has deficient fibrinogen clotting activity, it can attain full activity toward protein C in the presence of thrombomodulin so it must be able to properly structure its catalytic residues under some conditions (22).
Conclusions
For some time now it has been known that no large conformational changes accompany the conversion of zymogen forms of serine proteases to active proteases. The DSC experiments show that none of the zymogen forms differ appreciably from active thrombin in overall stability. By comparing prothrombin, prethrombin-2, meizothrombin and active thrombin the origin of the partial activity of meizothrombin was discerned. By comparing prethrombin-2 to active thrombin, we were able to observe the vast and striking dynamic changes that occur throughout the thrombin molecule when the single peptide bond is cleaved upon activation. Indeed, it appears that half of the loops surrounding the active site become more dynamic and half become less dynamic when the zymogen is converted to the active serine protease. A dynamic compensation model is most consistent with all of the data. In this model, the binding loops including ABE1 and the substrate-specificity pocket (90sCT loop) become more dynamic. In contrast, the loops that surround the catalytic machinery, including the autolysis loop and the Na+-binding site, which must be precisely positioned for catalytic activity, become less dynamic.
Supplementary Material
Melting profiles of prothrombin, prethrombin-2 and active thrombin from DSC. Duplicate scans gave similar results.
Acknowledgments
Financial support for this work was provided by NIH grant RO1-HL070999 to EAK. JK acknowledges support from the Cell and Molecular Genetics Training Program.
Financial support for this work was provided by NIH grant RO1-HL070999 to EAK.
ABBREVIATIONS
- TFA
trifluoroacetic acid
- MALDI-TOF
matrix assisted laser desorption time-of-flight
- ABE 1
anion-binding exosite 1
- TM
thrombomodulin
- DSC
differential scanning calorimetry
- TCEP
tris-carboxyethylphosphine
Footnotes
Thrombin residues are conventionally numbered according to an alignment with chymotrypsin and this convention results in loop residues carrying numbers such as 60A, 60B etc. This convention is confusing with amide exchange data since information is lost regarding the length of the peptide fragment in which amide exchange is being measured. We therefore will give all thrombin residue information as sequential numbers first, followed by the chymotrypsin convention numbers in parentheses subscripted with CT to denote which are which.
References
- 1.Machovich R. CRC Press; Boca Raton, FL: 1984. p. 166. [Google Scholar]
- 2.Anderson PJ, Nesset A, Bock PE. Effects of activation peptide bond cleavage and fragment 2 interactions on the pathway of exosite I expression during activation of human prethrombin1 to thrombin. J Biol Chem. 2003;278:44482–44488. doi: 10.1074/jbc.M306917200. [DOI] [PubMed] [Google Scholar]
- 3.Khan AR, James MNG. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Prot Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stroud RM, Kossiakoff AA, Chambers JL. Mechanisms of zymogen activation. Ann Rev Biophys Bioeng. 1977;6:177–193. doi: 10.1146/annurev.bb.06.060177.001141. [DOI] [PubMed] [Google Scholar]
- 5.Arni RK, Padmanabhan K, Padmanabhan KP, Wu TP, Tulinksy A. Structures of the noncovalent complexes of human and bovine prothrombin fragment 2 with human PPACK-thrombin. Biochemistry. 1993;32:4727–4737. doi: 10.1021/bi00069a006. [DOI] [PubMed] [Google Scholar]
- 6.Vijayalakshmi J, Padmanabhan KP, Mann KG, Tulinksy A. The isomorphous structures of prethrombin2, hirugen-, and PPACK-thrombin: changes accompanying activation and exosite binding to thrombin. Prot Sci. 1994;3:2254–2271. doi: 10.1002/pro.5560031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malkowski MG, Martin PD, Guzik JC, Edwards BF. The co-crystal structure of unliganded bovine alpha-thrombin and prethrombin-2: movement of the Tyr-Pro-Pro-Trp segment and active site residues upon ligand binding. Prot Sci. 1997;6:1438–1448. doi: 10.1002/pro.5560060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens WK, Nesheim ME. Structural changes in the protease domain of prothrombin upon activation as assessed by N-bromosuccinimide modification of tryptophan residues in prethrombin-2 and thrombin. Biochemistry. 1993;32:2787–2794. doi: 10.1021/bi00062a008. [DOI] [PubMed] [Google Scholar]
- 9.Lentz BR, Zhou CM, Wu JR. Phosphatidylserine-containing membranes alter the thermal stability of prothrombin’s catalytic domain: a differential scanning calorimetric study. Biochemistry. 1994;33:5460–5468. doi: 10.1021/bi00184a015. [DOI] [PubMed] [Google Scholar]
- 10.Ni F, Konishi Y, Scheraga HA. Thrombin-bound conformation of the C-terminal fragments of hirudin determined by transferred nuclear Overhauser effects. Biochemistry. 1990;29:4479–89. doi: 10.1021/bi00470a030. [DOI] [PubMed] [Google Scholar]
- 11.Rosing J, Zwaal RFA, Tans G. Formation of meizothrombin as intermediate in Factor Xa-catalyzed prothrombin activation. J Biol Chem. 1986;261:4224–4228. [PubMed] [Google Scholar]
- 12.Schoen P, Lindhout T. The in Situ inhibition of prothrombinase-formed human alpha thrombin and meizothrombin (des F1) by anithrombin III and heparin. J Biol Chem. 1987;262:11268–11274. [PubMed] [Google Scholar]
- 13.Han JH, F CHC, M TD. Inhibition of meizothrombin and meisothrombin(desF1) by heparin cofactor II. J Biol Chem. 1997;272:28660–28665. doi: 10.1074/jbc.272.45.28660. [DOI] [PubMed] [Google Scholar]
- 14.Fenton JW. Thrombin. Ann N Y Acad Sci. 1986;485:5–15. doi: 10.1111/j.1749-6632.1986.tb34563.x. [DOI] [PubMed] [Google Scholar]
- 15.Croy CH, Koeppe JR, Bergqvist S, Komives EA. Allosteric Changes in Solvent Accessibility Observed in Thrombin upon Active Site Occupation. Biochemistry. 2004;43:5246–55. doi: 10.1021/bi0499718. [DOI] [PubMed] [Google Scholar]
- 16.Mandell JG, Falick AM, Komives EA. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal Chem. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 17.Mandell JG, Falick AM, Komives EA. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc Nat Acad Sci U S A. 1998;95:14705–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandell JG, Baerga-Ortiz A, Akashi S, Takio K, Komives EA. Solvent accessibility of the thrombin-thrombomodulin interface. J Mol Biol. 2001;306:575–89. doi: 10.1006/jmbi.2000.4416. [DOI] [PubMed] [Google Scholar]
- 19.Hughes CA, Mandell JG, Anand GS, Stock AM, Komives EA. Phosphorylation causes subtle changes in solvent accessibility at the interdomain interface of methylesterase CheB. J Mol Biol. 2001;307:967–76. doi: 10.1006/jmbi.2001.4523. [DOI] [PubMed] [Google Scholar]
- 20.Koeppe JR, Seitova A, Mather T, Komives EA. Thrombomodulin tightens the thrombin active site loops to promote protein C activation. Biochemistry. 2005;44:14784–91. doi: 10.1021/bi0510577. [DOI] [PubMed] [Google Scholar]
- 21.Johnson DJ, Adams TE, Li W, Huntington JA. Crystal structure of wild-type human thrombin in the Na+-free state. Biochem J. 2005;392(Pt 1):21–8. doi: 10.1042/BJ20051217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cote HCF, Bajzar L, Stevens WK, Samis JA, Morser J, MacGillivray RTA, Nesheim ME. Functional characterization of recombinant human meizothrombin and meizothrombin(desF1) J Biol Chem. 1997;272:6194–6200. doi: 10.1074/jbc.272.10.6194. [DOI] [PubMed] [Google Scholar]
- 23.DiBella EE, Maurer MC, Scheraga HA. Expression and folding of recombinant bovine rethrombin-2 and its activation to thrombin. J Biol Chem. 1995;270:163–169. doi: 10.1074/jbc.270.1.163. [DOI] [PubMed] [Google Scholar]
- 24.Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Prot Sci. 1992;1:426–71. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Melting profiles of prothrombin, prethrombin-2 and active thrombin from DSC. Duplicate scans gave similar results.