

Abstract
The p53 tumor suppressor is a tetrameric transcriptional enhancer, and its activity is compromised by mutations that cause amino acid substitutions in its tetramerization domain. Here we analyze the biochemical and biophysical properties of peptides corresponding to amino acids 319 to 358 of wild-type human p53, which includes the tetramerization domain, and that of a cancer-derived mutant with valine substituted for glycine 334. Unlike the wild-type peptide, the G334V peptide forms amyloid fibrils by a two step process under physiological conditions of temperature and pH. Nevertheless, the G334V peptide is capable of forming hetero-oligomers with a wild-type peptide. Computational modeling of the G334V peptide structure suggests that substitution of valine for glycine 334 causes a local distortion that contributes to a β-dominated structural transition leading to amyloid formation. Since the distortion is mostly on the surface, the mutant peptide is still able to form a pseudo-native tetramer complex at higher concentrations and/or lower temperatures. Our study suggests a new potential mechanism by which mutations that compromise tetramer formation inactivate p53 as a tumor suppressor.
The tumor suppressor protein p53 is a 393 amino acid phosphoprotein that acts as a transcriptional enhancer in tetrameric form and suppresses cell cycle progression or induces apoptosis in response to DNA damage (1, 2). Inactivation of p53 through mutation of its gene or interaction with cellular or viral proteins appears to be a critical step in the etiology of many cancers. The tetramerization domain of p53 is essential for efficient site-specific DNA binding and contributes to the ability of p53 to activate transcription from natural promoters (3). The core tetramerization domain is located in the carboxy-terminal region just distal to the site-specific DNA binding domain (2). The structure of the p53 tetramerization domain has been determined by both NMR and X-ray crystallography (4-7). Each subunit consists of a β-strand (residues 326−333) and an α-helix (residues 335−356) that are separated by a sharp hairpin at residue Gly334. As shown in Figure 1, the tetramer is best described as a dimer of two primary dimers. The core of the tetramer is a four-helix bundle (i.e., one helix from each monomer), and the two anti-parallel β-sheets of each primary dimer are on opposing faces of the surface.
Figure 1.

p53 tetramerization domain. (A) Amino acid sequence of the human p53 tetramerization domain (Lys319-Glu358); Gly334 is highlighted using a larger font. (B) Top and (C) side schematic views of the 3D structure of the tetramer (6: 1C26.pdb). One primary dimer is color-coded red and blue to indicate each peptide strands, and the second dimer is color-coded gray. The two Gly334 residues of the first primary dimer are represented as green spheres. The α-helices are shown as cylinders, and the β-strands are shown as wavy rectangular boxes.
Mutations in the p53 gene have been found in more than 50% of human tumors (8). The majority of cancer-associated p53 mutations is located within the DNA-binding domain and disrupts DNA binding activity. However, it has been reported that many mutations within the tetramerization domain are also associated with human cancer (9). To date, 206 mutations have been found in 26 positions among 31 residues of the tetramerization domain (IARC TP53 Mutation Database, release R9). A mutation of Gly334 to Val was reported to be linked to small- and non-small cell lung cancer and to squamous cell carcinoma (10, 11). Interestingly, this mutation in exon 10 represents a G to T transversion, a type of mutation that is frequently found in lung cancers from patients who smoke (12). However, the biophysical properties of p53 with Gly334 changed to Val have not been studied.
Amyloidosis is a group of diseases characterized by abnormal protein folding and assembly that result in the accumulation of insoluble protein fibrils (13-16). Deposition of amyloid causes cell and organ dysfunction, and in many cases, apoptosis. At least 20 different, non-homologous proteins and polypeptides are known to form amyloid deposits in vivo that are associated with human diseases. In addition, many other proteins have also been found to form amyloid fibrils in vitro under more extreme environmental conditions (e.g., sub-physiologic pH or organic solvents), suggesting that it is a shared potential of all proteins (17). The exact structure of amyloid fibrils has been hard to determine, due at least in part to their insolubility. However, they can generally be characterized as hollow-tubes formed by several β-sheet-rich protofibrils, with the hydrogen bonds parallel and the β-strands perpendicular to the fibril axis (18). That it is primarily inter-backbone hydrogen bonding that holds each β-sheet together and not interactions between the side chains explains why it is a potential structure of all proteins (17). Substantial effort has gone into understanding the molecular and physiochemical factors that transform a natively folded protein into amyloid fibrils (17, 19-24). Protein aggregation and amyloid formation is governed by the interplay of sequence and environmental conditions. An important first step is destabilization of the native protein structure to an at least partially unfolded state, which may be accomplished by energetically unfavorable site mutations (19). This partially denatured, “ molten globule” state provides an alternate environment that may stabilize “ non-native” secondary structure elements along different regions of the sequence. At this stage, the specific sequence and physiochemical properties of the amino acids, such as hydrophobicity, secondary structure propensity and electrostatic charge, are very influential in determining aggregation of the protein strands, and subsequent fibril formation (25).
In the present study, we characterized the biophysical properties of the tetramer domain of p53 with substitution of Val for Gly334 (G334V) by circular dichroism (CD), ultracentrifugation, chemical dye binding and atomic force microscopy (AFM). The mutant tetramerization domain clearly showed amyloid formation and hetero-tetramerization at physiological pH and temperature, which suggests a new potential mechanism for the inactivation of p53 in tumor cells. We also did theoretical sequence and structural modeling to better understand how the G334V single-site mutation can increase the physiochemical propensity for amyloid fibril formation.
MATERIALS AND METHODS
Peptide Synthesis and Purification
Peptides were synthesized using Fmoc-chemistry on Rink amide resin with an Applied Biosystems 431A peptide synthesizer (Applied Biosystems Inc., Foster City, CA). Fmoc-amino acids were activated with HBTU in the presence of HOBt and DIEA. Peptide resin cleavage and peptide deprotection were accomplished in a single step using reagent K (TFA:H2O:thioanisole:ethanedithiol:phenol = 85:5:5:2.5:2.5) to yield crude peptide amide. All synthetic peptides were purified using a Beckman HPLC system with a Vydac C-8 column, eluted with a linear gradient of water containing 0.05% TFA and acetonitrile containing 0.04% TFA. Molecular weights were confirmed by electron spray ionization mass spectroscopy performed with a Finnigan MAT SSQ 7000 (Finnigan MAT, San Jose, CA). Peptide concentrations were measured spectrophotometrically, using an extinction coefficient of ε280 = 1280 M−1 cm−1 (26). All buffer solutions were degassed and purged with Argon gas.
Circular Dichroism
The CD spectra were recorded on a JASCO J-710 spectropolarimeter using a 1-mm path length quartz cell. The instrument was calibrated with recrystallized D(+)-10-camphorsulfonic acid. A Lauda water bath was used to control the temperature of the cell. CD spectra were recorded in 50 mM sodium phosphate buffer containing 100 mM NaCl, pH 7.5 or 25 mM sodium acetate buffer, pH 3.6 as indicated. Peptide concentrations were 10 μM obtained by quantitative serial dilution of a 500 μM stock solution. All spectra were the average of 4 to 8 repeats obtained by collecting data from 260 nm to 190 nm at 0.2 nm intervals and at a rate of 50 nm/min with a response time of 2 s for each point. For GdnHCl denaturation studies, the prepared samples were incubated at room temperature for 1 h before acquisitions of CD spectra were started. For thermal denaturation studies, spectra were recorded at discrete temperatures from 10°C to 90°C in 5°C intervals. CD spectra were obtained after 10 min of equilibration of the sample at each temperature point. For mixing experiments, a mixture of two peptide stock solutions was boiled for 5 min, the same volume of acetonitrile was added, and the mixture then was lyophilized to denature the peptides. The mixtures of lyophilized peptides were reconstituted in 50 mM sodium phosphate buffer. Then, the peptide mixtures were incubated for 1 hr at room temperature before obtaining the CD spectra. Analysis of the mixing experiments was performed by comparing the spectra of the two peptides mixed in the solution (experimental spectrum) to the sum of the individual spectra of each peptide. The predicted content of secondary structure was calculated using the SELCON3 program (27).
Congo Red Assay
The G334V mutant (10 μM) was pre-incubated in 50 mM sodium phosphate buffer, pH7.5, 100 mM NaCl at 37°C for 10 days. Congo red was added to the sample solutions at the final concentration of 0.2% (w/v) (28). After incubation with Congo red at 37°C for 3 hours, the samples were centrifuged at 15,000 rpm for 10 minutes. The pellets were resuspended in 100 μl of the phosphate buffer and spread onto glass slides for examination with a light microscope using cross polarizers.
Atomic Force Microscopy
A 2 μL drop of sample solution was spotted on a freshly prepared oxidized Si(111) surface at room temperature, and the surface was dried in air until the solvent was completely removed. Tapping mode AFM measurements in air were performed with a Nanoscope IIIa system (Digital Instruments, Santa Barbara, CA). Standard etched silicon probes (nominal spring constant ∼40 N/m, resonance frequency ∼300 kHz) were used. Typical imaging parameters were: drive amplitude 50−200 kHz with set points of 0.2−0.3 V, scan frequencies of 1.0−1.2 Hz, image resolution 512 by 512 points.
Thioflavin T Assay
The assay was performed in a Shimadzu RF-5000 spectrofluorometer. Excitation and emission wavelengths were 444 and 485 nm, respectively. Peptides at the concentration of 10 μM were incubated in 50 mM sodium phosphate buffer, pH 7.5, 100 mM NaCl at 37°C. Then the sample solutions (0.1 ml) were added into 1.9 ml of the same buffer containing thioflavin T at a final concentration of 5 μM, which had been pre-incubated at 37°C with stirring (29). Fluorescence change of the sample solutions was measured at each incubation time.
Analytical Ultracentrifugation
Analytical centrifugation was carried out using a Beckman XL-A analytical ultracentrifuge with a four-place titanium rotor and cells with carbon-filled epoxy double-sector centerpieces. Rotor speeds were in the range of 30,000 rpm. With column lengths of approximately 5 mm, 48 hours were allowed for the initial equilibrium and 24 hours were allowed for re-equilibration following temperature changes. All scans were performed at 230 nm. Samples were prepared with appropriate buffers as indicated in the Results section. The data were edited using the XL-A software and analyzed by mathematical modeling using MLAB (Civilized Software, Bethesda, MD) to perform non-linear least-squares curve-fitting of the data with mathematical models as described (30, 31).
Precipitation of biotinylated peptides with avidin beads
Wild-type or G334V peptides were mixed with an equal concentration of biotinylated p53(319−393) with a final acetonitrile concentration of 50%, and the solution was boiled for 5 min. After lyophilizing, the samples were dissolved in 50 mM sodium phosphate buffer, pH 7.5, containing 100 mM NaCl to give a final concentration of 10 μM for each peptide; then they were incubated for 1 hr. at room temperature. The peptides mixtures were incubated with UltraLink Immobilized NeutrAvidin beads (Pierce, Rockford, IL) for 30 min, and the beads were washed with 50 mM sodium phosphate buffer. The peptides then were extracted with 20% acetic acid and analyzed by analytical HPLC on a C-18 reverse-phase column. For G334V peptide forming a hetero-oligomer with the biotinylated WT peptide, a peak containing the mutant peptide was identified by HPLC elution and by mass spectrometry.
Molecular modeling and sequence analysis
The p53 tetramerization domain crystal structure of Jeffrey et al. (6) was used as the starting point for all 3D molecular modeling. This is file 1C26.pdb in the Protein Data Bank (32: http://www.pdb.org/). Formation of the G334V mutant and energy minimization were done with the CHARMM software package (33) using the all hydrogen top_all22_prot and par_all22_prot residue topologies and parameters (34). The minimization procedures were carried out in two phases: first with all but the hairpin residues fixed in space (i.e., residues 333−335 for the left-handed α-helix and 333−337 for the antiparallel β-sheet forced conformations), and then with all atoms allowed to move. Each phase employed 200 steps of the Steepest Descents algorithm, and then 1000 steps of the Adopted Basis Newton-Raphson algorithm. The dihedral constraint facility was used through-out both phases to maintain the amide bonds of the most effected residues in idealized trans (180°) conformations: i.e., residues 331−336 for the left-handed α-helix and 331−338 for the antiparallel β-sheet forced conformations. Additionally, the (φ, ψ) backbone dihedral angles of residue V334, and sometimes Arg333, were also constrained to idealized secondary structure values as indicated in the text. The shielding effect of the solvent was represented by using a 1/r distant-dependent dielectric term. The pairwise non-bonded atom list was truncated by the shift algorithm for the electrostatic interactions (cutnb 8.0 Å) and by the switch algorithm for the van der Waals' interactions (ctonnb 6.5 Å and ctofnb 7.5 Å). The non-bonded list was updated every 25 steps of the minimization. The constraints were applied identically to each monomer of the tetramer complex, which was minimized as a whole.
The p53 tetramerization domain WT and G334V mutant sequences (residues 319−358) were subjected to three secondary structure prediction computer programs that use different algorithmic strategies. The programs were accessed over the World Wide Web at the given addresses. These were: 1) the Chou and Fasman method (35: http://fasta.bioch.virginia.edu/fasta_www/chofas.htm), which uses statistically derived structural propensities for each amino acid type; 2) PROF (36) through the PredictProtein server (37: http://cubic.bioc.columbia.edu/predictprotein/), which calculates a divergent profile and uses neural networks; and 3) CSSP (38, 39: http://askb.umdnj.edu/), which uses an artificial neural network to quantify the influence of tertiary contacts on the secondary structure propensities.
RESULTS
Conformational change of the mutant peptide G334V
Wild-type (WT) and G334V mutant p53 peptides corresponding to residues 319−358 were chemically synthesized (Figure. 1A), and their secondary structures were characterized by circular dichroism (CD) in sodium phosphate buffer, pH 7.5 at 20°C over a range of concentrations (Figure 2A). Under these conditions, the WT peptide exhibited double minimum bands at 208 and 222 nm, which is a characteristic of the p53 tetramer structure (30). The helicity of the WT peptide reached a plateau at 10 μM peptide concentration with negative [θ]222 values. Deconvolution of the CD spectra of the WT peptide revealed a secondary structure content of 45% α-helix, 23% β-sheet, 6% turn and 26% random, which agreed with the one based on NMR and crystal structures (55% α-helix, 20% β-sheet, 7.5% turn and 17.5% random). The spectrum of the G334V peptide was almost identical to that of the WT peptide at a concentration of 10 μM or higher (Figure 2A). However, at low concentrations, the CD spectrum of G334V exhibited a weak band at 200 nm, indicating a non-ordered structure.
Figure 2.

Conformational changes in WT and G334V mutant tetramer-domain peptides. (A) Concentration dependencies of WT and G334V. CD spectra were recorded in 50 mM sodium phosphate buffer (pH 7.5), 100 mM NaCl at 20°C as described in Materials and Methods. (B) Thermal denaturation of WT and G334V. All spectra were recorded with 10 μM peptide in 50 mM sodium phosphate buffer (pH 7.5), 100 M NaCl. CD spectra were obtained after 10 min of equilibration at each temperature point. (C) Time-dependent secondary structural transition of WT and G334V. CD spectra were measured with 10 μM peptides in 50 mM sodium phosphate buffer (pH 7.5), 100 mM NaCl. Samples were incubated for 1 min (line,——), 10 min (dots,......), 30 min (dash, ------) and 2 hrs (dash and dots, −.−.−) at 30°C, 33°C and 37°C.
Figure 2B shows the temperature-dependent conformational change of WT and G334V peptides at physiological ionic strength and pH. With increasing temperature at pH 7.5, both peptides underwent a conformational transition from p53 tetramer to a largely disordered structure. The spectra of the WT peptide showed a clear isodichroic point near 206 nm, indicating that the thermal unfolding process of the WT peptide occurred as a one state process from tetramer to disordered monomer. On the other hand, the G334V spectra did not have an isodichroic point and showed increased content of a β-structure at temperatures higher than 35°C. This result indicated that the thermal conformational change of G334V is distinct from the wild-type peptide and occurs through a β-dominant structural change.
Upon close examination, we noticed that prolonged incubation of G334V peptide at 37°C induced a significant change in the CD spectra although the mutant peptide showed a wild-type conformation in the early time period (Figure 2C). After 10 min, the double minima at 208 nm and 222 nm disappeared, and the spectra exhibited a new broad band with a minimum at 217 nm (25% α-helix, 28% β-sheet, 27% turn and 20% random). After 120 min, the G334V peptide displayed a spectrum with a broad band at 215 nm and a shoulder at 203 nm, which is typical of a β-sheet conformation. The structural conversion of the mutant peptide was effectively irreversible under these conditions. Interestingly, the G334V peptide showed few structural changes at lower temperatures, whereas the WT peptide did not exhibit any time-dependent conformational changes (data not shown).
Under acidic conditions, the spectrum of the G334V peptide indicated a predominantly β-sheet structure with the typical minimum around 215 nm, while the WT peptide had a native conformation (see Figure S1 in Supporting Information). The β-sheet structure content increased with prolonged incubation time at 37°C. The spectrum also had a shoulder around 203 nm, which is a characteristic of a fine precipitate suspension (40).
These results indicate that the mutant G334V peptide structure is converted to a β-structure dominant conformation from the wild-type tetramer-like conformation under physiological conditions of 37°C and pH 7.5 as well as at acidic pH.
Formation of amyloid-like fibrils and globular aggregation of the G334V peptide
The formation of a β-structure dominant conformation is consistent with the formation of amyloid. Characteristics of amyloid fibrils include strong binding by the chemical dyes Congo red (28) and thioflavin T (29). As shown in Figure 3A, the G334V peptide displayed a strong apple-green birefringence under a polarizing microscope after staining with Congo red, an effect that is characteristic of amyloid fibers. Examination by AFM detected not only amyloid-like fibrils but also globular aggregates. Representative atomic force microscopy (AFM) images of G334V aggregates after incubation in the pH 7.5 buffer for 10 days at 37°C are shown in Figure 3B. The height of the fibrils rarely exceeded 1 nm, and their apparent width typically was 25 nm with a length of 150 − 300 nm. The size of the aggregates was 60 nm in diameter and their height was 3 nm. In contrast, no fibrils were obseved with the WT peptide under the same conditions (Figure 3C).
Figure 3.
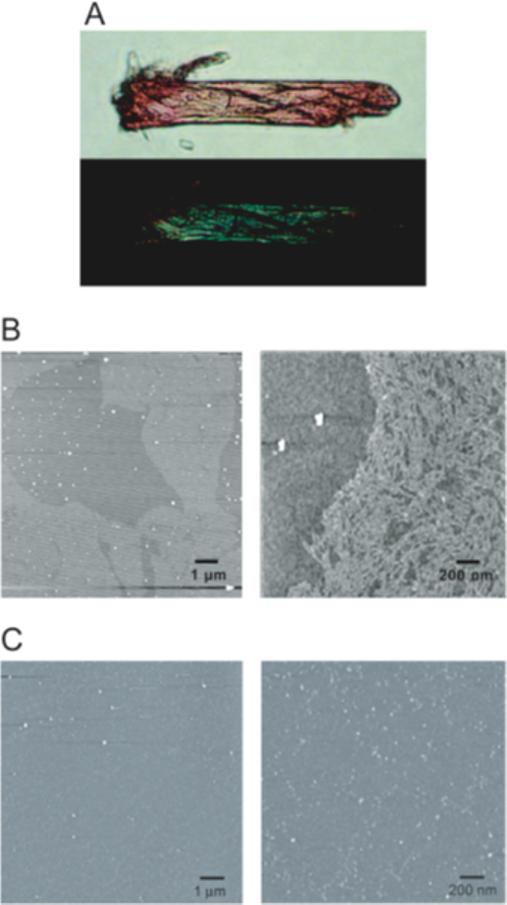
Characterization of aggregates formed by G334V mutant peptide. (A) Optical microscope image obtained under cross-polarized light and containing G334V mutant aggregates stained with Congo red. The photograph shows the blots of green bifringence coming from regions rich in amyloid fibrils. (B) AFM images of G334V aggregates. Amyloid-like fibrils (bright parts) were present in left panel. Right panel is the magnified image of G334V amyloid-like fibrils. Small globular aggregates (white dots) also were present. (C) AFM images of WT peptide. No fibrils were observed with the WT peptide.
To further examine the kinetics of the G334V peptide fibril formation, we performed a fluorescent assay using thioflavin T (ThT). Samples of the peptides were incubated in sodium phosphate buffer, pH 7.5, at 37°C and then with ThT. The fluorescence intensity of ThT gradually increased and reached a maximum after 5 days (Figure 4A); amyloid formation could be detected with this assay at concentrations as low as 2 μM (Figure S3, Supporting Information). Such an increase in ThT fluorescence is diagnostic of amyloid formation (29). In contrast, the WT peptide and the monomeric mutant L330A did not show any change of fluorescence intensity even after 10 days. These results indicated that the G334V peptide forms amyloid-like molecular ribbons when incubated under physiological conditions. However, the increase in fluorescence was accelerated at elevated temperatures (Figure 4B) and at acidic pH (Figure 4C).
Figure 4.
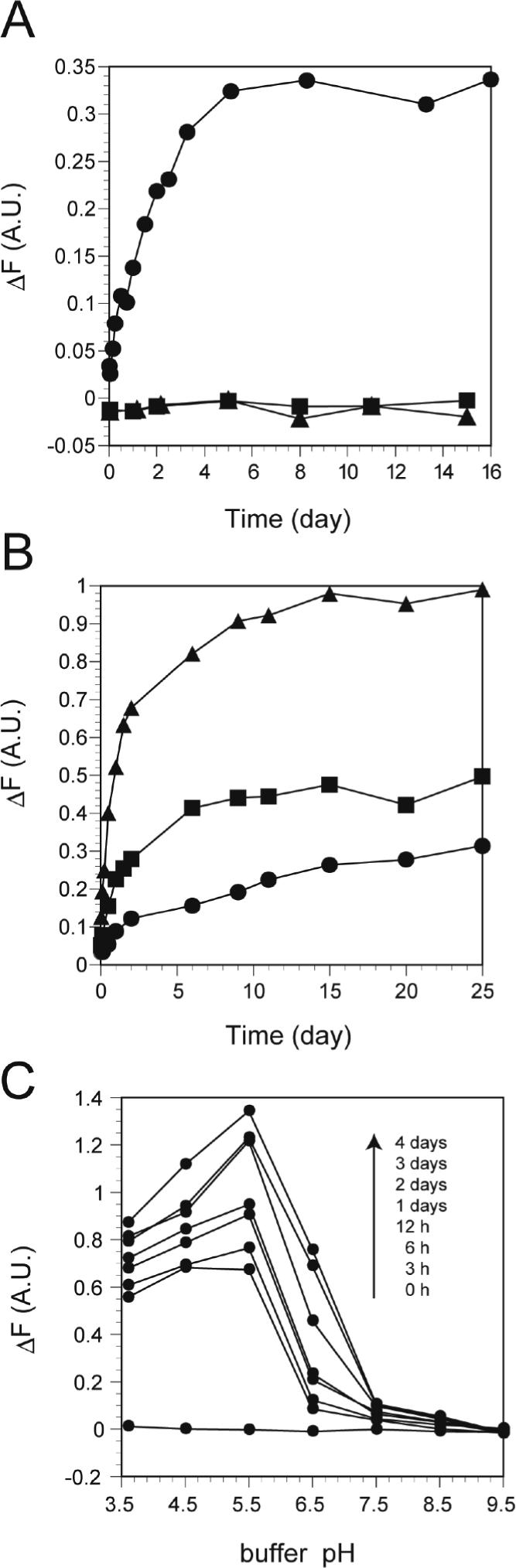
Formation of amyloid-like fibrils by G334V mutant peptide. (A) The time courses of the increase in ThT fluorescence: G334V (Circle), L330A (square), and WT (triangle). (B) Temperature dependency for amyloid formation of G334V. Temperatures used are: 30 (circle), 37 (square), and 45°C (triangle). (C) Amyloid formation at various pHs. Experimental conditions are described in Material and Methods.
Conformational Stability of the G334V peptide
Figure 5A shows the change in ellipticity at 222 nm as plotted against temperature for the WT and G334V peptides. The WT peptide had a transition temperature (Tm) of 72°C while this value for G334V dropped to 48°C. The conformational stability of the mutant peptide also was determined by monitoring changes in the CD spectra at various concentrations of GdnHCl (Figure 5B). The midpoint of the chemical denaturation curve for the WT peptide was observed at 2.2 M GdnHCl while that for the G334V peptide was observed at 0.6 M GdnHCl. The denaturation curve of the WT peptide exhibited the characteristic sigmoid shape associated with cooperative unfolding of proteins. In contrast, the curve of the G334V peptide did not show a typical sigmoid shape, suggesting a lack of cooperativity for the GdnHCl-dependent denaturation, which is consistent with results obtained for other aggregated structures comprised of β-strands (41).
Figure 5.
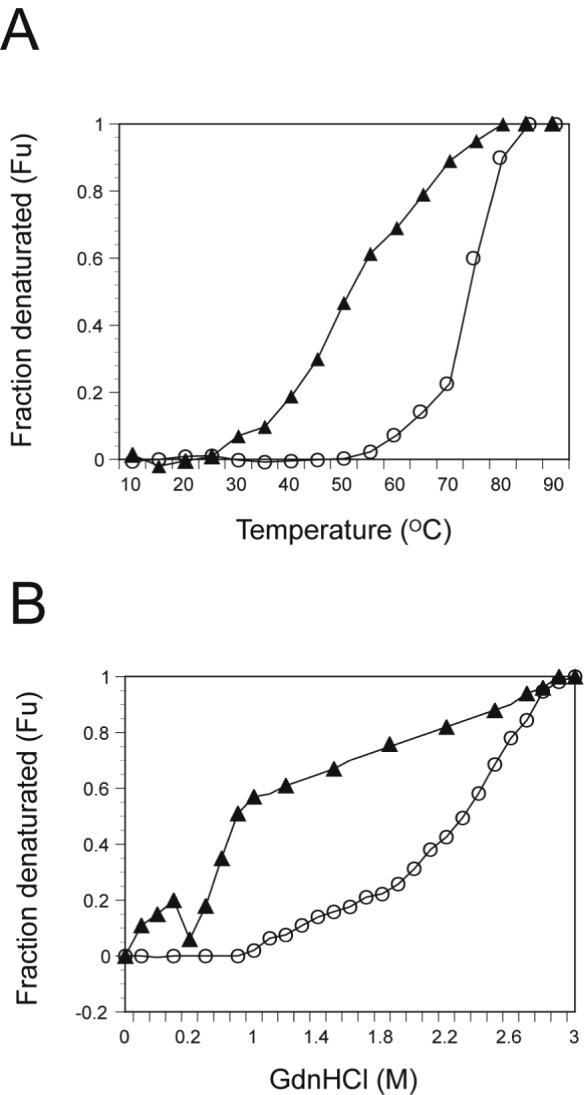
Thermal and GdnHCl denaturation of WT and G334V mutant peptides. All spectra were recorded with peptides at 10 μM in 50 mM sodium phosphate buffer (pH 7.5), 100 mM NaCl. CD ellipticities at 222 nm were plotted, and these values were used to calculate the mol fraction of denatured molecules (Fu) at each temperature (10−90°C) (A) or at each GdnHCl concentrations (B): G334V (Filled triangle) and WT (circle).
Effects of TFE on G334V secondary structure and oligomerization state
TFE is a co-solvent known to destabilize hydrophobic interactions within polypeptide chains and to stabilize local hydrogen bonds between residues close in the amino acid sequence, particularly those forming α-helices and β-hairpins (42). Therefore, we analyzed the effect of TFE addition on structural changes of p53 peptides at pH 7.5 and pH 3.6. At pH 7.5, the WT peptide showed a decreased ellipticity at 208 nm upon the addition of TFE (Figure 6A). The ellipticity ratio of [θ]222 to [θ]208 decreased steadily from 1.26 to 0.82 over the range 0−50% TFE, suggesting that the peptide might become monomeric at high TFE concentrations (43). The G334V peptide showed a broad negative ellipticity at 217 nm in the presence of 5−20% TFE, suggesting that TFE induced more β-sheet structure in the peptide (Figure 6B). The G334V peptide showed the highest content of β-sheet (>40%) in 20% TFE. At higher TFE concentrations, the β-sheet spectra were replaced by spectra typical for an α-helix similar to that of the WT peptide. At pH 3.6 in presence of 20% TFE, the WT peptide displayed the characteristics of a monomer-tetramer and/or a monomer-dimer-tetramer by sedimentation equilibrium (Table 1). Addition of 20% TFE did not significantly affect the free energy of WT tetramer formation. The WT peptide was monomeric in 50% TFE and exhibited an increased α-helical content. In contrast, the G334V peptide formed a high-degree polymer in 0 and 20% TFE solution at pH 3.6 (see Figure S2 in Supporting Information). In the presence of 50% TFE, the G334V peptide formed a monomeric helical structure. Similar results also were obtained in the case of methanol addition (data not shown).
Figure 6.

TFE-induced CD conformational change of WT (A) and G334V mutant (B) peptides. All spectra were measured with peptides at 10 μM in 50 mM sodium phosphate buffer, pH 7.5 in the presence of TFE (0−50 %) at 20°C.
Table 1.
Oligomeric state of WT and G334V in the presence of TFE.
| p53 peptides | TFE (%) | Oligomeric State |
|---|---|---|
| 319−393 (WT) | 0 | monomer-tetramer |
| 20 | monomer-tetramer* | |
| 50 | monomer | |
| G334V | 0 | polymer n=37.5 |
| 20 | polymer n=28 | |
| 50 | monomer |
All ultracentrifugation scans were performed at 230 nm, in 25 mM sodium acetate, pH 3.6 with 0−50% TFE as described in Materials and Methods. All peptide concentrations were 10 μM.
The curve also could be modeled as a monomer-dimer-tetramer.
To determine the oligomeric state of the p53 tetramerization peptides, we performed analytical ultracentrifugation of the p53 tetramer in the presence or absence of TFE. Since measurement of the oligomeric state of G334V was difficult due to rapid aggregation of the peptide in phosphate buffer, pH 7.5, we used a sodium acetate buffer, pH 3.6, for this experiment. In the absence of TFE, the WT peptide self-associated via a monomer-tetramer equilibrium as it did in the pH 7.5 buffer (Table 1). In contrast, the G334V peptide formed a higher-order polymer in phosphate buffer (pH 7.5) with a polymeric value of 20 to 30 (data not shown).
As noted above, the WT peptide was monomeric in 50% TFE concentrations. The G334V peptide, however, formed a high-degree polymer in 0 and 20% TFE solution at pH 3.6 (see Figure S2 in Supporting Information). In the presence of 50% TFE, the G334V peptide formed a monomeric helical structure. These results suggest that the G334V mutant preferentially forms a β-dominant structure and aggregates through β-strand formation.
Hetero-oligomerization of G334V with WT
We investigated whether the G334V mutant peptide could form a hetero-oligomer with the wild-type sequence. When biotinylated p53(319−393) was mixed with the G334V peptide and precipitated with avidin beads, the G334V peptide was co-precipitated with the biotinylated WT peptide (Figure 7A). The amount of co-precipitated G334V peptide was comparable to the amount recovered from the WT peptide under the conditions employed. The C-terminal basic peptide p53(361−393), which does not contain a tetramerization domain, was not co-precipitated. These results indicated that the G334V peptide can interact and form hetero-oligomers with the wild-type tetramerization domain sequence. Figure 7B shows the spectrum of WT and G334V mixed in solution at room temperature. At this temperature, the G334V peptide shows a wild-type conformation. Interestingly, the mixture of WT and G334V displayed the spectrum of a β-structure. In addition, the experimental spectrum of the peptide mixture differed from a simple summation of the individual spectra of the WT and mutant peptides. This result suggests that the structural changes in G334V and/or WT were induced by the hetero oligomerization of G334V with the WT peptide.
Figure 7.
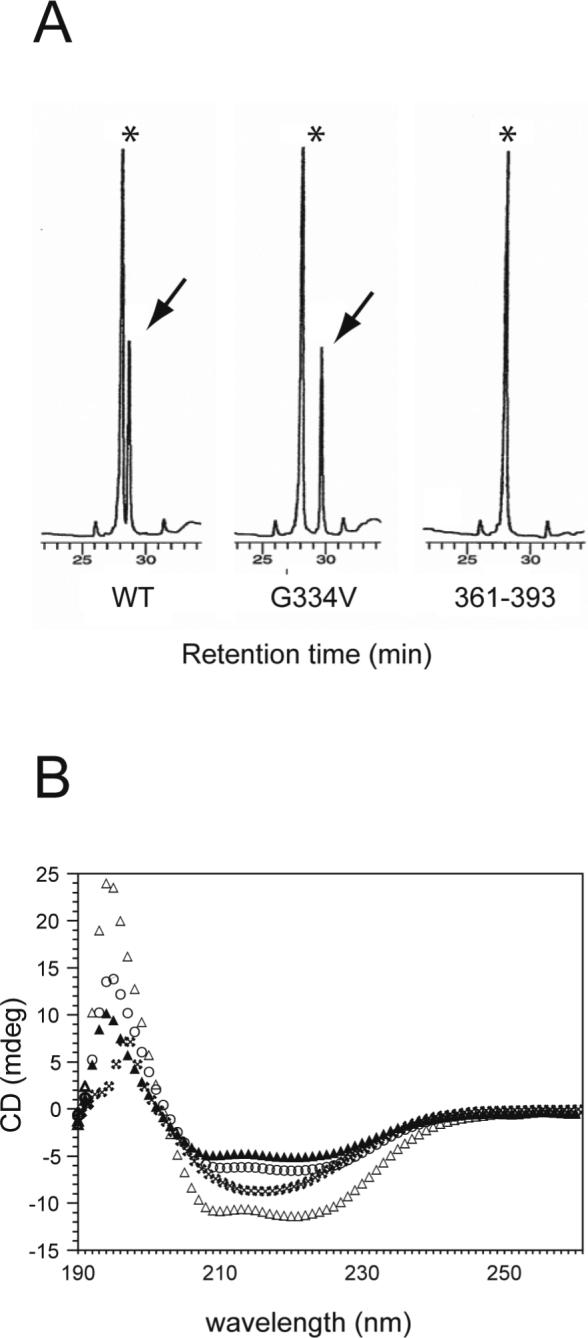
Formation of hetero-oligomers between wild-type and G334V peptides. (A) p53 peptides were co-precipitated with biotinylated p53(319−393) using avidin beads as described in Materials and Methods. The extracted peptides were analyzed by C-18 HPLC. Asterisks indicate the eluting position of biotinylated p53(319−393). Arrows indicate the eluting position of each p53 peptide. (B) CD spectra of a mixture of the p53(319−358) WT and G334V peptides. The mixtures of peptides were prepared as described in Materials and Methods, reconstituted in 50 mM sodium phosphate buffer, and incubated for 1 hr at room temperature before CD spectra were obtained. Experimental spectrum of 10 μM WT (open circle), experimental spectrum of 10 μM G334V (filled triangle), experimental spectrum of a mixture of 10 μM WT and 10 μM G334V (asterisk), and calculated sum of the spectrum of the WT and G334V peptide mixture (open triangle).
Structural modeling of the G334V mutant
As seen in Figure 1B and C, the Gly334 turn regions are on the outside surface of the core helical bundle, where they form the tight, V-shaped backbone connections with the outer, 2-stranded β-sheets. For each subunit, the amide nitrogen of Gly334 makes the last β-strand hydrogen bond, and its carbonyl oxygen makes the first hydrogen bond of the α-helix (6). This tight hairpin turn over a single residue requires backbone dihedral angles (φ, ψ) that are sterically accessible only to a glycine residue, which lacks a side chain. As has been noted, this explains the conservation of glycine at this position among all species of the p53 protein (5, 6). For the Jeffrey et al. (6) and Mittl et al. (7) crystal structures (1C26.pdb & 1AIE.pdb), these values are (99°, 130°) and (101°, 125°), respectively. For the minimized average solution NMR structures of Clore et al. (Clore et al. 1995), the values are (86°, 140°) and (97°, 136°) using two different sets of effective van der Waals' atomic radii for non-bonded contacts (1SAK.pdb & 1SAL.pdb). For the NMR structure of Lee et al. (4) the values are (156°, −136°) (1PES.pdb). The small variation among the first four structures, and the larger difference with the last structure, likely reflect the use of different energy/scoring functions in the refinement process, the number and types of constrains, and differences between the crystal and solution environments.
In the absence of X-ray diffraction and NMR data on the G334V mutant tetramer, we used atomic-scale molecular modeling to reveal the broad outline of the conformational effects. Unfortunately, the current state of theoretical energy functions and computational power are insufficient to accurately determine the structure de novo using molecular dynamics simulations and/or other computer methods. Instead, we used limiting assumptions by constraining the backbone dihedral angles of the substituted Val334 residue into two idealized, energetically favorable conformations. The first was the left-handed α-helix (57°, 47°), which is the closest minimum in the Ramachandran plot (44) to the wild type in the crystal and Clore et al. (5) NMR structures. The results are shown in Figure 8A, where the distorted portion of the minimized structure is superimposed on the wild-type crystal structure. As seen in the companion Figure 8B, the Val334 side chain extends freely on the surface of the protein, where it is predominantly exposed to the solvent. Figure 8A also shows that this distortion of the hairpin results in fraying of the β-sheet on the N-terminal side, and tightening of the α-helix on the C-terminal side. However, in globular proteins valine is most often found in β-sheet structures (45). Thus, the backbone dihedral angles of Val334 were also constrained into an idealized antiparallel β-sheet conformation (−139°, 135°) as a second possible limiting assumption. The backbone of the preceding residue (Arg333) was also constrained to the same values to continue the β-strand-structure of the N-terminal portion of the peptide through to Val334. As seen in Figure 8C, the shift to a more distant region of the Ramachandran map leads to even larger disruptions of the β-sheet and α-helix structures near the hairpin region. The companion Figure 8D shows that the Val334 side chain again ends-up in an unencumbered, solvent exposed position on the protein surface.
Figure 8.

Molecular modeling of the G334V mutation. (A) The backbone dihedral angles (φ, ψ) of the 334V residue were constrained to an idealized left-handed α-helix (57°, 47°) during the minimization procedure. The backbone of the mutant peptide and the valine side chain are shown as thin sticks of green (carbon) and blue (nitrogen) superimposed on the WT native structure in gray (carbon), blue (nitrogen), red (oxygen) and white (hydrogen). Hydrogen bonds are shown in the native structure as white lines for the unaffected ones and magenta lines for those lost in the minimized mutant structure. (B) Space-filling representation of the Val334 residue in the left-handed α-helical minimized structure. (C) and (D) are analogous to (A) and (B), except that the backbone dihedral angles of Val334 and the preceeding Arg333 residues were constrained to an idealized antiparallel β-sheet (−139°, 135°).
Secondary structure analyses
Recently, Kallberg et al. (45) identified “ α-helix/ β-strand-discordance” as a predictor of proteins that will form β-strand-rich, amyloid fibrils (21). A discordant segment is one that forms an α-helix in the native state of a protein despite having a predominant prediction for β-strand by theoretical methods. Analysis of the tetramerization domain peptide by the Chou and Fasman and PROF methods failed to indicate any discordance for the α-helix in the C-terminal half of the native peptide structure. Rather, both methods predicted the same secondary structure for the WT sequence as is present in the native crystal and NMR structures of the tetramer complex (results not shown). For the G334V mutant sequence, both programs predicted extension of the N-terminal β-strand through the hairpin turn region as would be expected by the predominant β-strand propensity of valine (45). However, as shown in Figure 9, analysis of the sequences for “ hidden β propensity” by the CSSP method of Yoon and Welsh (38, 39) predicted that the C-terminal α-helix will transform to β-strand--loop-- β-strand as the number of tertiary contacts increase. Such an increase in tertiary contacts is assumed to occur to peptides as they go from their native structure to a disordered, aggregated state. Additionally, like the other two methods, the CSSP method predicted enhanced β-strand propensity at the hairpin region for the G334V mutant compared to the WT.
Figure 9.
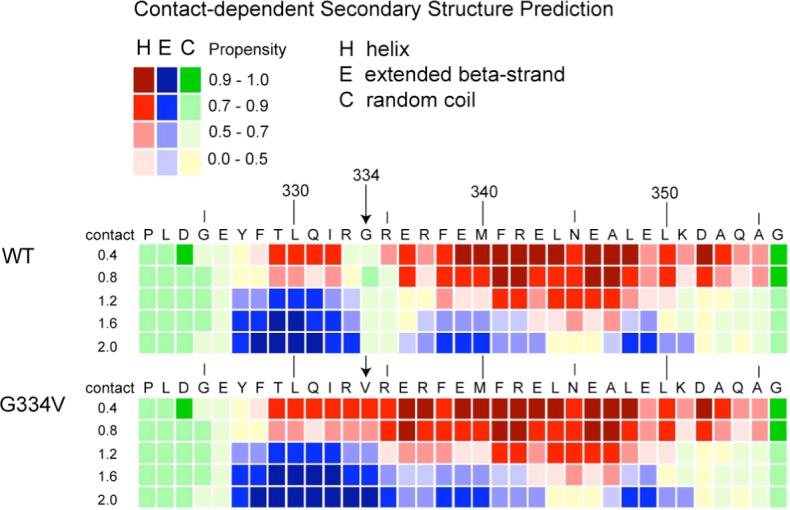
Analysis of the WT and G334V peptide sequences by the CSSP method. Secondary structure predictions are presented as a function of amino acid sequence and the number of tertiary contacts in 0.4 unit bins centered from 0.4 to 2.0. At each matrix position, the most likely of the three secondary structure classes is presented as a red, blue or green-yellow rectangle for α-helix, β-strand or random coil, respectively. The shade of the colors varies to indicate the strength of the predicted propensity, which goes from 0.0 to 1.0 (see key).
DISCUSSION
The p53 gene is mutated in about half of all human tumors (8, 9). Although the vast majority of cancer associated mutations occur in codons specifying the central sequence-specific DNA binding domain and disrupt DNA binding, a significant number of mutations associated with human cancer occur in other regions, including the tetramerization domain. The formation of p53 tetramers is known to be important for p53’s activity as a transcription factor (3), but few cancer-associated mutants outside the DNA-binding domain have been characterized biochemically or biophysically. In this study, we demonstrate that a p53 peptide (319−358) corresponding to the tetramerization domain of a lung cancer-associated mutant (10) formed amyloid-like fibrils under physiological pH and temperature conditions. At concentrations below 10 μM, CD analysis revealed rapid and significant changes in structure caused by the G334V substitution. The conversion from a wild-type-like structure started within 5 min and was accompanied by an increase in β-strand-containing structure (Figure 2C). After 30 min, the intensity of the CD signal began to decrease, probably due to the formation of very fine aggregates. The subsequent slower formation of amyloid fibrils was then detected by the Congo red and thioflavine T assays. These results clearly indicate that amyloid formation by the G334V peptide goes through at least two different stages.
We are aware of only two other studies in which p53 was shown to specifically form amyloid fibrils. One is of the DNA-binding domain of p53, which forms fibrils after partial denaturation by both elevated hydrostatic pressure and temperature (46). This process was found to be enhanced for the “ hot-spot” R248Q contact and structural mutant, which is one of many changes in this domain known to destabilize the native structure (47). The other is for the R337H mutation in the tetramerization domain (48, 49), which is associated with adrenocortical carcinoma (ACC) in children from southern Brazil (50). Both the WT and mutant peptides formed amyloid-like fibrils when incubated at pH 4.0 and elevated temperatures, with the mutant being susceptible at a lower temperature. The pH-dependence of the process was attributed to protonation of Asp352 under acidic conditions, which disrupts the two salt bridges with Arg337 (His337 in the mutant) in the native conformation of the primary dimer. Interestingly, unlike many other amyloidgenic systems, the process was found to be reversible with elevation of the pH from 4.0 to 8.5.
In contrast to these other cases, the present study demonstrates that the G334V mutant peptide can undergo amyloid fibril formation under relatively mild, physiological conditions of temperature, pressure, solvent polarity and pH. This is because, irrespective of environmental conditions, valine is sterically prohibited from assuming the same backbone dihedral angles as the highly conserved glycine that forms the tight hairpin of the native p53 monomer structure (5, 6). Mutations that changed G334 to other amino acids with bulky side chains also would disrupt the tight hairpin of the native structure, but they would not necessarily increase the propensity for amyloid formation. As shown in Figure 8 A and C, the substitution is consistent with a variable degree of local distortion and loss of hydrogen bonds depending on which of the allowed regions of the Ramachandran map the backbone dihedral angles assume. As shown in Figure 8 B and D, the G334V mutation also is consistent with exposure of the hydrophobic valine side chain to the aqueous solvent, which also would help destabilize the native conformation. These observations are the same for all nearby conformations resulting from modeling. However, even with a distorted conformation, the G334V mutant peptide is able to maintain a predominantly native tetramer structure under the conditions of lower temperature and/or higher concentration (Figure 2). This ability is explained by the fact that the locally distorted hairpin segments are on the surface of the complex and are not part of the stabilizing, four-helix bundle of the hydrophobic core (Figure 1). As shown in Figure 2C, at 10 μM monomer concentration and pH 7.5 the distorted conformation of the mutant does not disrupt the tetramer until it reaches approximately normal body temperature (37°C). This thermodynamic balance at physiological conditions places the G334V mutant tetramerization domain peptide in league with the 20 or so other proteins known to form amyloid fibrils in vivo (14).
It should be noted, however, that it is not simply disruption of the quaternary structure that causes the G334V mutant peptide to form amyloid fibrils. For example, previous investigations have revealed a number of naturally occurring and engineered single site mutations that cause varying degrees of destabilization of the tetramer complex without subsequent fibril formation (51). Among these are substitutions of hydrophobic residues buried at the primary dimer and tetramer interfaces and polar residues participating in salt-bridges and/or hydrogen bond networks. We experimentally confirmed for the L330A mutant, which was one of the three most destabilizing to the tetramer (51), that it doesn't form fibrils or even aggregate under the conditions used in this present study (data not shown).
One possible explanation for the observed fibrillogenic potential of the G334V mutant is that, unlike glycine, valine has a high propensity for forming β-strands (45), which is the primary secondary structure component of amyloid fibrils. This is consistent with the experimental findings presented here that the tetramer structure must unfold for β-structure to appear (Figure 2). However, the location of this mutation only covers the hinge region. What then explains the conversion of the C-terminal α-helix in the native monomer structure to β-strand? Previous experimental studies have demonstrated the importance of α-helical intermediates and the α-helix to β-sheet transition to amyloid fibril formation (52-54). As a theoretical explanation, it recently was demonstrated that α-helical segments in the native structures of many physiological amyloidgenic proteins actually have a high propensity for β-sheet formation as determined by secondary structure prediction methods (21, 45). As described above, for the both the WT or G334V mutant, the Chou and Fasman and PROF prediction methods did not detect significant β-strand potential for the C-terminal helix. However, using the new CSSP method of Yoon and Welsh, two regions of “ hidden β-strand propensity” separated by three residues of random coil were identified in this segment under the condition of high tertiary contact (Figure 9). This is interpreted to mean that in an environment of enhanced protein-protein contact, such as in an aggregated state, the helix in the native structure is less thermodynamically stable than a β-strand--loop-- β-strand conformation. That the G334V mutant is more prone to aggregation than the WT in aqueous solution at pH 7.5 was demonstrated by analytical ultracentrifugation experiments (described above). This may, at least in part, be due to the greater hydrophobic character of valine in the mutant compared to glycine in the WT sequence.
Several groups have proposed a two-stage model for the mechanism of amyloid formation (15, 55, 56). We found that while there was a relatively rapid change in the G334V peptide secondary structure at 37°C over about 2 hr toward a β-like conformation (Figure 2C), the rate of ThT fluorescence increase (Figure 4A, indicating amyloid formation) and the rate of peptide precipitation (data not shown) were much slower. These findings suggest a two-stage model for amyloid formation by the G334V p53 mutant. In the first stage, the G334V peptide, which folds into a native-like tetramer structure under physiological conditions at elevated concentrations, forms a partially unfolded intermediate with an extended region of β-structure. When conditions are suitable, this first intermediate is converted to a second intermediate, the structure of which should be similar to that of protofibrils. At this stage, the oligomerization process is achieved by conformational reorganization to slowly form the mature amyloid-like fibril. The first stage happens relatively quickly under physiological conditions and is accelerated at acidic pH. Globular aggregates are formed directly from the first intermediate.
Interestingly, Stucki et al. (57) reported that a mutation that changes Gly292 in human fibrinogen to Val, which is observed in the Fibrinogen St. Gallen I and the Fibrinogen variant Baltimore I, led to an opening of a loop in the backbone chain compared to the structure of normal fibrinogen and the loss of a hydrogen bond, resulting in defective polymerization and fibrinogenolysis. However, this amino acid change is in the middle of an extended turn rather than between an α-helix and a β-strand. A mutation that causes a valine-for-glycine change also is associated with Gerstmann-Straussler-Scheinker syndrome, a rare form of prion disease (58). Our study thus provides complementary further evidence for the existence of a linkage between mutations that change Gly to Val and the formation of misfolded amyloid proteins, and it highlights the importance of a molecular classification of disorders associated with protein misfolding.
In contrast to the results of Atz et al. (59), we found that the G334V mutant p53 peptide formed homotetramers, but these were only stable at high concentrations (>10 μM) that most likely were much higher than those explored by Atz et al. We also found that the G334V peptide interacts with wild-type p53 to form heterotetramers (Figure 7A), confirming, in this case, the findings of Atz et al. (59). Hetero-oligomerization of the G334V mutant with wild-type p53 would be expected to destabilize the wild-type p53 tetramer, resulting in a loss of efficient site-specific DNA binding and a consequent compromise of activity as a transcription factor for either activation or repression. It should be noted, however, that in vivo the ability to form homo- or hetero-oligomers also may be modulated by the present of DNA binding sites and by the presence of several proteins that recently have been shown to specifically interact with the tetramerization domain. Mutant p53 with Arg337 changed to Cys, a change which is associated with Li-Fraumeni-like syndrome, also forms hetero-tetramers with wild-type p53; however, the hetero-tetramer retains some functional activity including DNA binding and an ability to activate transcription of some genes (49, 59, 60). However, the thermal stability of the R337C mutant is much lower than that of wild-type p53, and at physiological temperatures, less than half of this mutant is tetrameric. Another tetramerization mutant, L330H, also forms heterotetramers with the wild-type p53 protein (unpublished data). Thus, hetero-oligomerization could be a common mechanism for inactivation of wild-type p53 by mutations occurring in the tetramerization domain. However, it remains to be determined if p53G334V forms amyloid fibrils in vivo, either on its own or with wild-type p53, which might further reduce the available functional p53 within precancerous cells harboring this mutation.
In summary, our findings show a mutation which changes Gly334 to Val induced formation of amyloid fibrils through an α−β transition conformational change; the mutant peptide also formed hetero-tetramers with wild-type p53 under physiological conditions. Mutations in at least 23 of 30 codons that specify the p53 tetramerization domain are associated with tumors. Biochemical characterization of tetramerization domain mutants, such as that reported here, will be important for the development of drugs that restore functionality to p53 with these mutations.
Supplementary Material
ABBREVIATIONS
- CD
circular dichroism
- DIEA
N,N-diisopropylethyl amine
- G334V
mutant p53 peptide with Val in place of Gly at p53 amino acid position 334
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HOBt
1-hydroxybenzotriazole
- L330A
mutant p53 peptide with Ala at position 330
- GdnHCl
guanidine hydrochloride
- TFA
trifluoroactic acid
- TFE
trifluoroethanol
- ThT
thioflavin T
- WT
wild-type p53 peptide
Footnotes
KS was supported in part by Grants-in-Aid for Scientific Research on Priority Areas 16041202 and Scientific Research (B)(2) 14380290 from the Ministry of Education, Culture, Sports, Science and Technology of Japan. YH was supported in part by Grants-in-Aid for Young Scientists (B) 15770073. CWA was supported in part by a Laboratory Directed Research and Development Award at the Brookhaven National Laboratory under contract with the U.S. Department of Energy. SRD and EA were supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
REFERENCES
- 1.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 3.Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Biol. 1994;1:877–890. doi: 10.1038/nsb1294-877. [DOI] [PubMed] [Google Scholar]
- 5.Clore GM, Ernst J, Clubb R, Omichinski JG, Kennedy WMP, Sakaguchi K, Appella E, Gronenborn AM. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat. Struct. Biol. 1995;2:321–333. doi: 10.1038/nsb0495-321. [DOI] [PubMed] [Google Scholar]
- 6.Jeffrey PD, Gorina S, Pavletich NP. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science. 1995;267:1498–1502. doi: 10.1126/science.7878469. [DOI] [PubMed] [Google Scholar]
- 7.Mittl PRE, Chène P, Grütter MG. Crystallization and structure solution of p53 (residues 326−356) by molecular replacement using an NMR model as template. Acta Crystallogr. D Biol. Crystallogr. 1998;54:86–89. doi: 10.1107/s0907444997006550. [DOI] [PubMed] [Google Scholar]
- 8.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 9.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 10.D'Amico D, Carbone D, Mitsudomi T, Nau M, Fedorko J, Russell E, Johnson B, Buchhagen D, Bodner S, Phelps R, Gazdar A, Minna JD. High frequency of somatically acquired p53 mutations in small-cell lung cancer cell lines and tumors. Oncogene. 1992;7:339–346. [PubMed] [Google Scholar]
- 11.Fujita T, Kiyama M, Tomizawa Y, Kohno T, Yokota J. Comprehensive analysis of p53 gene mutation characteristics in lung carcinoma with special reference to histological subtypes. Int. J. Oncol. 1999;15:927–934. doi: 10.3892/ijo.15.5.927. [DOI] [PubMed] [Google Scholar]
- 12.Hainaut P, Pfeifer GP. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–374. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 13.Dumoulin M, Dobson CM. Probing the origins, diagnosis and treatment of amyloid diseases using antibodies. Biochimie. 2004;86:589–600. doi: 10.1016/j.biochi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y, Ueno A, Mihara H. Optimization of hydrophobic domains in peptides that undergo transformation from α-helix to β-fibril. Bioorg. Med. Chem. 1999;7:77–185. doi: 10.1016/s0968-0896(98)00236-3. [DOI] [PubMed] [Google Scholar]
- 16.Sipe JD, Cohen AS. Review: history of the amyloid fibril. J. Struct. Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 17.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 18.Serpell LC. Alzheimer's amyloid fibrils: structure and assembly. Biochim. Biophys. Acta. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 19.Dobson CM. The structural basis of protein folding and its links with human disease. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:133–145. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohnishi S, Takano K. Amyloid fibrils from the viewpoint of protein folding. Cell. Mol. Life Sci. 2004;61:511–524. doi: 10.1007/s00018-003-3264-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson J. Molecular determinants for amyloid fibril formation: lessons from lung surfactant protein. C. Swiss Med. Wkly. 2003;133:275–282. doi: 10.4414/smw.2003.09987. [DOI] [PubMed] [Google Scholar]
- 22.Thirumalai D, Klimov DK, Dima RI. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 23.Zerovnik E. Amyloid-fibril formation. Proposed mechanisms and relevance to conformational disease. Eur. J. Biochem. 2002;269:3362–3371. doi: 10.1046/j.1432-1033.2002.03024.x. [DOI] [PubMed] [Google Scholar]
- 24.Rochet JC, Lansbury PT., Jr. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 25.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 26.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 27.Sreerema N, Woody RW. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- 28.Klunk WE, Pettegrew JW, Abraham DJ. Quantitative evaluation of congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J. Histochem. Cytochem. 1989;7:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 29.Levine H. Thioflavin-T interaction with amyloid β-sheet structures. Amyloid. 1995;2:1–6. [Google Scholar]
- 30.Sakamoto H, Lewis MS, Kodama H, Appella E, Sakaguchi K. Specific sequences from the carboxyl terminus of human p53 gene product form anti-parallel tetramers in solution. Proc. Natl. Acad. Sci. U.S.A. 1994;91:8974–8978. doi: 10.1073/pnas.91.19.8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaguchi K, Sakamoto H, Lewis MS, Anderson CW, Erickson JW, Appella E, Xie D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry. 1997;36:10117–10124. doi: 10.1021/bi970759w. [DOI] [PubMed] [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983;4:187–217. [Google Scholar]
- 34.MacKerell AD, Jr., Bashford D, Bellott M, Dunbrack RL, Jr., Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics Studies of proteins. J. Physical Chemistry B. 1998;102:586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 35.Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry. 1974;13:222–245. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 36.Rost B. Review: Protein secondary structure continues to rise. J. Struct. Biol. 2001;134:204–218. doi: 10.1006/jsbi.2001.4336. [DOI] [PubMed] [Google Scholar]
- 37.Rost B, Yachdav G, Liu J. The PredictProtein Server. Nucleic Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon S, Welsh WJ. Detecting hidden sequence propensity for amyloid fibril formation. Protein Sci. 2004;13:2149–2160. doi: 10.1110/ps.04790604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon S, Welsh WJ. Rapid assessment of contact-dependent secondary structure propensity: relevance to amyloidogenic sequences. Proteins. 2005;60:110–117. doi: 10.1002/prot.20477. [DOI] [PubMed] [Google Scholar]
- 40.Lawless MK, Barney S, Guthrie KI, Bucy TB, Petteway SR, Jr., Merutka G. HIV-1 membrane fusion mechanism: structural studies of the interactions between biologically-active peptides from gp41. Biochemistry. 1996;35:13697–13708. doi: 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- 41.Bothner B, Lewis WS, DiGiammarino EL, Weber JD, Bothner SJ, Kriwacki RW. Defining the molecular basis of Arf and Hdm2 interactions. J. Mol. Biol. 2001;314:263–277. doi: 10.1006/jmbi.2001.5110. [DOI] [PubMed] [Google Scholar]
- 42.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 43.Lau SYM, Taneja AK, Hodges RS. Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded alpha-helical coiled-coils. J. Biol. Chem. 1984;259:13253–13261. [PubMed] [Google Scholar]
- 44.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 45.Kallberg Y, Gustafsson M, Persson B, Thyberg J, Johansson J. Prediction of amyloid fibril-forming proteins. J. Biol. Chem. 2001;276:12945–12950. doi: 10.1074/jbc.M010402200. [DOI] [PubMed] [Google Scholar]
- 46.Ishimaru D, Andrade LR, Teixeira LSP, Quesado PA, Maiolino LM, Lopez PM, Cordeiro Y, Costa LT, Heckl WM, Weissmüller G, Foguel D, Silva JL. Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry. 2003;42:9022–9027. doi: 10.1021/bi034218k. [DOI] [PubMed] [Google Scholar]
- 47.Friedler A, Veprintsev DB, Hansson LO, Fersht AR. Kinetic instability of p53 core domain mutants: implications for rescue by small molecules. J. Biol. Chem. 2003;278:24108–24112. doi: 10.1074/jbc.M302458200. [DOI] [PubMed] [Google Scholar]
- 48.Lee AS, Galea C, DiGiammarino EL, Jun B, Murti G, Ribeiro RC, Zambetti G, Schultz CP, Kriwacki RW. Reversible amyloid formation by the p53 tetramerization domain and a cancer-associated mutant. J. Mol. Biol. 2003;327:699–709. doi: 10.1016/s0022-2836(03)00175-x. [DOI] [PubMed] [Google Scholar]
- 49.Davison TS, Yin P, Nie E, Kay C, Arrowsmith CH. Characterization of the oligomerization defects of two p53 mutants found in families with Li-Fraumeni and Li-Fraumeni-like syndrome. Oncogene. 1998;17:651–656. doi: 10.1038/sj.onc.1202062. [DOI] [PubMed] [Google Scholar]
- 50.Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, DeLacerda L, Rabin M, Cadwell C, Sampaio G, Cat I, Stratakis CA, Sandrini R. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9330–9335. doi: 10.1073/pnas.161479898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mateu MG, Fersht AR. Nine hydrophobic side chains are key determinants of the thermodynamic stability and oligomerization status of tumour suppressor p53 tetramerization domain. EMBO J. 1998;17:2748–2758. doi: 10.1093/emboj/17.10.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi Y, Ueno A, Mihara H. Mutational analysis of designed peptides that undergo structural transition from α-helix to β-sheet and amyloid fibril formation. Structure. 2000;8:915–925. doi: 10.1016/s0969-2126(00)00183-0. [DOI] [PubMed] [Google Scholar]
- 53.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in Amyloid _-protein fibrillogenesis. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 54.Fezoui Y, Teplow DB. Kinetic studies of amyloid β-protein fibril assembly. Differential effects of α-helix stabilization. J. Biol. Chem. 2002;277:36948–36954. doi: 10.1074/jbc.M204168200. [DOI] [PubMed] [Google Scholar]
- 55.Modler AJ, Gast K, Lutsch G, Damaschun G. Assembly of amyloid protofibrils via critical oligomers--a novel pathway of amyloid formation. J. Mol. Biol. 2003;325:135–148. doi: 10.1016/s0022-2836(02)01175-0. [DOI] [PubMed] [Google Scholar]
- 56.Gorman PM, Yip CM, Fraser PE, Chakrabartty A. Alternate aggregation pathways of the Alzheimer β-amyloid peptide: Ab association kinetics at endosomal pH. J. Mol. Biol. 2003;325:743–757. doi: 10.1016/s0022-2836(02)01279-2. [DOI] [PubMed] [Google Scholar]
- 57.Stucki B, Schmutz P, Schmid L, Haeberli A, Lämmle B, Furlan M. Fibrinogen St. Gallen I (g 292 Gly--> Val): evidence for structural alterations causing defective polymerization and fibrinogenolysis. Thromb. Haemost. 1999;81:268–274. [PubMed] [Google Scholar]
- 58.Panegyres PK, Toufexis K, Kakulas BA, Cernevakova L, Brown P, Ghetti B, Piccardo P, Dlouhy SR. A new PRNP mutation (G131V) associated with Gerstmann-Sträussler-Scheinker disease. Arch. Neurol. 2001;58:1899–1902. doi: 10.1001/archneur.58.11.1899. [DOI] [PubMed] [Google Scholar]
- 59.Atz J, Wagner P, Roemer K. Function, oligomerization, and conformation of tumor-associated p53 proteins with mutated C-terminus. J. Cell Biochem. 2000;76:572–584. doi: 10.1002/(sici)1097-4644(20000315)76:4<572::aid-jcb6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 60.Lomax ME, Barnes DM, Hupp TR, Picksley SM, Camplejohn RS. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene. 1998;17:643–649. doi: 10.1038/sj.onc.1201974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.