

Abstract
The endonuclease dicer cleaves RNAs that are 100% double stranded and certain RNAs with extensive but <100% pairing to release ∼21-nucleotide (nt) fragments. Circular 1,679-nt genomic and antigenomic RNAs of human hepatitis delta virus (HDV) can fold into a rod-like structure with 74% pairing. However, during HDV replication in hepatocytes of human, woodchuck, and mouse origin, no ∼21-nt RNAs were detected. Likewise, in vitro, purified recombinant dicer gave <0.2% cleavage of unit-length HDV RNAs. Similarly, rod-like RNAs of potato spindle tuber viroid (PSTVd) and avocado sunblotch viroid (ASBVd) were only 0.5% cleaved. Furthermore, when a 66-nt hairpin RNA with 79% pairing, the putative precursor to miR-122, which is an abundant liver micro-RNA, replaced one end of HDV genomic RNA, it was poorly cleaved, both in vivo and in vitro. In contrast, this 66-nt hairpin, in the absence of appended HDV sequences, was >80% cleaved in vitro. Other 66-nt hairpins derived from one end of genomic HDV, PSTVd, or ASBVd RNAs were also cleaved. Apparently, for unit-length RNAs of HDV, PSTVd, and ASBVd, it is the extended structure with <100% base pairing that confers significant resistance to dicer action.
There is an extensive literature on the interaction of double-stranded RNA-specific proteins with highly structured single-stranded RNAs. There are host proteins that bind to and are activated by such RNAs. Examples include protein kinase R (PKR) and oligo(A) synthetase (3, 24). In addition, there is a family of adenosine deaminases, ADAR, that recognize such RNA structure and can convert adenosine to inosine (5). Finally, there is a family of RNase III-related endoribonucleases that can cleave such structured RNAs (15). At another level, many viruses produce proteins that will bind to structured regions on their viral RNAs and protect them against interactions with such host proteins. Alternatively, some viruses express small structured RNAs that act as decoys; for example, such RNAs can bind PKR but fail to activate its kinase activity (45).
Our studies are concerned with the structure and replication of hepatitis delta virus (HDV). The 1,679-nucleotide (nt) single-stranded circular RNA genome of HDV is replicated by RNA-directed RNA synthesis, most probably using host RNA polymerase II (49). Three discretely sized HDV RNA species accumulate during replication: the genome; its exact complement, the antigenome; and relatively small amounts of an 800-nt polyadenylated RNA (of the same polarity as the antigenome), which is translated to produce a 195-amino-acid protein known as the small delta antigen, or δAg-S. Both the genome and antigenome of HDV are predicted to fold into an unbranched rod-like structure, with intramolecular base pairing of 74% of all nucleotides (25).
Previous studies have shown that regions of these structured RNAs of HDV are substrates for activation of PKR (14, 44) and for deamination by ADAR proteins. It is also known that the one viral protein produced by HDV replication, the delta antigen, is a basic protein with some specificity for binding to the structure of the genomic and antigenomic RNAs (9). Furthermore, this essential protein is known to reduce the ability of ADAR proteins to edit HDV RNA (13).
In this study, we focus on the question of whether the endonuclease dicer, an important member of the RNase III family, acts on HDV RNAs during their replication. Dicer is central to the phenomenon of RNA interference (RNAi) (2). It cleaves RNA substrates that are 100% double stranded into double-stranded RNA fragments of about 21 nt called small interfering RNAs (siRNAs) (4, 42, 59). It is known that these siRNAs can then be incorporated into an RNA-induced silencing complex to generate a specific endonuclease activity that cleaves related mRNA species (18).
However, dicer sometimes cleaves RNAs that possess intramolecular secondary structure with <100% base pairing. It thus releases single-stranded fragments, also of ∼21 nt, that are called micro-RNAs (miRNAs) (23, 30, 42). These miRNAs can be shown to interfere with the translation of related mRNAs (58) and sometimes with mRNA stability (57). At least a few of the miRNAs are known to play important roles in cell differentiation and developmental patterning in eukaryotic organisms ranging from nematodes to humans (27, 36, 38). It has recently been estimated that the human genome encodes as many as 255 different miRNA species (32).
Table 1 summarizes data not only for HDV RNA but also for other RNAs with similar levels of double-strandedness that may be valuable precedents for our study of dicer sensitivity. The first is the putative precursor to miR-122, which is possibly the most abundant miRNA in the mammalian liver (27). This precursor is 66 nt long and is predicted to have a hairpin structure with 79% base pairing (see Fig. 4A). The very existence of miR-122 supports the hypothesis that dicer is both present and active in the mammalian liver. Our studies provide the first in vitro evidence for the expected release of the 22- to 23-nt miR-122 from this precursor.
TABLE 1.
Features of RNAs with <100% intramolecular base pairinga
| RNA species | No. of nucleotides | % Predicted base pairing | Presence of ≈21-nt RNA speciesb |
|---|---|---|---|
| miR-122 precursor | 66 | 79 | + |
| PSTVd | 359 | 70 | + |
| ASBVd | 247 | 67 | − |
| HDV genomic RNA | 1,679 | 74 | − |
The sources for data listed in this table are cited in the text.
This refers to whether or not such species have been detected in vivo. The datum for HDV refers to this study.
FIG. 4.

Predicted secondary structures for four different 66-nt hairpin RNAs. (A) Putative precursor to miR-122, as previously reported (27), with a 22-nt miR-122 cleavage product indicated by shading. (B) Structure for nt 760 to 825, one end of the predicted rod-like structure for a published sequence of genomic HDV RNA (25). The boxed sequence on HDV indicates the site at which we removed 13 nt of HDV sequence and replaced it with the 66 nt of the miR-122 precursor. (C) One end of the rod-like folding of PSTVd RNA (22). (D) A similar region from ASBVd RNA (35).
Also shown Table 1 are data for the RNAs of two plant viroids. We have previously seen numerous analogies between viroids and HDV (48). These analogies include genomes that are small single-stranded RNAs in a circular conformation and, of particular interest here, the ability of these RNAs to fold into an unbranched rod-like structure via significant levels of intramolecular base pairing. As indicated in the table, potato spindle tuber viroid (PSTVd) and avocado sunblotch viroid (ASBVd) have 70 and 67% predicted base pairing, respectively (22). Recent reports have shown that ∼21-nt RNAs can be detected during the replication of PSTVd (21, 41) but not that of ASBVd (34). The result for PSTVd is puzzling in that PSTVd is considered to replicate in the nucleus (19), while dicer has been reported to be largely cytoplasmic (4). HDV RNAs are also considered to be transcribed in the nucleus (17), but there are also reports that after transcription, these RNAs can shuttle between the nucleus and cytoplasm (47) or even be rapidly transported to the cytoplasm (33).
Therefore, the initial aim of our studies was to determine whether in vivo, during HDV replication, or in vitro, in the presence of purified recombinant human dicer, the RNAs of HDV might act as substrates for dicer-mediated cleavage to ∼21-nt fragments. Our initial studies indicated that under both circumstances, ∼21-nt HDV fragments were not detectable (<1%). Further studies were then undertaken to understand the basis for this phenomenon. We thus found that 66-nt hairpin RNAs based on the sequences and predicted structures of HDV, PSTVd, and ASBVd RNAs, as well as those of the putative precursor to miR-122, had a significantly increased sensitivity to dicer action. From these results, we propose that it is the extended structure with <100% pairing that confers significant resistance against dicer to full-length RNAs.
MATERIALS AND METHODS
Plasmids.
Plasmids pDL553 and pDL448 were described previously (28, 29). Briefly, pDL553 transcribes 1.2× the unit-length HDV genomic sequence under control of a simian virus 40 (SV40) promoter. pDL448 expresses a truncated form of the HDV small antigen. pJC145 was constructed by using the 66-nt miR-122 precursor (see Fig. 4A) to replace 13 nt of HDV sequence in pDL553 (Fig. 4B) by a previously described strategy (56). pJC144 was constructed by inserting unit-length PSTVd sequence into the BamHI site of pcDNA3 vector (Invitrogen), in the genomic orientation; similarly, pJC125 carries the genes for a dimer of the ASBVd sequence. In both cases, RNA was then transcribed in vitro using T7 RNA polymerase. pSVTVA expresses both T antigen and VA genes and enhances in vivo transcription from transfected SV40-based vectors (1).
Cell culture and transfection.
Huh7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Transfection was performed by using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen).
Northern blot analysis.
Total RNA was extracted with Tri Reagent (Molecular Research Center) and loaded onto gels of either 2% agarose-formaldehyde or 15% polyacrylamide with 7 M urea. Electrophoresis and electrotransfer were performed as described previously (6). HDV RNA probes were prepared by T7 transcription with [α-32P]UTP (Perkin-Elmer). miR-122 was detected by use of a 5′-32P-labeled 22-nt DNA containing the sequence complementary to miR-122. High-stringency hybridization was performed at 65°C in Ekono hybridization solution (Research Products International). Lower-stringency hybridization was performed at 50°C; materials were washed at 50°C, with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) plus 1% sodium dodecyl sulfate.
Western blot analysis.
Protein from transfected cells was extracted and analyzed by a standard procedure (26), with immunoblots carried out by use of rabbit anti-delta virus antibody followed by 125I-staph A protein (Perkin-Elmer) and detection by a bioimager (Fuji).
In vitro RNA transcription.
Most in vitro RNA transcriptions were performed with the RiboMAX large-scale RNA production system T7 (Promega). Double-stranded RNA (700 nt) was made by using a PCR-amplified template with a T7 promoter at both ends. Unit-length HDV genomic and antigenomic RNAs were transcribed by expression PCR as previously described (8). HDV unit-length genomic RNA with an insertion of miR-122 precursor was transcribed by expression PCR, with pJC145 as template. PSTVd and ASBVd RNAs were transcribed by use of pJC144 and pJC125 as templates. [α-32P]UTP was added to the reaction when radioactively labeled RNA was needed.
For the experiments shown in Fig. 6, labeled RNAs of 66 nt, corresponding to the predicted precursor of miR-122 and sequences from HDV, PSTVd, and ASBVd (see Fig. 4), were made by use of a Silencer kit (Ambion) along with chemically synthesized DNA templates.
FIG. 6.

Action of dicer on full-length and 66-nt hairpin forms of HDV and other RNAs. Seven different species of 32P-labeled RNA were analyzed on gels of 15% acrylamide-7 M urea following incubation without (−) or with (+) dicer. The RNAs used were as follows: lanes 1 and 2, 100% double-strand (ds) RNA; lanes 3 and 4, unit-length genomic HDV RNA; lanes 5 and 6, unit-length modified HDV genomic RNA; lanes 7 and 8, 66-nt miR-122 precursor; lanes 9 and 10, 66-nt HDV genomic hairpin; lanes 11 and 12, 66-nt PSTVd RNA hairpin; lanes 13 and 14, 66-nt ASBVd RNA hairpin. After electrophoresis, the RNAs were electrotransferred to a nylon membrane, after which 32P was quantitated by use of a bioimager. Size markers p and q were as described for Fig. 1.
In vitro dicer reactions.
Typically, each digestion (10 μl) contained 1 μg of 100% double-stranded unlabeled RNA as a positive control for the dicer reaction, together with trace amounts of radiolabeled RNA. With or without the addition of 1 U of recombinant human dicer (Gene Therapy Systems), the mix was then incubated for 16 h at 37°C. (An exception was that for Fig. 3, lanes 11 and 12, 1 μg of each unlabeled double-stranded RNA and HDV RNA was used.) Reactions were terminated with the provided stop solution, after which aliquots were analyzed on gels of 3% nondenaturing agarose and 15% polyacrylamide gels with 7 M urea. Unlabeled RNA was detected by staining with ethidium bromide followed by illumination with short-wave UV light and image capture with a Kodak digital system, after which the image intensities were inverted.
FIG. 3.

Action of recombinant dicer on HDV and other RNAs. Lanes 1 to 12, RNA samples either with (+) or without (−) dicer during incubation for 16 h at 37°C. Each sample contained 1 μg of double-stranded RNA. To each sample was added a 32P-labeled RNA as follows: lanes 1 and 2, genomic HDV RNA; lanes 3 and 4, antigenomic RNA; lanes 5 and 6, PSTVd RNA; lanes 7 and 8, ASBVd RNA; and lanes 9 and 10, modified genomic HDV RNA. For lanes 11 and 12, we added 1 μg of modified genomic RNA that was not labeled. The modification, as described in the text and in the legend for Fig. 4, was to replace some HDV sequences with those of the predicted precursor to miR-122. (A) Aliquots of each sample were analyzed in nondenaturing gels of 3% agarose followed by ethidium bromide staining and detection by digital imaging. (B) Aliquots were analyzed in gels of 15% polyacrylamide-7 M urea, after which the RNAs were electrotransferred to a nylon membrane. In lanes 11 and 12, miR-122 sequences were detected by low-stringency hybridization using a radiolabeled oligonucleotide probe. 32P was detected by use of bioimager. p, 1-kb DNA ladder; q, 21-bp RNA.
RESULTS
Can siRNA be detected during HDV replication?
In a search for siRNA species associated with HDV replication, we tested seven different cellular RNA samples, as summarized in Table 2. These included RNAs from livers of woodchucks isolated during chronic and acute infections, RNAs from Huh7 cells (39) following both transient transfection and the generation of a stably transfected cell line, and liver RNAs from mice that were transfected with HDV DNA (7). Northern analyses of these seven RNAs are shown in Fig. 1. For panels A to D, the RNAs were first separated by electrophoresis on 2% agarose gels in the presence of formaldehyde. Panels A and B show the detection of genomic and antigenomic HDV RNAs, respectively. In both cases, the major species was unit-length HDV RNA.
TABLE 2.
Sources of RNAs tested for presence of ≈21-nt RNA
| Sample no. | Sourcea |
|---|---|
| 1 | Human Huh7 cells, HDV replication initiated by transient DNA transfection, day 4 |
| 2 | Human Huh7 cells, HDV replication initiated by transient DNA transfection, day 15 |
| 3 | Human Huh7 cells, HDV replication initiated by stable DNA transfection |
| 4 | Woodchuck liver, acute infection, day 21 |
| 5 | Woodchuck liver, chronic infection |
| 6 | Mouse liver, replication initiated by transient DNA transfection, day 5 |
| 7 | Mouse liver, replication initiated by transient DNA transfection, day 20 |
Samples 1 and 2 were obtained by transfection with pDL553. Sample 3 was from a cell line stably transfected with HDV cDNA (12). Samples 4 and 5 were obtained from infected woodchucks (40). Samples 6 and 7 were obtained from mice that had been transfected with HDV cDNA by a hydrodynamics-based procedure (7).
FIG. 1.

Northern gel analyses of RNAs from various sources of HDV genome replication. The seven sources of RNA are those listed in Table 2. As indicated, RNAs were separated in gels of either 2% agarose-formaldehyde (A to D) or 15% acrylamide-7 M urea (E and F). (A and B) Analysis to detect HDV genomic and antigenomic RNAs, respectively. (C to F) The amount of RNA analyzed was increased from 1 to 20 μg and the exposure times were increased 10-fold. Panels D and F show hybridization to detect antigenomic RNA, but under low-stringency conditions. Panels C and E show hybridization at low stringency to detect miR-122 (27). Lanes p to r represent end-labeled markers as follows: p, 1-kb ladder; q, 21-nt RNA; and r, 42-nt DNA. Lane s, 21-nt antigenomic RNA sequence used as a positive control for the ability of our low-stringency hybridization conditions to detect such a species. In panels E and F, the greater sample mass for lanes 1 to 7 slowed migration in 15% acrylamide relative to the standards q and s.
In order to detect possible ∼21-nt species, two steps were taken. First, we increased the amount analyzed by Northern blotting 20-fold, from 1 to 20 μg. Second, we lowered the stringency of hybridization for the Northern blot from 65 to 50°C in order to better allow stability of short hybrids. As shown in panel D, we were still unable to detect ∼21-nt fragments of antigenomic RNA. Similarly, no such genomic fragments were detected (data not shown). As a positive control for hybridization, we were able to detect a 21-nt HDV RNA sequence (lanes s), as transcribed in vitro. In addition, as shown in panel C, when the filter was rehybridized to detect miR-122, we could detect this species in all samples, especially the woodchuck and mouse liver samples. In the mouse, miR-122 is known to be the most abundant miRNA in the liver (27), but until this study, there have been no corresponding data for miR-122 in either human or woodchuck livers. It should also be noted that the species of ≥100 nt, as detected for the seven samples in panel D, were also detected in RNA samples lacking HDV sequences (data not shown). Therefore, we speculate that these species of ≥100 nt probably represent hybridization at lower stringency to fragments of abundant host rRNA sequences.
In order to increase the electrophoretic separation and resolution of ∼21-nt RNA species, we moved from using agarose gels to using 15% polyacrylamide gels in the presence of 7 M urea, as shown in panels E and F. As for panels C and D, the amount of RNA sample analyzed was 20 μg. Again, under conditions of lowered hybridization stringency, we were unable to detect ∼21-nt RNAs related to HDV (panel F), and yet we were able to detect hybridization to a 21-nt HDV RNA control (lane s). Also, as shown in panel E, by rehybridization at the same lower stringency, we were able to detect miR-122. It should also be mentioned that we used size controls (lanes p to s in panels A to D and lanes q in panels E and F) to demonstrate the separation and transfer of low-molecular-weight species. We also tested a method developed by others, in which prior to electrophoresis, a step is used to enrich for low-molecular-weight species (16). This step also failed to achieve the detection of ∼21-nt HDV RNAs (data not shown).
Does HDV genome replication induce RNAi against HDV mRNA species?
While the above experiments were unable to detect HDV-specific ∼21-nt fragments during HDV replication, it remained possible that an RNAi effect was present but not detected by our assay. We hypothesized that if such an activity were present, it would inhibit the accumulation of the HDV δAg-S, either by destroying the HDV mRNA, as for siRNA, or by blocking the translation of that mRNA, as for miRNA. A prior observation that could be considered supportive of this hypothesis is that the HDV mRNA species is about 50 times less abundant than unit-length antigenomic RNAs (11), so we considered a second hypothesis, that this mRNA, being cytoplasmic, might have been the remainder after some form of RNAi-like attack. To test these two hypotheses, we devised the following experiment.
We expressed from a DNA construct an mRNA with a small in-frame deletion in the open reading frame for δAg-S. This construct, pDL448, as previously described (29), has a deletion in the code that corresponds to the coiled-coil domain. It produces a protein, δAg-ΔS, that is stably expressed but neither supports nor inhibits HDV replication. We then determined whether prior transfection or cotransfection with HDV DNA to achieve HDV genome replication could inhibit expression of the mutated protein. That is, we asked whether HDV replication could induce an RNAi effect, via either siRNA or miRNA, that would act to reduce the expression of δAg-ΔS.
As shown in Fig. 2, we assayed by immunoblotting at day 3 for the accumulation of δAg-ΔS as well as of δAg-S that arises during HDV replication. No inhibition of the accumulation of δAg-ΔS was detected when HDV replication was initiated at the time of transfection (lane 3) or even two days earlier (lane 2). (Similar data were obtained from cells harvested at days 2 and 4 [data not shown].) We estimate that we could have readily detected as little as 20% inhibition of δAg-ΔS expression relative to the control (lane 1).
FIG. 2.
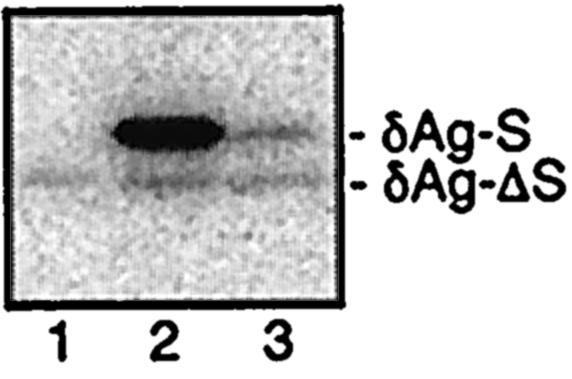
HDV genome replication did not lead to silencing of a related mRNA species. At day 0, cells were transfected with pDL448 to express δAg-ΔS, a form of δAg with a deletion. At days −2 and day 0, some of these cells (lanes 2 and 3, respectively) were transfected with pDL553 to initiate HDV genome replication and the expression of δAg-S. Another culture was not transfected with pDL553 (lane 1). Total protein was harvested at day 3 and examined by immunoblot to detect the two delta protein species.
From the results of these experiments, we reject the hypothesis that HDV replication induces an RNAi effect. Furthermore, we discount the related hypothesis that the relatively low levels of HDV mRNA detected during replication represent the consequences of siRNA activity.
Will dicer act in vitro on HDV RNAs?
Since the above in vivo studies provided no evidence that HDV RNAs were being degraded by dicer in vivo, we set about to test in vitro whether dicer could act on HDV RNAs. For these studies, we prepared unit-length radioactively labeled genomic and antigenomic HDV RNAs. To these, as an internal positive control for the action of recombinant human dicer, we added unlabeled 700-bp double-stranded RNA, made from an unrelated nucleotide sequence. As shown in Fig. 3A, lanes 1 to 4, treatment with dicer converted most of the double-stranded RNA to fragments with mobilities as expected for approximately double-stranded ∼21-nt siRNA (lanes 2 and 4). (In separate control studies, as in Fig. 6, lane 2, using 100% double-stranded RNA that was radiolabeled, we quantitated the extent of digestion to be 85%.) Panel B shows that in contrast, <0.2% of the genomic (lane 2) or antigenomic (lane 4) RNAs were converted to ∼21-nt species by such treatment. Moreover, we did not detect the creation of RNA fragments of sizes intermediate between uncut and 21 bp, which might have been indicative of partial cleavage.
As a follow-up to these negative results, we used similar in vitro assays to determine whether dicer would act on viroid RNAs. We tested RNAs of PSTVd and ASBVd. Only a small fraction of these RNAs (0.5%) underwent digestion to release a band of ∼21 nt (lanes 6 and 8, respectively).
Given these results, we next asked whether the putative precursor to miR-122 would be cleaved with efficiency by dicer. To do this, we modified one end of HDV genomic RNA, with the intent of being able to test for dicer action both in vivo and in vitro and with the expectation that the precursor sequences would make HDV sensitive to dicer. As indicated in Fig. 4, we removed 13 nt from one end of the HDV genomic RNA (panel B) and replaced it with the 66-nt miR-122 precursor (panel A).
Consider first the ability of the modified HDV RNA to be cleaved in vitro by dicer. As can be seen in Fig. 3, lanes 9 and 10, we did detect the release of small amounts (0.7% ± 0.2%) of a band consistent with being just longer than 21 nt. The expected size of miR-122 is 22 to 23 nt. To test whether this was in fact the miR-122 sequence, we digested a corresponding unlabeled RNA (lanes 11 and 12) and examined it by Northern blotting using a probe for miR-122. As shown, we thus detected relatively small amounts (0.3% ± 0.1%) of a species with the expected mobility of miR-122. When these same samples (lanes 11 and 12) were hybridized at low stringency with a probe to detect HDV sequences, no ∼21-nt species were detected (data not shown). Thus, we interpret that dicer cleaved miR-122 from the modified HDV but that it did not release detectable levels of HDV-specific ∼21-nt RNA. It should be noted for panel B, lane 12, that since we used an miR-122 oligonucleotide probe, 0.3% ± 0.1% represents the molar amount rather than the mass amount of miR-122 that was released.
Because the efficiency of this cleavage was so low in vitro, further studies were carried out to determine the efficiency in vivo. To do this, we transfected Huh7 cells with DNA constructs to express HDV RNA modified to contain the miR-122 precursor sequences. Northern blot analyses to detect HDV genomic and antigenomic RNAs showed that this modified HDV RNA did not replicate significantly (Fig. 5A, lane 2), unlike the unmodified RNA (lane 1). This inhibition of replication was not totally unexpected, since we have previously found that even small changes to the HDV RNA sequence can inhibit replication (56). Therefore, as an alternative approach, in the absence of HDV replication and using an enhanced transient transfection procedure, we were able to achieve high levels of accumulation of the modified genomic RNA that was processed to unit length (Fig. 5A, lane 3). We asked whether any of the miR-122 sequences were being processed out of the modified HDV RNAs. Such processing could have created an increase from the endogenous level of miR-122. We therefore used analysis in both 2% agarose (Fig. 5B) and 15% polyacrylamide (Fig. 5C), followed by hybridization using an oligonucleotide probe specific for miR-122. As shown, we could readily detect the endogenous miR-122, but we found no significant changes in the amount of this species as a consequence of either HDV replication (Fig. 5B and C, lanes 1) or accumulation of nonreplicating modified HDV (Fig. 5B and C, lanes 3). In terms of the latter RNA, we also observed that there was a relatively large amount of uncleaved RNA and a 6% amount of RNA cleaved to about the size of the putative 66-nt miR-122 precursor. If this species (asterisk in the figure) is in fact the 66-nt precursor, it is clearly more abundant than the endogenous precursor (compare lanes 3 with lanes 1).
FIG. 5.

Assays for the replication and processing of an HDV genomic RNA modified to contain the putative precursor to miR-122. Huh7 cells were transfected with a DNA construct to express unmodified HDV (lanes 1) or with a construct containing the 66-nt putative miR-122 precursor (lanes 2 and 3). In lanes 3, the cells were additionally transfected with pSVTVA, a plasmid that creates a 16-fold increase in DNA-directed RNA transcription from the SV40-based expression vector (1). At day 4 after transfection, total RNA was extracted and aliquots were subjected to electrophoresis in gels of either 2% agarose or 15% acrylamide-7 M urea, as indicated. Northern analyses were then used to detect genomic and antigenomic HDV RNAs (A) or miR-122 (B and C). Size markers p and q were as described for Fig. 1. The position of the putative precursor to miR-122 is indicated by an asterisk.
Three conclusions can be made from these data. (i) While the insertion of the miR-122 precursor sequences onto HDV RNA interfered with HDV replication (Fig. 5A, lane 2), the nonreplicating genomic RNA was still relatively stable and not processed to form miR-122 (Fig. 5B and C, lanes 3). (ii) Neither the replication of unmodified HDV RNA nor the presence of δAg-S produced by that replication had any effect on the endogenous levels of miR-122 (Fig. 5B and C, lanes 1). (iii) The presence of the species of about 66 nt cleaved from the modified HDV RNA and containing the miR-122 sequences was not associated with a detectable increase in miR-122 relative to the endogenous level. We cannot distinguish whether this reflects resistance to dicer or inaccessibility to dicer. (Formally, we cannot exclude that a small fraction of this modified RNA was cleaved but the released miR-122 was not stabilized.)
Thus, both these in vivo studies and the earlier in vitro studies are consistent with the interpretation that the putative precursor in the context of flanking HDV sequences is not efficiently converted by dicer to miR-122. In our studies with this 66-nt precursor, we had to this point considered it to be a substrate, either in vivo or in vitro, but only when it was embedded at one end of the HDV rod-like RNA sequences (Fig. 3 and 5). We therefore considered it necessary to also test whether in the absence of this embedding sequence, the 66-nt precursor could be cleaved in vitro by dicer. As shown in Fig. 6, lane 8, we thus found to our surprise that the RNA was now >80% sensitive to dicer and that this released a major band of approximately the size expected for miR-122 (lane 8). This sensitivity even matched that of 100% double-stranded RNA (lane 2). As before (Fig. 3, lanes 2 and 10), we detected little, if any, cleavage of unmodified and modified genomic HDV RNAs (Fig. 6, lanes 4 and 6, respectively).
Given this observation that the putative 66-nt miR-122 precursor was actually cleaved efficiently, we made similar 66-nt RNA hairpins from HDV (Fig. 4B) and even from one end of both PSTVd (Fig. 4C) and ASBVd (Fig. 4D). We thus found that all three were digested by dicer, with the release of ∼21-nt species (lanes 10, 12, and 14, respectively). The HDV and ASBVd hairpins were digested 55 and 88%, respectively. The PSTVd only released 2.5% as ∼21-nt species (on a mass basis), but this was still significantly higher than the value of 0.5% for the corresponding full-length PSTVd RNA (Fig. 3B, lane 6).
Our interpretation of all of these in vitro studies is that the ability of dicer to cleave a short 66-nt RNA hairpin with <100% base pairing into ∼21-nt RNA largely depends upon whether the 5′ and 3′ ends of that RNA are embedded in HDV sequences or within PSTVd or ASBVd sequences. In all three cases, embedding dramatically reduced the sensitivity to cleavage by dicer.
DISCUSSION
Our studies support the interpretation that unit-length HDV genomic and antigenomic RNAs are intrinsically resistant to dicer. First, in vivo studies showed that HDV replication did not induce detectable siRNA species. At the same time, we could detect miR-122, which both demonstrates that our assay conditions were appropriate and supports the interpretation that dicer activity was present in these cells. Second, in vitro studies using purified dicer that was active against 100% double-stranded RNA were unable to detect digestion of HDV genomic and antigenomic RNAs that were unit-length and linear. While the in vitro studies demonstrate resistance of the RNAs per se, the resistance observed in vivo may have additional causes, such as the RNAs being inaccessible to dicer.
Our strategy of replacing one end of the rod-like genomic HDV RNA with the putative 66-nt hairpin precursor to miR-122 produced only a modified HDV RNA that, like the unmodified RNA, was not significantly cleaved in vivo or in vitro. Only trace amounts of miR-122 were released. In contrast, and to our surprise, we found that the 66-nt hairpin precursor by itself, with no attached HDV sequences, was readily cleaved in vitro (Fig. 6, lane 8). Furthermore, three other 66-nt hairpin RNAs were tested and found to be susceptible to dicer. These three RNAs were derived from the sequences and predicted folding patterns of the RNAs for HDV, PSTVd, and ASBVd (Fig. 6, lanes 10, 12, and 14, respectively). As indicated in Fig. 4, each was predicted to have <100% base pairing. This dicer sensitivity was dramatically higher than that we detected for the corresponding full-length RNA species (Fig. 3, lanes 2, 6, and 8, respectively). We thus deduce that somehow it is the extent of the regions of <100% pairing on the full-length RNAs of HDV, PSTVd, and ASBVd that confers significant resistance to dicer.
For in vitro studies with full-length RNAs of HDV, PSTVd, and ASBVd, we detected little, if any, digestion by dicer (<1%). Therefore, with such low yields, we have to allow that the substrate for such limited cleavage might not even have been the predominant RNA species (be it HDV, modified HDV with miR-122 precursor, or something else) but instead was a minor component in that RNA preparation. For example, the actual substrate might have been minor amounts of shorter RNAs that were the products of abortive transcription or full-length transcripts that had undergone posttranscriptional degradation. Certainly, based on our present findings of dicer preferentially acting on short RNA hairpins, we also speculate that the ∼21-nt RNAs associated with the replication of PSTVd (21, 41), but absent from ASBVd replication (34), might reflect whether or not such viroid replication has significant levels of an associated by-product of some form(s) of short RNA hairpins. We further speculate more generally that in some cases where RNAi is induced, the putative and mysterious aberrant RNAs (50) may in fact be short RNA hairpins derived by transcriptional pauses or products of partial posttranscriptional degradation, just like for the miRNA precursors (30).
Although each of the 66-nt hairpin RNAs was significantly digested by dicer (Fig. 6), we noted differences in the extent of cleavage and in the homogeneity of the cleavage products. The precursor to miR-122 was the most efficiently cleaved RNA, and the products so released were largely of a discrete size. Consistent with this, it can be readily seen in Fig. 4 that this miR-122 precursor is predicted to have more extensive base pairing than the other three RNAs. In addition, in the unpaired regions of the miR-122 precursor, the mismatches are shorter and more likely to be unpaired bases rather than bulges produced by insertions or deletions on one strand relative to the other. In this respect, it is notable that the 1,679-nt unit-length HDV genomic RNA is predicted to have 145 pairing disruptions, 88% of which represent insertions or deletions (25, 53).
We believe that our results provide yet another example of how the HDV RNAs and their replication sit on the edge of recognition by host mechanisms as being double-stranded. In terms of avoiding recognition, they are not attacked by dicer (see Results). In addition, HDV replication does not induce interferons (20, 37). In contrast, in terms of being recognized, they may activate PKR (10, 44), and as an essential part of the replication cycle, the antigenomic rod-like structure is recognized and edited at a specific site by ADAR-1, an adenosine deaminase acting on RNA (46, 55).
Incidental to the primary aim of this study, we provide here the first evidence that miR-122 is present not only in the mouse liver (27) but also in human and woodchuck livers (Fig. 1C and E). In addition, we provide the first direct evidence that the 66-nt putative precursor to miR-122 can actually be cleaved by dicer in vitro (Fig. 6, lane 8).
Recent studies by others with proteins of plant viruses have in some cases demonstrated an ability of such proteins to interfere with one or more aspects of dicer action and/or siRNA targeting (31, 43, 51, 52, 54). Since we knew that δAg-S is an RNA-binding protein with some specificity for double-stranded RNAs (9), we considered whether it could confer protection of HDV RNAs against dicer action. During in vivo expression of nonreplicating HDV genomic RNA, in the absence of any δAg-S, we failed to detect ∼21-nt HDV RNA (Fig. 5 and data not shown). More specifically, in the in vitro studies, the HDV RNA was resistant to dicer in the absence of δAg-S. In addition, recombinant δAg-S did not interfere with dicer action in vitro on 100% double-stranded RNA (data not shown). It is also worth noting that δAg-S, when expressed in vivo either transiently or stably, did not lead to any alterations in the processing and accumulation of miR-122 (Fig. 1 and 5; data not shown). That is, δAg-S did not interfere with dicer activity on this host RNA.
Acknowledgments
Joao Monjardino provided the Huh7 cell line stably expressing HDV. Clones of PSTVd and ASBVd were provided by Robert Owens and Ricardo Flores, respectively. Long DNA oligonucleotides were obtained from the DNA Synthesis Facility at Fox Chase. Constructive comments on the manuscript were given by Glenn Rall, Eric Moss, Severin Gudima, and Chi Tarn.
This work was supported by grants AI-26522 and CA-06927 from the NIH and by an appropriation from the Commonwealth of Pennsylvania.
REFERENCES
- 1.Asselbergs, F. A. M., and P. Grand. 1993. A two-plasmid system for transient expression of cDNAs in primate cells. Anal. Biochem. 209:327-331. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein, E., A. A. Caudy, S. M. Hammond, and G. J. Hannon. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363-366. [DOI] [PubMed] [Google Scholar]
- 3.Bevilacqua, A., M. C. Ceriani, S. Capaccioli, and A. Nicolin. 2003. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J. Cell. Physiol. 195:356-372. [DOI] [PubMed] [Google Scholar]
- 4.Billy, E., V. Brondani, H. Zhang, U. Muller, and W. Filipowicz. 2001. Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc. Natl. Acad. Sci. USA 98:14428-14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cattaneo, R. 1994. Biased (adenosine to inosine) hypermutation in animal virus genomes. Curr. Opin. Genet. Dev. 4:895-900. [DOI] [PubMed] [Google Scholar]
- 6.Chang, J., G. Moraleda, and J. Taylor. 2000. Limitations to the replication of hepatitis delta virus in avian cells. J. Virol. 74:8861-8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, J., L. J. Sigal, A. Lerro, and J. Taylor. 2001. Replication of the human hepatitis delta virus genome is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J. Virol. 75:3469-3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, J., and J. Taylor. 2002. In vivo RNA-directed transcription, with template switching, by a mammalian RNA polymerase. EMBO J. 21:157-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao, M., S.-Y. Hsieh, and J. Taylor. 1991. The antigen of hepatitis delta virus: examination of in vitro RNA-binding specificity. J. Virol. 65:4057-4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, C.-W., Y.-G. Tsay, H.-L. Wu, C.-H. Lee, D.-S. Chen, and P.-J. Chen. 2002. The double-stranded RNA-activated kinase, PKR, can phosphorylate hepatitis D virus small delta antigen at functional serine and threonine residues. J. Biol. Chem. 277:33058-33067. [DOI] [PubMed] [Google Scholar]
- 11.Chen, P.-J., G. Kalpana, J. Goldberg, W. Mason, B. Werner, J. Gerin, and J. Taylor. 1986. Structure and replication of the genome of hepatitis D virus. Proc. Natl. Acad. Sci. USA 83:8774-8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng, D., A. Yang, H. Thomas, and J. Monjardino. 1993. Characterization of stable hepatitis delta expressing hepatoma cell lines: effect of HDAg on cell growth. Prog. Clin. Biol. Res. 382:149-153. [PubMed] [Google Scholar]
- 13.Cheng, Q., G. C. Jayan, and J. L. Casey. 2003. Differential inhibition of RNA editing in hepatitis delta virus genotype III by the short and long forms of hepatitis delta antigen. J. Virol. 77:7786-7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Circle, D. A., O. D. Neel, H. D. Robertson, P. A. Clarke, and M. B. Mathews. 1997. Surprising specificity of PKR binding to delta agent genomic RNA. RNA 3:438-448. [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad, C., and R. Rauhut. 2002. Ribonuclease III: new sense from nuisance. Int. J. Biochem. Cell Biol. 34:116-129. [DOI] [PubMed] [Google Scholar]
- 16.Di Serio, F., H. Schob, A. Iglesias, C. Tarina, E. Bouldoires, and F. Meins, Jr. 2001. Sense- and antisense-mediated gene silencing in tobacco is inhibited by the same viral suppressors and is associated with accumulation of small RNAs. Proc. Natl. Acad. Sci. USA 98:6506-6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gowans, E. J., B. M. Baroudy, F. Negro, A. Ponzetto, R. H. Purcell, and J. L. Gerin. 1988. Evidence for replication of hepatitis delta virus RNA in hepatocyte nuclei after in vivo infection. Virology 167:274-278. [DOI] [PubMed] [Google Scholar]
- 18.Hammond, S. M., E. Bernstein, D. Beach, and G. J. Hannon. 2000. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404:293-296. [DOI] [PubMed] [Google Scholar]
- 19.Harders, J., N. Lukacs, M. Robert-Nicoud, T. M. Jovin, and D. Riesner. 1989. Imaging of viroids in nuclei from tomato leaf tissue by in situ hybridization and confocal laser scanning microscopy. EMBO J. 8:3941-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilan, Y. M., A. Klein, J. Taylor, and R. Tur-Kaspa. 1992. Resistance of hepatitis delta virus replication to alpha interferon treatment in transfected human cells. J. Infect. Dis. 166:1164-1166. [DOI] [PubMed] [Google Scholar]
- 21.Itaya, A., A. Folimonov, Y. Matsuda, R. S. Nelson, and B. Ding. 2001. Potato spindle tuber viroid as inducer of RNA silencing in infected tomato. Mol. Plant-Microbe Interact. 14:1332-1334. [DOI] [PubMed] [Google Scholar]
- 22.Keese, P., and R. H. Symons. 1987. The structure of viroids and virusoids, p. 1-47. In J. S. Semancik (ed.), Viroids and viroid-like pathogens. CRC Press, Boca Raton, Fla.
- 23.Ketting, R. F., S. E. J. Fischer, E. Bernstein, T. Sijen, G. J. Hannon, and R. H. A. Plasterk. 2001. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 15:2654-2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar, M., and G. G. Carmichael. 1998. Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev. 62:1415-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo, M. Y.-P., J. Goldberg, L. Coates, W. Mason, J. Gerin, and J. Taylor. 1988. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: sequence, structure, and applications. J. Virol. 62:1855-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 227:680-685. [DOI] [PubMed] [Google Scholar]
- 27.Lagos-Quintana, M., R. Rauhut, A. Yalcin, J. Meyer, W. Lendeckel, and T. Tuschl. 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12:735-739. [DOI] [PubMed] [Google Scholar]
- 28.Lazinski, D. W., and J. M. Taylor. 1994. Expression of hepatitis delta virus RNA deletions: cis and trans requirements for self-cleavage, ligation, and RNA packaging. J. Virol. 68:2879-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazinski, D. W., and J. M. Taylor. 1993. Relating structure to function in the hepatitis delta virus antigen. J. Virol. 67:2672-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, Y. H., K. Jeon, J.-T. Lee, S. Kim, and V. N. Kim. 2002. MicroRNA maturation, stepwise processing and subcellular localization. EMBO J. 21:4663-4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lichner, Z., D. Silhavy, and J. Burgyan. 2003. Double-stranded RNA-binding proteins could suppress RNA interference-mediated antiviral defences. J. Gen. Virol. 84:975-980. [DOI] [PubMed] [Google Scholar]
- 32.Lim, L. P., M. E. Glasner, S. Yekta, C. B. Burge, and D. P. Bartel. 2003. Vertebrate microRNA genes. Science 299:1540. [DOI] [PubMed] [Google Scholar]
- 33.Macnaughton, T. B., and M. M. Lai. 2002. Genomic but not antigenomic hepatitis delta virus RNA is preferentially exported from the nucleus immediately after synthesis and processing. J. Virol. 76:3928-3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez De Alba, A. E., R. Flores, and C. Hernandez. 2002. Two chloroplastic viroids induce the accumulation of small RNAs associated with posttranscriptional gene silencing. J. Virol. 76:13094-13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McInnes, J. L., and R. H. Symons. 1991. Comparative structure of viroids and their rapid detection using radioactive and nonradioactive nucleic acid probes, p. 21-58. In K. Maramorosch (ed.), Viroids and satellites: molecular parasites at the frontier of life. CRC Press, Boca Raton, Fla.
- 36.McManus, M. T., and P. A. Sharp. 2002. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 3:737-747. [DOI] [PubMed] [Google Scholar]
- 37.McNair, A. N. B., J. Monjardino, D. Cheng, H. C. Thomas, and I. M. Kerr. 1993. Hepatitis delta virus and the interferon system. Prog. Clin. Biol. Res. 382:161-164. [PubMed] [Google Scholar]
- 38.Moss, E. G. 2002. MicroRNAs: hidden in the genome. Curr. Biol. 12:R138-R140. [DOI] [PubMed] [Google Scholar]
- 39.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 40.Netter, H. J., T.-T. Wu, M. Bockol, A. Cywinski, W.-S. Ryu, B. C. Tennant, and J. M. Taylor. 1995. Nucleotide sequence stability of the genome of hepatitis delta virus. J. Virol. 69:1687-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papaefthimiou, I., A. Hamilton, M. Denti, D. Baulcombe, M. Tsagris, and M. Tabler. 2001. Replicating potato spindle tuber viroid RNA is accompanied by short RNA fragments that are characteristic of post-transcriptional gene silencing. Nucleic Acids Res. 29:2395-2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Provost, P., D. Dishart, J. Doucet, D. Frendewey, B. Samuelsson, and O. Radmark. 2002. Ribonuclease activity and RNA binding of recombinant human dicer. EMBO J. 21:5864-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qu, F., T. Ren, and T. J. Morris. 2003. The coat protein of turnip crinkle virus suppresses posttranscriptional gene silencing at an early initiation step. J. Virol. 77:511-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson, H. D., L. Manche, and M. B. Mathews. 1996. Paradoxical interactions between human hepatitis delta agent RNA and the cellular protein kinase PKR. J. Virol. 70:5611-5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robertson, H. D., and M. B. Mathews. 1996. The regulation of the protein kinase PKR by RNA. Biochimie 78:909-914. [DOI] [PubMed] [Google Scholar]
- 46.Sato, S., S. K. Wong, and D. W. Lazinski. 2001. Hepatitis delta virus minimal substrates competent for editing by ADAR1 and ADAR2. J. Virol. 75:8547-8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tavanez, J. P., C. Cunha, M. C. A. Silva, E. David, J. Monjardino, and M. Carma-Fonseca. 2002. Hepatitis delta virus ribonucleoproteins shuttle between the nucleus and the cytoplasm. RNA 8:637-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor, J. M. 1999. Replication of human hepatitis delta virus: influence of studies on subviral plant pathogens. Adv. Vir. Res. 54:45-60. [DOI] [PubMed] [Google Scholar]
- 49.Taylor, J. M. 2003. Replication of human hepatitis delta virus: recent developments. Trends Microbiol. 11:185-190. [DOI] [PubMed] [Google Scholar]
- 50.Tuschl, T. 2001. RNA interference and small interfering RNAs. Chembiochem 2:239-245. [DOI] [PubMed] [Google Scholar]
- 51.Vance, V., and H. Vaucheret. 2001. RNA silencing in plants—defense and counterdefense. Science 292:2277-2280. [DOI] [PubMed] [Google Scholar]
- 52.Voinnet, O., Y. M. Pinto, and D. C. Baulcombe. 2003. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc. Natl. Acad. Sci. USA 96:14147-14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, K.-S., Q.-L. Choo, A. J. Weiner, J.-H. Ou, C. Najarian, R. M. Thayer, G. T. Mullenbach, K. J. Denniston, J. L. Gerin, and M. Houghton. 1986. Structure, sequence and expression of the hepatitis delta viral genome. Nature 323:508-513. [DOI] [PubMed] [Google Scholar]
- 54.Waterhouse, P. M., M. B. Wang, and T. Lough. 2001. Gene silencing as an adaptive defence against viruses. Nature 411:834-842. [DOI] [PubMed] [Google Scholar]
- 55.Wong, S. K., and D. W. Lazinski. 2002. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 99:15118-15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu, T.-T., H. J. Netter, D. W. Lazinski, and J. M. Taylor. 1997. Effects of nucleotide changes on the ability of hepatitis delta virus to transcribe, process and accumulate unit-length, circular RNA. J. Virol. 71:5408-5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie, Z., K. D. Kasschau, and J. C. Carrington. 2003. Negative feedback regulation of dicer-like 1 in Arabidopsis by microRNA-guided mRNA degradation. Curr. Biol. 13:784-789. [DOI] [PubMed] [Google Scholar]
- 58.Zeng, Y., and B. R. Cullen. 2003. Specific requirements for micro RNA processing and function in human cells. RNA 9:112-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang, H., F. A. Kolb, V. Brindani, E. Billy, and W. Filipowicz. 2002. Human dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J. 21:5875-5885. [DOI] [PMC free article] [PubMed] [Google Scholar]