

Abstract
The chemical methylating agents methylmethane sulfonate (MMS) and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) have been used for decades as classical DNA damaging agents. These agents have been utilized to uncover and explore pathways of DNA repair, DNA damage response, and mutagenesis. MMS and MNNG modify DNA by adding methyl groups to a number of nucleophilic sites on the DNA bases, although MNNG produces a greater percentage of O-methyl adducts. There has been substantial progress elucidating direct reversal proteins that remove methyl groups and base excision repair (BER), which removes and replaces methylated bases. Direct reversal proteins and BER thus counteract the toxic, mutagenic and clastogenic effects of methylating agents. Despite recent progress, the complexity of DNA damage responses to methylating agents is still being discovered. In particular, there is growing understanding of pathways such as homologous recombination, lesion bypass, and mismatch repair that react when the response of direct reversal proteins and BER is insufficient. Furthermore, the importance of proper balance within the steps in BER has been uncovered with the knowledge that DNA structural intermediates during BER are deleterious. A number of issues complicate elucidating the downstream responses when direct reversal is insufficient or BER is imbalanced. These include inter-species differences, cell-type specific differences within mammals and between cancer cell lines, and the type of methyl damage or BER intermediate encountered. MMS also carries a misleading reputation of being a ‘radiomimetic,’ i.e., capable of directly producing strand breaks. This review focuses on the DNA methyl damage caused by MMS and MNNG for each site of potential methylation to summarize what is known about the repair of such damage and the downstream responses and consequences if not repaired.
Keywords: MMS, MNNG, methyl damage, DNA repair, DNA strand breaks
1. Introduction
1.1 Chemical view of the DNA structure and nucleophilic sites
The biological view of DNA is traditionally a four-letter alphabet where ‘A’ pairs with ‘T’ and ‘G’ pairs with ‘C’. The chemical view of DNA acknowledges the unique shape of each base and position of the functional groups that distinguish the four ‘letters’ of the alphabet. It is the shape and electrostatics of each base that allow proteins to recognize specific sequences. Yet these same functional groups also provide a susceptibility to modification that lies at the heart of understanding mutagenesis. The types of DNA base modifications possible via oxidation, deamination, and alkylation reactions are surprisingly numerous, given the importance of DNA. Furthermore, the ribose-phosphodiester backbone is susceptible to modification and breakage. In a broad sense, the deleterious biological consequences of DNA modifications are toxicity, mutagenicity, or clastogenicity. Yet the specific responses can be as complex as the types of modifications possible. This review will specifically focus on methyl DNA damage, as opposed to larger alkylations, and are not to be confused with enzymatic methylation reactions (e.g., 5-methylcytosine, N6-methyladenine). Many of the early discoveries in the repair of methylation damage were made in E. coli and S. cerevisiae. Where appropriate, the repair sections begin with the bacterial and/or yeast paradigm, although descriptions will concentrate on known mammalian repair activities and responses.
Electrophilic methylating agents are capable of reacting with a number of nucleophilic sites on DNA (Figure 1). The exocyclic amino groups of guanine, cytosine, and adenine are poor nucleophiles in methylation reactions. Although each ring nitrogen and exocyclic oxygen is potentially capable of acting as a nucleophile, their reactivity towards electrophiles substantially varies by position and whether the DNA is single or double stranded. Molecular electrostatic potentials of the nucleophilic sites on the bases and base pairs are helpful in understanding the reactions of electrophiles with DNA (1). The N7 position of guanine possesses the highest negative electrostatic potential in DNA. In double stranded DNA, O6-guanine and N3-adenine have the next highest potential, followed by N3-guanine, O2-cytosine, N7-adenine, O4-thymine and O2-thymine (1). The negative electrostatic potential is greatest for a guanine: cytosine base pair on the major groove face, while it is greatest for an adenine: thymine base pair on the minor groove face. In single stranded DNA, it is worth noting that N1-cytosine has similar negative electrostatic potential to that of N7-guanine while N1-adenine has similar potential to that of O6-guanine (1). Consideration of electrostatic potentials and steric hinderances help in part explain the reaction preferences seen with methylating agents, but the type of agent also influences the distribution of adducts.
Figure 1.

Potential sites of chemical methylation in double strand DNA. The arrows point to each methyl adduct and whether the adduct is known to be predominantly toxic or mutagenic. The open arrows represent sites that are methylated by MMS, MNNG, and MNU. The filled arrows point to sites that are methylated by MNNG and MNU, but not detectably by MMS. Note that methylation of different sites on the same base at the same time is extremely rare. The size of the arrows roughly represent the relative proportion of adducts. In single strand DNA, the N1-adenine and N3-cytosine positions display a greater reactivity.
1.2. Prototypical methylating agents
Methyl methane sulfonate (MMS)1, dimethyl sulfate (DMS), N-methyl-N-nitrosourea (MNU), and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) have been used extensively as direct-acting methylating agents. The common perception is that MMS and DMS are SN2-type (biomolecular nucleophilic substitution) agents while MNNG and MNU are SN1-type (unimolecular nucleophilic substitution) agents. However, it is not accurate to say that MNNG and MNU react by an SN1 mechanism, because this would require a diffusible methyl carbocation. MNNG decomposes to form a methyl-diazonium cation that methylates DNA (2). The term ‘oxyphilic’ has been suggested instead of SN1 (3). Although not relevant for this review, it is interesting to note that MNNG decomposition also forms a nitrocyanamide anion, which can further decompose to form reactive nitrogen species. Lastly, there are a couple of examples of methylating agents that have been used as chemotherapeutics, including more recently Temozolomide (TMZ) (4). TMZ is thought to behave similarly to MNNG and MNU regarding adduct distribution.
The predominant adduct in double stranded (ds) DNA resulting from MMS or MNNG exposure is 7-methylguanine (N7-MeG), which comprise 67% and 82% of the MNNG and MMS induced dsDNA damage, respectively (Figure 1) (5). Indeed, the guanine-specific sequencing reaction devised by Maxam and Gilbert exploited the reactivity of DMS for the N7 position of guanine (6). The difference in reactivity between MMS and MNNG lies in the proportion of oxygen adducts. MMS produces 11% 3-methyladenine (N3-MeA) and 0.3% O6-methylguanine (O6-MeG), whereas MNNG produces 12% N3-MeA and 7% O6-MeG (5). The remaining base positions (N1-adenine, N7-adenine, N1-guanine, N3-guanine, N3-cytosine, O2-cytosine, N3-thymine, O2-thymine, and O4-thymine) combined comprise <5% of the adduct burden in dsDNA produced by MMS or MNNG (5). The percentages, a compilation from five different references, show that MMS does not detectably produce other O-methyl pyrimidine adducts while MNU produces 0.1 to 0.7% each of O2-pyrimidines and O4-thymine (MNNG was not determined) (5). Some of the positions that represent only a minor percentage of methylation participate in hydrogen bonding in dsDNA, including N1-adenine, N1-guanine, O6-guanine, N3-cytosine, O2-cytosine, N3-thymine, and O4-thymine. In light of the recent discovery of the function of the AlkB family of proteins (Section 2.2), it is worth briefly revisiting what is known about methylating single stranded DNA and RNA. The methylation (and ethylation) of O6-guanine, O2-cytosine, and O4-thymine is not influenced by hydrogen bonding, presumably because of the extra pair of electrons available (7). In contrast, N1-adenine and N3-cytosine are much more reactive as nucleophiles in the absence of hydrogen bonding participation, i.e., these two sites are much more susceptible to methylation in single stranded DNA. N1-adenine and N3-cytosine have a reactivity characteristic of amidine groups due to the adjacent amino groups on the neighboring carbons, whereas the N1-guanine and N3-thymine are secondary amines that have adjacent carbonyl groups and are thus less reactive (Figure 1). Lastly, methyl phosphodiesters comprise 0.8% of the adducts from MMS, but are predicted to comprise ~12–17% of the adducts from MNNG based on data with MNU (5). When considering the relative concentrations used, MNU and MNNG are substantially more toxic and mutagenic than MMS. The higher potency of MNU and MNNG infers that O-methyl adducts are more deleterious; evidence points to O6-MeG in particular and will be discussed.
It is important to briefly discuss the stability of N-methylpurine adducts for biological and experimental reasons. Methylating the N3 or N7 position of purines destabilizes the N-glycosidic bond, thus rendering these modified bases more susceptible to being hydrolyzed into an abasic site. The half lives vary from less than 3 h for N7-MeA to approximately 30 and 70 h, respectively, for N3-MeA and N7-MeG at 39° C (8), which for E. coli is many generations but might represent only one or two rounds of replication in mammalian cells. The depurination of N3- and N7 adducts is accelerated with heat under neutral pH, which is relevant for experimental procedures that might subject methylated DNA samples to heat. Alkali treatment converts abasic sites into single strand breaks via a β-elimination reaction. However, alkaline conditions convert the N7-MeG adduct into a ring-opened methyl-formamidopyrimidine adduct, which is resistant to strand breakage (9, 10). Alkyl-formamidopyrimidine adducts can be quantitatively converted to a strand break in the presence of a secondary amine (e.g., piperidine) and heat (6). The influence of depurination in cell culture experiments following treatment with methylating agents has been difficult to assess. Sequence context can also influence adduct distribution, specifically a preference for guanines within runs of guanines, although the reaction pattern for simple methylating agents shows less of a preference than larger alkyl agents (11). Interestingly, the effects of sequence specificity on methylation seen in reactions with purified DNA appeared to be similar to in vivo methylation reactions (12).
1.3. N-methyl and O-methyl adducts
The structural and biological consequences of the different N- and O-methyl adducts have been studied to various extents. Studies using DNA templates treated with methylating agents and purified polymerases established the dogma that N3-MeA blocks replication, whereas N7-MeG does not block or miscode (10, 13). High-resolution structural studies of N3-MeA have not been performed because its susceptibility to depurination. However, structural studies of DNA polymerases suggest that minor groove N3-purine and O2-pyrimidine contacts are important during extension from newly synthesized base pairs, thus providing an explanation why methyl groups at these positions would impede polymerases (14). In murine embryonic stem cells, N3-MeA can slow S-phase progression in the absence of its repair (15). Although N3-MeA is largely thought of as a toxic lesion, there is evidence that low levels of A:T to T:A transversions can occur following treatment with MMS and an agent that specifically produces N3-MeA called methyl-lexitropsin (Me-Lex) (16). Lexitropsins are analogues of netropsin and distamycin, which bind non-covalently in the minor groove of DNA (17). Me-lex contains a methyl sulfonate ester tethered to a lexitropsin and appears to produce N3-MeA almost exclusively (18). Me-lex has been used in a number of studies on the biological effects of N3-MeA and has been reviewed elsewhere (16).
N7-MeG does not appear to block replication or miscode, but depurination produces an abasic site, which is known to have mutagenic and toxic consequences. Oligodeoxynucleotides containing N7-MeG have been synthesized using DNA polymerases; NMR structural studies did not indicate any significant perturbation of B-DNA (19). Studies have shown that the ring-opened methyl-formamidopyrimidine adduct of 7-MeG can block DNA polymerases in vitro (9, 10), although the in vivo relevance has not been established. The instability of N3 and N7 purine adducts has prevented more thorough studies of these adducts via site-specific approaches.
O6-MeG is well known to induce G:C to A:T mutations because it readily mispairs with thymine (20). An important early study in human cell lines established that unrepaired O6-MeG could be very toxic (21). However, the same study also identified an MNNG-resistant line (MT1) that ‘tolerated’ persistent O6-MeG but were hypermutable by MNNG, leading the authors to hypothesize that MT1 were mismatch repair deficient (21). These studies and others suggested that O6-MeG does not induce cell cycle arrest until a second round of replication, which implied that the biological consequences are not elicited until O6-MeG is paired opposite thymine (21–24). O6-MeG:T is known to be recognized by mismatch repair (MMR) in a process that is a potent signal of apoptosis (25–27). O6-MeG is therefore a toxic adduct in mammalian cells that are deficient in the repair of O6-MeG and proficient in MMR. The concept of MMR as a sensing mechanism for methyl damage will be discussed in sections 2.1 and 4.3.
The consequences of methyl groups at the remaining positions have received considerably less attention in mammalian cells, in part because of their lower occurrence. O4-MeT has been examined as a site-specific adduct in mammalian vectors and appears to be more mutagenic than O6-MeG (28, 29). Based on the above-mentioned structural studies of DNA polymerases, O2-MeC, O2-MeT, and N3-MeG are predicted to interfere with minor groove contacts, yet there have been very few studies of these modifications. The remaining sites are nitrogen atoms that participate in hydrogen bonding in dsDNA, but are now receiving renewed interest with the discovery of a repair mechanism for such modifications. N1-MeA and N3-MeC appear to be primarily toxic adducts, although they can produce low levels of mutagenesis (30). A more recent study using site-specific adducts in vectors passed through E. coli found that N3-MeC was more mutagenic than N1-MeA, while both adducts blocked replication (31). The rare N1-MeG and N3-MeT adducts were also shown to be mutagenic and replication-blocking (31). The repair of N1-methyl purines and N3-methyl pyrimidines will be discussed in section 2.2.
1.4. Methylating agents and DNA strand breaks
MMS has been used as a ‘radiomimetic’ in yeast studies for decades because many of the original radiation sensitive (rad) mutants were also found to be sensitive to MMS (32). The ability of MMS and MNNG to induce sister chromatid exchanges (SCEs) have been known for some time and were recently reviewed (33). SCEs are reciprocal exchanges of DNA between sister chromatids during S-phase and are thought to represent homologous recombination events. The ‘radiomimetic’ and SCE-inducing properties of MMS do not distinguish whether MMS directly causes DNA strand breaks, or whether strand breaks observed result from events subsequent to the methyl damage (i.e., repair intermediates that are strand breaks, or stalled/collapsed replication forks). It has been shown that histone H2AX becomes phosphorylated (γH2AX) in response to MMS (34, 35). H2AX phosphorylation seems to be accepted as a marker specific for double strand breaks, but does not identify the exact source of the breaks. Recently, it was shown that careful preparation of DNA to limit spontaneous depurination revealed that double strand breaks do not appear to be induced by MMS in yeast or mammalian cells (36). The issue of MMS and strand breaks will be discussed in the sections on base excision repair (BER). MNNG is a much more potent producer of SCEs than MMS. Specifically, O6-MeG can induce SCEs, chromosomal aberrations, and homologous recombination events in a MMR-dependent manner (37–41). These and other observations bearing on the topic of methylating agents and strand breaks will be discussed throughout the following sections on repair responses.
2. First line removal of methyl damage
The discovery of the adaptive response of E. coli to alkylation damage eventually revealed a multi-pronged approach to repairing methylated bases in DNA (42). The combined activities of five different polypeptides (Tag, Ogt, Ada, AlkA, AlkB) can remove eleven different methylated bases plus methyl-phosphate modifications (Figure 2) (43, 44). Ogt and Ada directly reverse O-methyl adducts via a suicide methyltransferase activity. AlkB directly reverses N-methyl adducts via dioxygenase activity. Tag and AlkA are DNA glycosylases that remove methylated bases as part of BER (Figure 4). E. coli deficient in these proteins display varying degrees of sensitivity to MMS or MNNG depending on the specificity of the repair protein and the distribution of N- versus O-methyl adducts. Mammalian cells contain different proteins with O6-MeG methyltransferase, N-methyl dioxygenase, and methylpurine N-glycosylase activity, which underscores the importance of removing methyl damage (Figure 3). However, there are a number of differences regarding these activities in mammalian cells that will be discussed. One broad difference is that mammals appear to possess only one O6-MeG methyltransferase (AGT, MGMT), one methylpurine DNA glycosylase (MPG, AAG), but multiple putative N-methyl dioxygenase homologues. The other difference is that Ada plays an important role in the adaptive response in E. coli, while mammalian cells do not appear to contain an analogous adaptive response system (45). The individual protein activities will be discussed next.
Figure 2.

Repair of methyl damage in E. coli. Arrows identify methyl groups or methyl bases removed by the specific proteins for each position. The arrows do not represent adduct proportions. Ogt, Ada, and AlkB directly remove the methyl group to reverse the damage. Tag and AlkA are DNA glycosylases that initiate BER. N7-MeA is a relatively rare adduct that depurinates rapidly and no repair mechanism is known.
Figure 4.
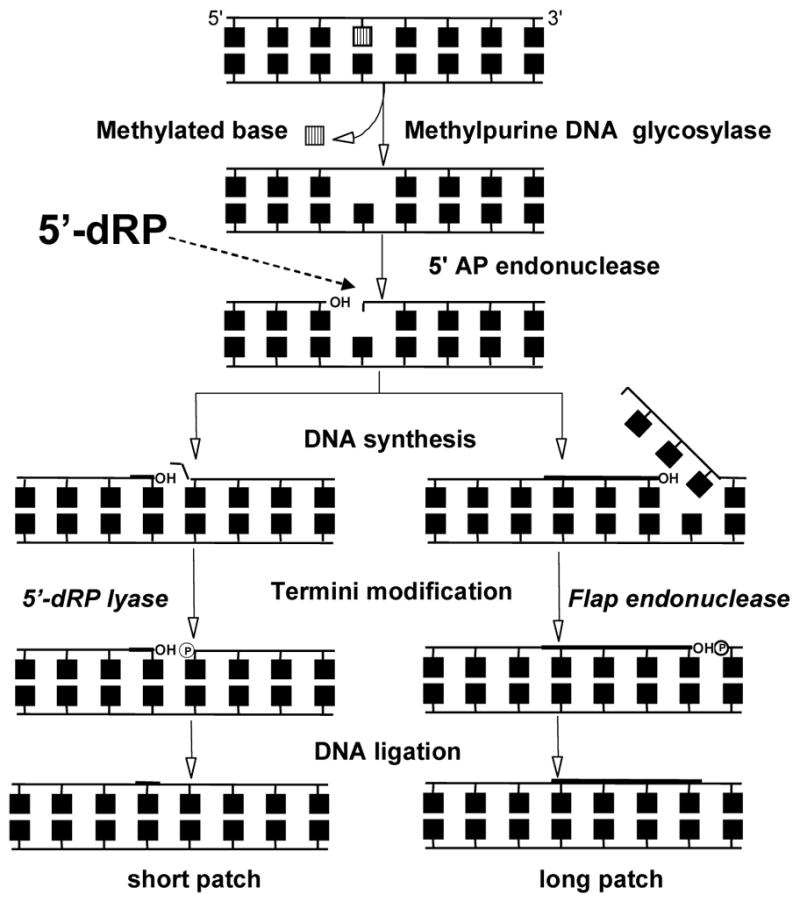
General schematic of base excision repair following methyl base damage. The first step is carried out by a DNA glycosylase, which produces an abasic site. The second step is carried out by an AP endonuclease, which creates a nick 5′- to the abasic site. DNA synthesis replaces a single nucleotide (short patch) or several nucleotides (long patch). The 5′-dRP group is removed by 5′-dRP lyase (short patch) or as part of an overhang by a ‘flap’ endonuclease (long patch). DNA ligase seals the nick to complete the pathway. Incomplete BER intermediates, in particular the 5′-dRP group, are thought to lead to downstream consequences such as chromosomal aberrations and toxicity.
Figure 3.

Repair of methyl damage in mammalian cells. Arrows identify methyl groups or methyl bases removed by the specific proteins for each position. MGMT and ABH2 directly remove the methyl group to reverse the damage. AAG is a DNA glycosylase that initiates BER. Question marks are shown where adduct repair has not yet been definitively identified.
2.1. Direct reversal of O-methyl adducts
The direct reversal of O6-MeG adducts is known to be carried out by proteins in a suicide reaction involving transfer of the Me group to a cysteine (45). In E. coli, Ogt removes O6-MeG, whereas ada removes O6-MeG, O4-MeT, and methyl phosphodiesters (Figure 2) (44). Ada has two distinct active site cysteines, one that removes a methyl group from O6-MeG or O4-MeT, and one that removes methyl phosphodiester adducts (44). The methyl transfer from phosphodiesters activates the ability of ada to up-regulate the expression of itself, AlkA, AlkB, and the aidB protein, the activity of which remains unresolved (44). The issue of how methyl phosphodiesters are repaired in mammalian cells is unresolved, although the general consensus is that they are relatively innocuous (44).
Mice and humans have a protein, MGMT (AGT), which appears to only remove O6-alkyl adducts in a similar suicide reaction to that seen for ogt and ada (Figure 3). There has been much interest in human MGMT in cancer chemotherapy because it removes O6-MeG adducts caused by TMZ and O6-chloroethylguanine adducts caused by chloroethyl nitrosoureas; MGMT can also be inactivated with O6-alkyl substituted guanine derivatives (46, 47). Mammalian cells deficient in MGMT (historically called Mer− or Mex−) are much more sensitive to killing by MNNG and MNU than proficient cells, which suggested that O6-MeG could directly cause cell death (45). Mice deficient in MGMT are hypersensitive to killing induced by agents that produce O6-MeG (48, 49). MGMT status has little effect on the sensitivity to killing by MMS, which indirectly confirms that MMS produces little O6-MeG and that MGMT does not repair other methylated bases. Human MGMT expression is susceptible to epigenetic silencing, a trait that is also shared by the gene encoding the MMR protein MLH1 (45).
Because of the specificity of mammalian MGMT for O6-MeG, studies modulating MGMT expression have proven useful in examining the downstream consequences of O6-MeG (26, 27, 37–40, 50). O6-MeG is also recognized by the MSH2:MSH6 heterodimer (MutSα), although MutSα recognition of O6-MeG leads to apoptosis, not repair. Mismatch or damage binding by MutSα is the initiating event in MMR, followed by the MLH1:PMS2 heterodimer recognizing bound MutSα (Figure 5) (51). Loss of MMR via epigenetic or genetic mechanisms therefore leads to methylation ‘tolerance’ and a higher probability of mutation following treatment with methylating agents that produce O6-MeG. The hypersensitivity of Mgmt-deficient mice to MNU toxicity is relieved when MLH1 is also inactivated (52). In addition to mutagenicity and cell death, O6-MeG can induce SCEs. MGMT expression studies strongly suggest that O6-MeG is the source of MNNG-induced SCEs (39). Furthermore, MNNG induces significantly more SCEs in MMR proficient cells and in the second mitosis following treatment, which suggests that recognition of O6-MeG paired opposite thymine leads to aberrant processing by MMR (37). Measurements of individual homologous recombination events induced by O6-MeG agree with measurements of SCEs (40, 41). The connections between O6-MeG, MMR, apoptosis and aberrant recombination in the second mitosis have been the subject of several reviews (51, 53, 54). Debate remains as to whether MMR recognition of O6-MeG:T leads to cell death due to reiterative repair attempts with O6-MeG in the template strand, termed ‘futile cycling’, or that some MMR proteins have a methyl damage sensing mechanism distinct from their roles in post-replicative mismatch repair; this will be discussed in Section 4.3.
Figure 5.
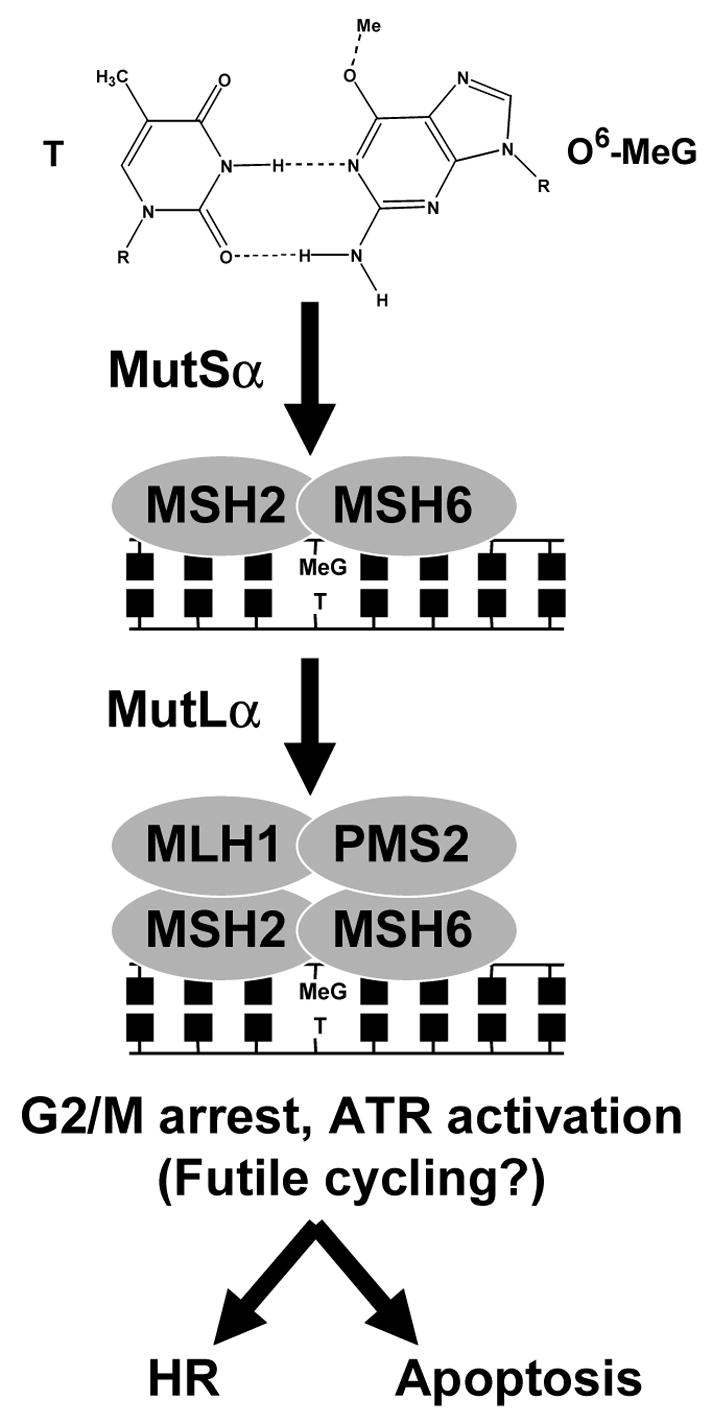
General schematic of the recognition of an O6-MeG:T mispair by the proteins of mismatch repair. In the first step, the MSH2:MSH6 heterodimer (MutSα) recognizes O6-MeG:T. In the second step, MutSα is recognized by the MLH1:PMS2 heterodimer (MutLα). The downstream consequences of these events include the induction of DNA damage signaling events, G2/M cell cycle arrest, induction of sister chromatid exchanges and apoptosis.
2.2. Direct reversal of N-methyl adducts
The function of the AlkB protein family was recently discovered to be a dioxygenase activity that oxidatively demethylates N1-MeA and N3-MeC and has been reviewed recently (55). Comparison of AlkB sequence to other protein domains suggested an enzymatic activity that was subsequently proven to be a dioxygenase requiring α-keto-glutarate and Fe2+ as cofactors (56–58). AlkB deficient bacterial cells were known to be more sensitive to killing by MMS than to MNNG, which suggested that AlkB was not likely to repair O-methyl adducts. Single-stranded phages were also known to be more sensitive to MMS modification than MNNG in AlkB deficient strains (30). An important piece of the puzzle was a study that showed E. coli AlkB could provide resistance to MMS when heterologously expressed in human cells, thus strongly suggesting that AlkB had an intrinsic activity (59). Note these results and others also suggest that MMS produces biologically significant amounts of N1-MeA and N3-MeC, at least in cells undergoing replication (30).
As mentioned earlier, there appear to be multiple mammalian homologues of AlkB, most of which have yet to be fully studied (55). One homologue, ABH3, can demethylate RNA, demonstrating an in vivo relevance for the repair of RNA base damage (57). Human ABH2 and ABH3 (AlkB homologues 2 and 3) have also been reported to remove the rare N1-MeG and N3-MeT adducts (43, 60). ABH2 appears to act more efficiently on dsDNA and has been suggested to act near replication forks (57). Recently, mice deficient for Abh2 and Abh3 were generated and are viable with no immediately obvious phenotypes (61). The Abh2 deficient cells were mildly sensitive to MMS and show accumulation of genomic N1-MeA (61). Taken together, the results suggest that reactions with bases in transiently single-stranded DNA are also important when evaluating the biological responses to methylating agents. Considering the ability to reverse N1-MeA and N3-MeC adducts, it would be interesting to reexamine S-phase progression following MMS treatment in the presence or absence of AlkB or ABH homologues. Full elucidation of the functions of ABH family members should provide better insight into the biological consequences of the replication-blocking N1-MeA and N3-MeC adducts.
2.3. DNA glycosylase removal of N-methyl bases
DNA glycosylases that remove N3-MeA have been identified in all species examined for such an activity (Figure 4). As mentioned above, E. coli possesses two such enzymes, Tag and AlkA. Tag only removes N3-MeA and N3-MeG (62). Analogous to heterologous expression studies with MGMT, Tag has been used to draw conclusions on the biological consequences of N3-methyl purines (63–65). In stark contrast to Tag, AlkA removes a surprisingly wide variety of damaged purines and pyrimidines, including N3-MeA, N7-MeG, O2-MeC and O2-MeT (66). In fact, the profligacy of AlkA and the S. cerevisiae homologue Mag1 is harmful when these enzymes are over-expressed even in the absence of exogenous damage (63, 65). Mammals do not possess homologues of tag or AlkA, but possess members of an AAG (AAG, alkyladenine; MPG, methylpurine; APNG, N-alkylpurine) family of N-methylpurine DNA glycosylases (67). The names do not accurately represent the broad range of modified purines that this DNA glycosylase family excises (68). This review will not discuss the substrate range; however, it is noteworthy to briefly discuss the enzymatic activity of AAG for methylated bases. First, AAG does not appear to remove O2-MeC, O2-MeT, or any other damaged pyrimidine, despite being able to bind to mismatched or damaged pyrimidines (69–71). Second, when the enzymatic activity of human AAG for N3-MeA and N7-MeG was normalized to the spontaneous depurination reactions, the comparison revealed that they are relatively poor substrates (71). Interestingly, AAG and AlkA family members can remove normal bases at low but detectable levels (71–73). Indeed, the consensus seems to be that the active site of AAG serves more to reject normal purines than to specifically recognize damaged bases (71, 74). Thus, the enzymatic activity of AAG for N3-MeA and N7-MeG offers only a modest rate enhancement above spontaneous depurination, yet enough of an enhancement to reduce the half lives of N3-MeA and N7-MeG to minutes instead of hours (71, 73). The activity for N7-MeG is particularly intriguing when the innocuous nature of this adduct is considered. The activity of AAG for N7-MeG may simply be a byproduct of the evolution of the mammalian AAG enzyme active site for more biologically deleterious adducts. Alternatively, there might be a biological benefit to enzymatically removing N7-MeG if spontaneous depurination results in an unprotected AP site, a benefit that would not be detected with high doses of MMS or MNNG that also produce substantial amounts of N3-MeA. Sequence context in vitro and in cells has been reported to affect the removal rates of N3-MeA and N7-MeG by as much as 6-fold and 30-fold, respectively, suggesting that adduct persistence is heterogeneous (12).
Studies in E. coli and S. cerevisiae demonstrated the importance of N-methylpurine DNA glycosylases in protecting against the mutagenic and toxic effects of alkylating agents (75–77). These studies also demonstrated that AlkA and Mag1 are inducible by methylating agents, thus restricting their activity to when it is needed. However, loss of the S. pombe Mag1 homologue does not confer sensitivity to alkylating agents, demonstrating that different species utilize N3-MeA DNA glycosylase-initiated BER to different extents (78). Studies of N-methylpurine DNA glycosylases in mammalian systems provide a complex picture of the importance of AAG activity. It is first worth noting that excessive AAG activity in human cells in the absence of exogenous damage leads to microsatellite instability (79). Studies have shown in mammalian cells that N3-MeA DNA glycosylase over-expression during exposure to methylating agents can be detrimental (80–82). Aag−/− embryonic stem (ES) cells are more sensitive to MMS than Aag+/+ ES cells, which recapitulated the bacterial paradigm (15, 83). Mice homozygous null for murine Aag are viable and do not suffer from accelerated spontaneous tumorigenesis (84, 85). Although Aag−/− primary embryonic fibroblasts were more sensitive to N3-MeA specifically produced by Me-lex (84), the Aag−/− cells were only very modestly if at all sensitive to MMS (84, 85). In one study, the Aag+/+ and Aag−/− mice were equally sensitive to MMS-induced hematopoietic toxicity; MMS induced increased mutations in splenic T-lymphocytes from Aag−/− mice, which were predominantly AT to TA transversions (85). In another study, Aag−/− bone marrow (BM) cells of myeloid lineage display resistance to killing by methylating agents compared to Aag+/+ myeloid BM cells (86). The Aag−/− BM cells were also more resistant to chromosomal damage as measured by the micronucleus assay, suggesting that Aag activity contributes to chromosomal damage induced by MMS in these cell types (86). More recently, it has been reported that HeLa cells can be sensitized to methylating agents when AAG is silenced by siRNA (87). It was also shown that over-expression of AAG sensitizes breast cancer cells to methylating agents, which was proposed to be due to the excessive removal of N7-MeG (88). A series of site-specific mutants of AAG were generated that varied in their substrate specificity and were expressed in S. cerevisiae lacking endogenous Mag1 (74, 89). S. cerevisiae expressing an AAG mutant (N169D) that did not detectably remove N7-MeG showed greater survival than S. cerevisiae expressing wild-type AAG (89).
The simple conclusion from the above studies is that too little or too much AAG activity can be detrimental (68). Practically speaking, this makes interpretation of phenotypes more challenging. Two complexities in interpreting the relative importance of AAG-initiated BER in response to DNA methylating agents are the relative formation of N7-MeG versus N3-MeA, and the downstream components of BER in the systems studied. The results strongly suggest that predicting toxic outcomes of treatments with methylating agents solely based on AAG expression is insufficient without understanding the status of the downstream events in BER, which are discussed in the next section.
3. Downstream events in BER as they relate to methyl damage
Because the AAG class of methylpurine DNA glycosylases appears to be exclusively monofunctional, the processing of damage by bifunctional glycosylases will not be discussed. Briefly, the downstream enzymatic events from monofunctional DNA glycosylase activity are: 1) abasic site (AP) endonuclease activity that produces a strand break with a 3′-OH and 5′-deoxyribosephosphate (5′-dRP) group; 2) dRP lyase to remove the 5′-dRP group and produce a free 5′-phosphate; 3) polymerase activity for DNA resynthesis; 4) ligase to seal the nick (Figure 4). E. coli, S. cerevisiae, and mammals perform these biochemical functions slightly differently. The differences relevant to methylation damage in E. coli and S. cerevisiae will be mentioned in each subsection, while the following description refers to mammalian pathways. The AP endonuclease function is carried out by APE1 (HAP1, APEX1, REF1). In what is referred to as short-patch BER, DNA polymerase β (Pol β) performs two enzymatic functions, polymerase and 5′-dRP lyase. The final step is carried out by DNA ligase III in concert with a scaffold protein, XRCC1. The rate-limiting step in short-patch BER appears to be the 5′-dRP lyase activity (90). In long-patch BER, DNA synthesis displaces a strand of up to seven nucleotides that are terminated by the 5′-dRP group. The displaced nucleotides and 5′-dRP group are removed as a ‘flap’ by FEN1 (flap endonuclease) and DNA ligase I seals the nick. The consensus seems to be that short-patch BER is the predominant pathway in mammalian cells (45). Poly(ADP)-ribose polymerase I (PARP1) also plays a yet to be fully elucidated role in mammalian BER and strand break repair that will be discussed in section 3.3.
3.1. AP endonuclease
E. coli and S. cerevisiae have at least two functional enzymes that possess 5′-AP endonuclease activity. E. coli deficient in Nfo (endonuclease IV) are much more sensitive to MMS than endonuclease III deficient E. coli (45). APN1Δ and APN1Δ/APN2Δ S. cerevisiae are hypersensitive to MMS, but eliminating Mag1 DNA glycosylase substantially reduces the lethality, which suggests that the initial methyl damage is not as deleterious as abasic sites (91, 92). As mentioned earlier, Mag1 over-expression in the absence of methylating agents causes a dramatic increase in mutation frequency (63). Interestingly, Apn1 deficiency is more closely associated with toxicity and mutagenicity of Me-Lex than Mag1 deficiency in S. cerevisiae, suggesting that AP sites (N3-MeA depurination) in yeast are as problematic as N3-MeA (93, 94). Ape−/− mice die very early in embryogenesis and APE deficient mammalian cells have not yet been reported. Mammalian APEs also possesses a redox domain that can reactivate the transcription factor AP-1, which complicates consideration of APE function. Methoxyamine has been used to block BER progression because it reacts with the aldehydic form of abasic sites and inhibits APE activity (95–97). Use of methoxyamine sensitized cells to methylating agents, including the sensitization of MMR deficient cells to TMZ (96).
3.2. DNA polymerase and 5′-dRP end-trimming activity
E. coli, S. cerevisiae, and mammalian cells have somewhat different approaches to these two steps. DNA polymerase I serves as the major BER polymerase in E. coli, but a number of proteins might act to remove 5′-dRP groups. S. cerevisiae appear to lack an explicit 5′-dRP lyase and rely on the Rad27 protein to remove the 5′-dRP group as part of a flap parallel to the function of FEN1 in long-patch BER (Figure 4). RAD27 mutants are hypersensitive to MMS, but the loss of Apn1 largely restores resistance (98). This suggests that BER strand break intermediates are toxic compared to abasic sites. Chicken DT40 cells deficient in FEN1 are viable, which was surprising given that the primary function of FEN1 is thought to be processing Okazaki fragments during replication (99). The FEN1-deficient DT40 cells were hypersensitive to MMS and MNNG, suggesting that this cell type employs long-patch BER following methyl damage (99).
As mentioned above, mammalian cells possess a bifunctional Pol β. The polymerase domain of Pol β is not terribly processive and lacks a 3′ to 5′ proofreading exonuclease function, thus contributing to the relatively high error rates compared to replicative polymerases (100). The second enzymatic activity is a 5′-dRP lyase activity to remove the remaining sugar group left by AP endonuclease activity (101). Pol β−/− mice are embryonic lethal (102). Murine embryonic fibroblast (MEF) cells deficient in Pol β are sensitive to killing induced by MMS and to MNNG (103). The 5′-dRP lyase activity of Pol β, but not the polymerase activity, is required to protect against MMS toxicity (104). Initiating BER in the absence of Pol β leads to numerous negative consequences, including elevated SCEs following MMS treatment (105–108). Indeed, it was shown that knocking out AAG activity in Pol β deficient cells reverses the sensitivity to MMS killing and SCE induction (108). The results suggest that the 5′-dRP group is a toxic BER intermediate and AAG-mediated removal of N-methyl purines is more deleterious than the methyl bases in the absence of Pol β (106, 108). The toxicity and clastogenicity of BER intermediates in Pol β deficient cells appear to be independent of p53 and MMR activity (108). In the absence of Pol β, single strand breaks are seen in cells in G1, while γH2AX is seen in S-phase cells, suggesting that the 5′-dRP group creates a double strand break when encountered by replication (34). Taken together, the data strongly suggest that single strand BER intermediates and the 5′-dRP group in particular are toxic and clastogenic.
3.3. Ligation step and PARP
In mammalian cells, DNA ligase I and ligase III appear biochemically capable of participating in BER (Figure 4). DNA ligase I appears to be limited to long-patch BER, which as mentioned earlier is thought to be a minor branch in mammalian BER. Yet ‘46BR’ cells from a patient with a defect in DNA ligase I were reported to be hypersensitive to killing and SCE induction by MMS, MNU, and MNNG (109, 110). The overexpression of ligase III, but not ligase I, in HeLa cells provided resistance to MNNG (111). DNA ligase III appears to participate in short-patch BER via its physical association with a second protein called XRCC1 (x-ray cross complementation group 1). Xrcc1-deficient mice are embryonic lethal (112). XRCC1-deficient Chinese hamster ovary (CHO) cells are dramatically (12-fold) more sensitive to MMS while only 2-fold more sensitive to MNNG and x-rays (113). In addition to MMS sensitivity, XRCC1 deficient cells are extremely sensitive to SCEs induced by methylating and ethylating agents. The contribution of AAG activity towards the MMS-induced SCEs and sensitivity in XRCC1 deficient cells has not been examined as it has with Pol β deficient cells. The MMS sensitivity of cells that cannot efficiently complete the ligation step is likely caused by the accumulation of BER intermediates similar to that seen in Pol β deficient cells. The latter half of BER, namely Pol β and Ligase III/XRCC1, also participate in a pathway to repair single strand breaks, discussed below (45).
The mystery surrounding the precise roles of poly(ADP-ribose) polymerases (PARPs) extends into interpreting observations regarding PARPs and methylating agents. Among the many PARP family members, PARP1 has been studied most extensively, while PARP2 has more recently been implicated in BER (114). The biochemical function of PARP enzymes is to post-translationally modify proteins including itself by using NAD+ to add ADP-ribose groups. This enzymatic activity is stimulated when PARP1 avidly binds to single-strand breaks, suggesting that PARP1 functions as a sensor of single strand breaks. PARP1 physically interacts with Pol β, Ligase III, and XRCC1, but does not appear to perform an essential catalytic role because BER can be fully reconstituted in its absence in vitro. As with other BER-deficient mammalian cells, PARP1 deficient cells are sensitive to killing by MMS (115). In contrast to Ape1−/−, Pol −/−, and Xrcc1−/− deficient mice, PARP1−/− mice are viable, although sensitive to methylating agents (115). Inhibitors of PARP render cells much more sensitive to alkylating agents such as MMS, although the precise mechanism by which this occurs is not clear (116, 117). Recent evidence suggests that PARP sensitization to MMS, particularly in Pol β deficient cells, occurs when PARP is inhibited during S-phase (118). Lastly, PARP1 has been suggested to play a role in cell death decisions via its ability to deplete cellular NAD+ (119–121). Streptozotocin (STZ) consists of a methyl-nitrosourea conjugated to a pyranose and potently induces diabetes by killing pancreatic β cells. PARP1 deficient mice are resistant to STZ-induced diabetes, suggesting that a PARP1-specific response to the methyl damage or BER intermediates led to pancreatic cell death (119–121). Aag−/− mice are more resistant to the immediate burst of cell death induced by high-dose STZ, suggesting that initiation of BER by Aag provides the strand breaks that activate PARP1 (122). However, the Aag−/− mice displayed a delayed onset of apoptosis and STZ-induced diabetes, which suggested that N3-MeA was capable of inducing cell death in vivo.
The enigmatic role of PARP1 does not diminish its apparent importance as a key player in resolving strand break events in the absence of homologous recombination (HR). Specifically, cells deficient in breast cancer susceptibility genes BRCA1 and BRCA2 are dramatically sensitive to PARP inhibitors in the absence of exogenous damage (123, 124). BRCA1 and BRCA2 are known to be involved in HR and S-phase responses to DNA damage. HR and S-phase responses to methyl damage will be discussed in Section 4. The intriguing connections between PARP, BER, and HR pathways remain to be fully elucidated.
An alternative branch of BER that does not involve AP endonuclease can be initiated by the NEIL family of mammalian DNA glycosylases. The relevance for this review is that this has been proposed as a way to repair single strand breaks (125). The alternative branch involves human polynucleotide kinase phosphatase (PNKP) removing 3′-phosphates to produce a ligatable 3′-OH. PNKP associates with PARP1, and includes Pol β and DNA ligase III/XRCC1 downstream (125). It would be interesting to see whether PNKP deficient cells recapitulate the MMS-sensitive phenotype seen for deficiencies in Pol β, XRCC1, and PARP1.
3.4 Methyl damage and BER intermediates: conclusions
Taken together, the data strongly suggests that BER intermediates are toxic and clastogenic. The data also suggest that the 5′-dRP group is the most problematic of the BER strand break intermediates (Figure 4). Although the 5′-dRP group certainly appears more toxic than N7-MeG, it is harder to compare with N3-MeA or other methyl damage due to the sheer abundance of N7-MeG formed by MMS. Nevertheless, the ability of MMS to produce strand breaks appears to largely depend on the generation of BER intermediates. This seems applicable when considering the MMS sensitivity of S. cerevisiae rad-deficient strains, given the inducibility of Mag1 and an almost reckless ability of Mag1 to remove bases once induced. There is growing evidence that the sensitivity of recombination defective cells likely represent an inability to deal with BER intermediates and methyl damage (Section 3.2). This includes the possibility that strand breaks result from replication forks encountering BER intermediates or methyl bases. The suspect methyl bases in this regard are N3-methyl purines and O2-methyl pyrimidines because of minor groove polymerase contacts, or N1-methyl purines and N3-methyl pyrimidines because of hydrogen bonding faces. Cell-type differences in the balance of BER steps and BER branch choice complicate simplistic conclusions in mammalian cells. However, one conclusion for chemotherapy that produces N-methylpurines is that cells with a high ratio of AAG to Pol β levels are more likely to be killed by methylating agents because of an accumulation of the 5′-dRP group. This would be true for tumor and host tissue, thus providing a potential prediction for unwanted side effects. Considering that tumors have genomic instability phenotypes associated with either microsatellite instability or chromosomal instability (rarely both simultaneously), a thorough elucidation of crosstalk between repair responses should provide better insight into the processes of tumorigenesis.
4. Cleaning up the wreckage: bypass, strand break repair, and cellular responses
Direct reversal proteins and AAG-initiated BER provide the initial defense against methyl damage. The phenotypic responses to MMS and MNNG that have been discussed thus far attempted to focus on specific methyl adducts and BER intermediates. Insufficient direct reversal activity or an imbalance in AAG-initiated BER clearly causes the activation of additional pathways following exposure to MMS or MNNG. In this context, the ‘wreckage’ might be methyl damage, abasic sites, or the 5′-dRP group. The wreckage might also include replication forks that have encountered methyl damage or BER intermediates. There is substantial phenotypic evidence that MMR, polymerase bypass, and homologous recombination pathways are involved following methylating agent exposure. However, ascribing a particular phenotypic response to a single adduct is somewhat problematic when higher doses of MMS and MNNG were used in the absence of manipulating MGMT, AlkB (ABHs in mammalian cells), or AAG levels. It becomes even more challenging when the response and pathway importance is different between species and cell-types within an organism. Nevertheless, some observations regarding downstream consequences are worth placing in context of known adduct-specific responses and differences between MMS and MNNG or MNU.
Nucleotide excision repair (NER) is only briefly mentioned because it is not thought to play a significant role in the response to methylating agents. Studies in S. cerevisiae and mammalian cells, including those lacking AAG-initiated BER, indicate that N3-MeA and N7-MeG appear to be substrates for global genomic nucleotide excision repair but not transcription coupled NER (126–129). There is little phenotypic evidence thus far that NER plays an active role following methyl damage in mammalian cells.
4.1. Lesion bypass polymerases and methyl damage
Discoveries in the past decade have illuminated roles of specialized DNA polymerases, some of which relate to methyl damage and BER. DNA polymerase ι (Pol ι) is one such polymerase, in which nature coincidently provided a natural knockout in the 129 strain of mice (130). Pol ι appears to have a 5′-dRP lyase domain analogous to Pol β, although there is some debate as to whether this domain removes 5′-dRP groups (131, 132). Pol ι can complement biochemically for Pol β in BER in Pol β deficient murine cell extracts (133). However, Pol ι deficient murine cells do not appear to be sensitive to MMS or TMZ, suggesting that Pol ι does not influence survival following methyl damage (35).
The Rev1 and Rev3/7 (Pol ζ) proteins function in an error-prone DNA damage response (45). S. cerevisiae deficient in Rev1, Rev3 (catalytic subunit of Pol ζ), and Pol η (RAD30) are sensitive to killing by MMS; double mutants of Rev1/Pol η and Rev3/Pol η are even more sensitive, suggesting overlapping functions (134). The sensitivity to MNNG or MNU was not reported, which might have been interesting in light of a previous report that yeast and human Pol η can bypass O6-MeG (135). The strong mutator phenotype seen with Mag1 DNA glycosylase overexpression is dependent on Rev1 and Rev3/7, which suggests that the REV proteins participate in the bypass of abasic sites produced by methylpurine DNA glycosylases in S. cerevisiae (63). Mouse and chicken cell lines deficient in REV1, REV3, and Pol κ have been examined for sensitivity to MMS and MNNG. The Pol κ deficient murine cells were moderately sensitive to MMS, whereas the Pol κ deficient chicken cells were not (136). The REV1 and REV3 deficient chicken cells were significantly more sensitive to MMS and MNNG. Double knockouts for REV1/Pol κ and REV3/Pol κ were even more sensitive to MMS, suggesting that Pol κ can either act as a backup to Pol ζ or has non-overlapping functions in this cell type (136). The consensus is that REV1 and REV3/7 are important for the bypass of abasic sites, which should encourage further studies on the interactions between these proteins and BER in mammalian cells. The issue of MMS-induced SCEs should also be examined in cells deficient in different error-prone polymerases to determine whether MMS resistance gained in their presence is at the cost of increased mutagenic or clastogenic events. Lastly, a recent report that S. cerevisiae REV1 is selectively expressed in G2/M goes against the thought that translesion synthesis occurs during S-phase to bypass blocks to replication (137). More work remains to fully understand how error-prone polymerases interact with methyl damage and BER intermediates as part of the cellular response to methylating agents.
4.2. The response of recombinational repair mechanisms to methyl damage
As mentioned earlier, many of the radiation-sensitive (rad) recombinational repair mutants in S. cerevisiae are sensitive to MMS, which is conveniently introduced into the growth medium. The MMS sensitive phenotypes of recombination deficient cells from across many species logically implicate these processes as responding to methyl damage or downstream consequences of the damage. Because recombinational repair responds to double strand breaks (DSBs), it seemed reasonable to presume that MMS causes DSBs. Recombination-deficient E. coli are as sensitive to MMS and MNNG as Tag and AlkA deficient strains, while mutants deficient in recombination and BER were further sensitized (138). The increased sensitization of double mutants indicates that BER and recombination act in independent pathways and suggests that recombination is necessary to repair methyl damage or BER strand breaks that block replication in E. coli (138). Modulation of Tag and Mag1 levels in S. cerevisiae showed that too little glycosylase activity results in N3-MeA induced recombination, while too much Mag1 activity results in recombination induced by BER strand break intermediates (64).
There are two primary repair pathways for processing DSBs in eukaryotes, the nonhomologous end-joining (NHEJ) recombination pathway and the homologous recombination (HR) pathway. In mammalian cells, NHEJ is generally thought to be responsible for resolving DSBs in G1, while HR is responsible for resolving DSBs in S and G2/M (139). Defects in either pathway lead to radiation sensitivity. However, phenotypic evidence suggests that HR plays a more significant role in the response to damage resulting from MMS and MNNG exposure (Figure 6). Based on sequence homology, a number of recombinational repair genes were identified from the mammalian genome, permitting generation of knockout mutants in vertebrate cell lines and in mouse, reviewed in (140). These tools added to the already existing cell lines isolated by screening for recombination repair-deficient CHO cells generated by random mutagenesis (141, 142). Vertebrates possess seven members of the RAD51/RecA family of HR genes (RAD51, RAD51B, RAD51C, RAD51D, DMC1, XRCC2, and XRCC3). Their functions diverged during eukaryotic evolution with RAD51 primarily retaining the RecA-like functions in mitotically dividing cells (143) and DMC1 being exclusive to meiosis (144, 145). RAD51 is an essential gene in mammalian cell lines, thus limiting studies of sensitivity to DNA damaging agents (146–148).
Figure 6.

Events downstream of methyl base damage can lead to homologous recombination repair. BER intermediates or unrepaired methyl damage encountered by replication, or MMR recognition of O6-MeG:T mispairs can induce homologous recombination events. Homologous recombination promotes strand invasion onto a sister chromatid template to initiate repair. DNA synthesis and resolution of the Holliday junction completes the process. If left unrepaired, double strand breaks lead to chromosomal rearrangements.
Even though mutations in each of the remaining RAD51/RecA genes are also embryonic lethal in mice, vertebrate cell lines carrying mutations in four of the RAD51 paralog genes are available. The deficient cells examined share phenotypes of being extremely sensitive to DNA cross-linking agents and having high levels of spontaneous chromosomal instability that resembles chromosome rearrangements seen in the absence of BRCA1 and BRCA2 (149). Where tested, each repair-deficient cell line is sensitive to methylating agents. For example, RAD51D-deficient CHO and MEF cell lines are 5 and 6.3-fold more sensitive to MMS, respectively (150, 151). Xrcc2-deficient MEFs and XRCC3-deficient CHO cells are also highly sensitive to MNNG, suggesting that HR may process secondary lesions resulting from O6-MeG (36, 152). Indeed, several recent studies raise the intriguing possibility of a link between MMR recognition of O6-MeG and HR. In yeast, MMR deficiency rescues the MNNG-sensitive phenotype of HR deficient strains, providing evidence that MMR recognition of O6-MeG causes events that require HR (153). The connections between O6-MeG, MMR, and damage signaling will be discussed in section 4.3.
Despite being a major pathway for dealing with DSBs, NHEJ appears to play a lesser role following methyl damage as inferred by phenotypes in NHEJ-deficient cells. Ku80 (hdf2) mutant yeast strains are much less sensitive to MMS than RAD52 HR mutants (154). XRCC5 (Ku80) deficient CHO cell lines are modestly (2-fold) more sensitive to MMS (155). A DNA PKcs-deficient cell line was not sensitive to MNNG and only mildly sensitive to MMS (36). The thought that NHEJ predominates in G1, coupled with weak phenotypic evidence, suggests that methylating agents do not directly cause DSBs. In fact, MMS failed to produce cellular DSBs as measured by pulsed-field gel electrophoresis, and no evidence was observed for replication fork-associated DSBs following exposure to MNNG and MMS (36). Alternatively, methyl damage and BER intermediates may not be processed into ‘traditional’ DSBs that can be acted upon by NHEJ. In summary, there is little evidence that MMS or MNNG directly induce double strand breaks. Yet, repair of BER intermediates clearly requires HR proteins. The idea that HR is involved in ‘non-DSB’ repair networks has been proposed and is likely to be the case in the response to methylating agents (156). The thought that HR is important during S-phase and G2/M agrees with evidence that methylating agents exert cytotoxic effects during S-phase or G2/M and with the sensitivity of HR deficient cells to the methylating agents (Figure 6). Signaling responses in S-phase and G2/M following MMS and MNNG treatment appear to be closely related to HR and are discussed below.
4.3. Methyl DNA damage signaling responses
The ATM, ATR, and p53 proteins have received much attention as key players in DNA damage signaling pathways. Full discussion of these pathways is beyond the scope of this review. Although ATM and p53 can be activated following treatment with MNNG, studies have shown that apoptosis induced by O6-MeG is independent of ATM and p53 (26, 157). ATM deficient cells are hypersensitive to killing by ionizing radiation and radiomimetics, although their phenotype to methylating agents is not as clear. Atm−/− MEFs (also p53−/−) were more sensitive to MNNG than wild-type, but only modestly sensitive to MMS (157). Interestingly, the Atm−/− murine fibroblasts showed more SCEs in the second mitosis following MNNG, reminiscent of the observations in MMR proficient cells (157).
There is emerging evidence that some MMR proteins participate as a damage surveillance mechanism that is a distinct function from the post-replicative repair of mismatches (Figure 5). Mutants of MSH2 and MSH6 have been generated that retain the ability to bind to mismatches, but cannot carryout completion of mismatch repair (158, 159). When treated with MNNG, the MSH6 mutant cells remain as sensitive to apoptotic death as MSH6 wild-type cells, suggesting that the mismatch repair and damage-sensing functions of MMR are distinct (159). The fact that MLH1-deficient cells become resistant to apoptotic death induced by MNNG and MNU suggests that the damage surveillance mechanism includes the MLH1:PMS2 heterodimer, but there is no evidence that the MSH2:MSH3 heterodimer (MutSβ) participates. In S. cerevisiae, inactivating the exonuclease implicated in mismatch repair (EXO1) does not rescue sensitivity to MNNG, thus suggesting that the exonuclease step of MMR is not involved in the damage surveillance pathway (153). An earlier report suggested that MMR has a role in G2 arrest (160). MNNG and MNU at low doses induce a G2/M arrest in the second round of replication that is dependent on MutSα and MLH1 (22, 161). The amount of damage is important in this regard, because high doses of MNNG induce different signaling responses, presumably because excessive BER intermediates result from AAG excision of N7-MeG (162). The MMR-dependent G2/M arrest by low dose MNNG was shown to require ATR (23). More recently, the recognition of O6-MeG:T, but not O6-MeG:C or G:T, by MutSα was shown to recruit ATR-ATRIP (163). Taken together, the suggestion is that MutSα binding to O6-MeG:T serves to provide a signal for G2 arrest via an ATR-dependent signaling process. There was recently published biochemical evidence for reiterative abortive attempts at repair when O6-MeG is located on the template strand (164). The authors note that a ‘futile cycling’ and ‘damage signaling’ model of MMR dependent apoptosis are not mutually exclusive (164). Another issue to be resolved is how the MMR-dependent G2 arrest results in aberrant recombination events that leads to SCEs (37, 50). A more explicit link between MMR and HR was recently demonstrated, namely that loss of MMR deregulates HR (165). The implication is that MMR recognition of O6-MeG:T creates an aberrant DNA structure that requires HR for resolution. It also raises the intriguing possibility that the recruitment of ATR is a response to catastrophic MMR-dependent recombination events.
More investigation is required to understand the interactions of replication forks with methyl damage and BER intermediates. Studies with S. cerevisiae deficient in Mec1, the yeast homologue of ATR, indicate that MMS damage reduces the rate of replication fork progression and exerts its toxicity specifically during S-phase; it is presumed that these strains contained wild-type MAG1 (166). Aag−/− cells progress through S-phase slower following Me-Lex treatment that specifically produces N3-MeA, which argues that N3-MeA can block replication in ES cells (15). A large multiprotein complex, named BASC (BRCA1-associated genome surveillance complex), is involved in S-phase DNA damage response and includes among others MLH1, MSH2, MSH6, BRCA1, ATM, and BLM (167). The putative complex is noteworthy in that deficiencies in each result in cancer predisposition (colon or breast) or rare, severe clastogenic syndromes. Cells from patients with defects in BLM (Bloom’s syndrome) show S-phase defects, elevated spontaneous SCEs, and also hypersensitivity to killing by MMS, MNU, and MNNG (109). BLM is a RecQ helicase family member that appears to play a role in modulating recombination events during replication. The sensitivity of BLM-deficient cells might result from an inability to process methyl damage or BER intermediates. Taken together, damage signaling events probably result from a combination of responses to the variety of damage that results from MMS or MNNG exposure. Methyl damage and/or BER intermediates are problematic in S-phase and G2/M, with the best evidence emerging for O6-MeG thus far. More work is clearly needed to understand damage response events in S-phase and G2/M. UV damage in S. cerevisiae appears to uncouple leading and lagging strand synthesis, creating long and short single strand gaps that can persist into G2 (168). The authors suggest that damage responses during replication combine the efforts of translesion synthesis and HR in order to overcome damage encountered by replication forks (168). These observations are relevant and intriguing when considering the consequences of methyl damage, most of which are capable of blocking replication. There have been studies that suggest direct reversal and BER proteins act near replication forks. More studies are necessary on the crosstalk between repair proteins and pathways in S-phase and G2/M, in particular.
4.4. Global transcriptional responses and phenotypic screening
The advent of global analyses of responses to damage has greatly broadened the picture of how cells respond to MMS and MNNG and was reviewed recently (169). MMS and MNNG can alter the gene expression pattern of >1000 genes in S. cerevisiae, revealing many more genes than those directly involved in a DNA damage response. MMS and MNNG can essentially react with any cellular component that possesses nucleophilic character, so it is not surprising that transcriptional responses to methylating agents suggest that damage to RNA and proteins is occurring (169). MMS and MNNG also produce distinct transcriptional responses from each other. Yet it is interesting to note that the transcriptional responsiveness of a gene following DNA damage does not predict whether the gene product influences survival (169).
Combining genomic phenotypic screening with expression profiling provides a more thorough examination of assessing network interactions (170, 171). Genomic phenotypic screening in S. cerevisiae utilizes arrays containing nearly all viable single gene knockout strains that can be tested for toxicity to agents or stress conditions. Comparison between MMS and other DNA damaging agents revealed methylation-specific responses versus generalized network stress responses (170, 171). Genomic screening revealed a subset of genes that specifically provided resistance to MMS during S-phase (172). A direct genomic phenotypic comparison between MMS and MNNG or MNU might uncover as yet unknown responses that are specific for individual methyl adducts. Obvious challenges remain to move global approaches into mice and humans, where it will be necessary to understand differences between many different cell types, including tissue specific biases in pathway selection and network response.
5. Summary
Our understanding of the first line recognition of methyl adducts has come quite a ways in 25 years (Figure 2 and 3). If not dealt with, some forms of methyl damage are toxic, likely dependent on replication forks encountering the damage, be it an immediate block or creation of a mismatched base pair capable of initiating cell death. BER-mediated removal of N-methyl purines presents a curious balance considering that N7-MeG, the single greatest adduct produced by most methylating agents, does not appear to block replication or miscode. The general consensus is that BER intermediates generated by removing N-methyl purines are toxic and clastogenic if encountered during replication. In particular, the 5′-dRP group appears significantly toxic and clastogenic in S. cerevisiae and mammalian cells. Single strand breaks seen during MMS treatment are likely BER intermediates, whereas DSBs result from replication forks encountering methyl damage or BER strand break intermediates.
Future research will provide better understanding of how multiple pathway interactions work, particularly in mammalian cells. One predicts that in order to therapeutically exploit the knowledge of BER intermediates and O6-MeG, a better understanding is required of the connections to HR and the damage surveillance proteins of MMR. Indeed, the knowledge that chemotherapeutic alkylating agents have an unfortunate track record of inducing secondary, therapy-related cancers should give serious pause (173). Loss of MMR has been proposed to cause the deregulation of HR as a step during chemotherapy-induced leukemia (165). A better understanding is also needed of the links between methyl adducts, BER intermediates, and replication forks. Cancer cells that contain alterations in repair and apoptotic pathways presumably tilt the balance between survival and death. The viability of Mgmt−/−, Aag−/−, and Abh2−/− strains of mice allow these deficiencies to be crossed into other repair deficient backgrounds to test pathway interactions (48, 49, 61, 84). Studies with such knockouts and heterologous add-back expression of MGMT, AlkB (ABHs), and AAG will contribute to ascribing phenotypic responses to specific adducts, or at least the range of adducts repaired by each protein. Lastly, better understanding the crosstalk between damage response and repair pathways should provide a better picture to the systematic cellular response to methyl damage.
Acknowledgments
M.D.W. is supported by NIH grant number CA100450 from NCI. D.L.P. is supported by American Cancer Society grant number RSG-030158-01-GMC. The authors thank Drs. Alan Waldman and J. Walter Sowell for helpful discussions, and to the reviewers for helpful comments.
Footnotes
Abbreviations: MMS, methylmethane sulfonate; DMS, dimethyl sulfate; MNNG, N-methyl-N′-nitro-N-nitrosoguanidine; MNU, N-methyl-N-nitrosourea; TMZ, temozolomide; MGMT, O6-methylguanine methyltransferase; MMR, mismatch repair; Me-Lex, methyl-lexitropsin; BER, base excision repair; AAG, methylpurine DNA glycosylase (MPG, APNG); AP site, abasic site; APE, AP endonuclease; pol, DNA polymerase; 5′-dRP, 5′-deoxyribosephosphate; XRCC, x-ray cross complementation; PARP, poly(ADP-ribose) polymerase; rad, radiation sensitive; HR, homologous recombination; NHEJ, non-homologous end-joining; SCE, sister chromatid exchange.
References
- 1.Pullman A, Pullman B. Molecular electrostatic potential of the nucleic acids. Q Rev Biophys. 1981;14:289–380. doi: 10.1017/s0033583500002341. [DOI] [PubMed] [Google Scholar]
- 2.Galtress CL, Morrow PR, Nag S, Smalley TL, Tschantz MF, Vaughn JS, Wichems DN, Ziglar SK, Fishbein JC. Mechanism for the Solvolytic Decomposition of the Carcinogen N-Methyl-N′-Nitro-N-Nitrosoguanidine in Aqueous-Solutions. J Am Chem Soc. 1992;114:1406–1411. [Google Scholar]
- 3.Loechler EL. A violation of the Swain-Scott principle, and not SN1 versus SN2 reaction mechanisms, explains why carcinogenic alkylating agents can form different proportions of adducts at oxygen versus nitrogen in DNA. Chem Res Toxicol. 1994;7:277–280. doi: 10.1021/tx00039a001. [DOI] [PubMed] [Google Scholar]
- 4.Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev. 1997;23:35–61. doi: 10.1016/s0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- 5.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 6.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 7.Bodell WJ, Singer B. Influence of hydrogen bonding in DNA and polynucleotides on reaction of nitrogens and oxygens toward ethylnitrosourea. Biochemistry. 1979;18:2860–2863. doi: 10.1021/bi00580a029. [DOI] [PubMed] [Google Scholar]
- 8.Osborne MR, Phillips DH. Preparation of a methylated DNA standard, and its stability on storage. Chem Res Toxicol. 2000;13:257–261. doi: 10.1021/tx990182e. [DOI] [PubMed] [Google Scholar]
- 9.O’Connor TR, Boiteux S, Laval J. Ring-opened 7-methylguanine residues in DNA are a block to in vitro DNA synthesis. Nucleic Acids Res. 1988;16:5879–5894. doi: 10.1093/nar/16.13.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boiteux S, Laval J. Imidazole open ring 7-methylguanine: an inhibitor of DNA synthesis. Biochemical & Biophysical Research Communications. 1983;110:552–558. doi: 10.1016/0006-291x(83)91185-3. [DOI] [PubMed] [Google Scholar]
- 11.Kohn KW, Hartley JA, Mattes WB. Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res. 1987;15:10531–10549. doi: 10.1093/nar/15.24.10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye N, Holmquist GP, O’Connor TR. Heterogeneous repair of N-methylpurines at the nucleotide level in normal human cells. J Mol Biol. 1998;284:269–285. doi: 10.1006/jmbi.1998.2138. [DOI] [PubMed] [Google Scholar]
- 13.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 14.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 15.Engelward BP, Allan JM, Dreslin JA, Kelly JD, Gold B, Samson LD. A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the mammalian genome. J Biol Chem. 1998;273:5412–5418. doi: 10.1074/jbc.273.9.5412. [DOI] [PubMed] [Google Scholar]
- 16.Fronza G, Gold B. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250–257. doi: 10.1002/jcb.10698. [DOI] [PubMed] [Google Scholar]
- 17.Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc Natl Acad Sci U S A. 1985;82:1376–1380. doi: 10.1073/pnas.82.5.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Encell L, Shuker DEG, Foiles PG, Gold B. The in vitro methylation of DNA by a minor groove binding methyl sulfonate ester. Chem Res Toxicol. 1996;9:563–567. doi: 10.1021/tx9501849. [DOI] [PubMed] [Google Scholar]
- 19.Ezaz-Nikpay K, Verdine GL. The effects of N7-methylguanine on duplex DNA structure. Chem Biol. 1994;1:235–240. doi: 10.1016/1074-5521(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 20.Loechler EL, Green CL, Essigmann JM. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci U S A. 1984;81:6271–6275. doi: 10.1073/pnas.81.20.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldmacher VS, Cuzick RA, Jr, Thilly WG. Isolation and partial characterization of human cell mutants differing in sensitivity to killing and mutation by methylnitrosourea and N-methyl-N′-nitro-N-nitrosoguanidine. J Biol Chem. 1986;261:12462–12471. [PubMed] [Google Scholar]
- 22.Aquilina G, Crescenzi M, Bignami M. Mismatch repair, G(2)/M cell cycle arrest and lethality after DNA damage. Carcinogenesis. 1999;20:2317–2326. doi: 10.1093/carcin/20.12.2317. [DOI] [PubMed] [Google Scholar]
- 23.Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhukovskaya N, Branch P, Aquilina G, Karran P. DNA replication arrest and tolerance to DNA methylation damage. Carcinogenesis. 1994;15:2189–2194. doi: 10.1093/carcin/15.10.2189. [DOI] [PubMed] [Google Scholar]
- 25.D’Atri S, Tentori L, Lacal PM, Graziani G, Pagani E, Benincasa E, Zambruno G, Bonmassar E, Jiricny J. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol Pharmacol. 1998;54:334–341. doi: 10.1124/mol.54.2.334. [DOI] [PubMed] [Google Scholar]
- 26.Hickman MJ, Samson LD. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proc Natl Acad Sci USA. 1999;96:10764–10769. doi: 10.1073/pnas.96.19.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meikrantz W, Bergom MA, Memisoglu A, Samson L. O6-alkylguanine DNA lesions trigger apoptosis. Carcinogenesis. 1998;19:369–372. doi: 10.1093/carcin/19.2.369. [DOI] [PubMed] [Google Scholar]
- 28.Pauly GT, Moschel RC. Mutagenesis by O(6)-methyl-, O(6)-ethyl-, and O(6)-benzylguanine and O(4)-methylthymine in human cells: effects of O(6)-alkylguanine-DNA alkyltransferase and mismatch repair. Chem Res Toxicol. 2001;14:894–900. doi: 10.1021/tx010032f. [DOI] [PubMed] [Google Scholar]
- 29.Altshuler KB, Hodes CS, Essigmann JM. Intrachromosomal probes for mutagenesis by alkylated DNA bases replicated in mammalian cells: a comparison of the mutagenicities of O4-methylthymine and O6-methylguanine in cells with different DNA repair backgrounds. Chem Res Toxicol. 1996;9:980–987. doi: 10.1021/tx960062w. [DOI] [PubMed] [Google Scholar]
- 30.Dinglay S, Trewick SC, Lindahl T, Sedgwick B. Defective processing of methylated single-stranded DNA by E. coli AlkB mutants. Genes Dev. 2000;14:2097–2105. [PMC free article] [PubMed] [Google Scholar]
- 31.Delaney JC, Essigmann JM. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:14051–14056. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. ASM Press; Washington, D.C: 1995. [Google Scholar]
- 33.Kaina B. Mechanisms and consequences of methylating agent-induced SCEs and chromosomal aberrations: a long road traveled and still a far way to go. Cytogenet Genome Res. 2004;104:77–86. doi: 10.1159/000077469. [DOI] [PubMed] [Google Scholar]
- 34.Pascucci B, Russo MT, Crescenzi M, Bignami M, Dogliotti E. The accumulation of MMS-induced single strand breaks in G1 phase is recombinogenic in DNA polymerase β defective mammalian cells. Nucleic Acids Res. 2005;33:280–288. doi: 10.1093/nar/gki168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005;65:6394–6400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]
- 36.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armstrong MJ, Galloway SM. Mismatch repair provokes chromosome aberrations in hamster cells treated with methylating agents or 6-thioguanine, but not with ethylating agents. Mutat Res. 1997;373:167–178. doi: 10.1016/s0027-5107(96)00234-5. [DOI] [PubMed] [Google Scholar]
- 38.Galloway SM, Greenwood SK, Hill RB, Bradt CI, Bean CL. A role for mismatch repair in production of chromosome aberrations by methylating agents in human cells. Mutat Res. 1995;346:231–245. doi: 10.1016/0165-7992(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 39.Kaina B, Fritz G, Coquerelle T. Contribution of O6-alkylguanine and N-alkylpurines to the formation of sister chromatid exchanges, chromosomal aberrations, and gene mutations: new insights gained from studies of genetically engineered mammalian cell lines. Environmental & Molecular Mutagenesis. 1993;22:283–292. doi: 10.1002/em.2850220418. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Tsujimura T, Bhattacharyya NP, Maher VM, McCormick JJ. O6-methylguanine induces intrachromosomal homologous recombination in human cells. Carcinogenesis. 1996;17:2229–2235. doi: 10.1093/carcin/17.10.2229. [DOI] [PubMed] [Google Scholar]
- 41.Zhang H, Marra G, Jiricny J, Maher VM, McCormick JJ. Mismatch repair is required for O(6)-methylguanine-induced homologous recombination in human fibroblasts. Carcinogenesis. 2000;21:1639–1646. doi: 10.1093/carcin/21.9.1639. [DOI] [PubMed] [Google Scholar]
- 42.Samson L, Cairns J. A new pathway for DNA repair in Escherichia coli. Nature. 1977;267:281–283. doi: 10.1038/267281a0. [DOI] [PubMed] [Google Scholar]
- 43.Falnes PO. Repair of 3-methylthymine and 1-methylguanine lesions by bacterial and human AlkB proteins. Nucleic Acids Res. 2004;32:6260–6267. doi: 10.1093/nar/gkh964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sedgwick B, Lindahl T. Recent progress on the Ada response for inducible repair of DNA alkylation damage. Oncogene. 2002;21:8886–8894. doi: 10.1038/sj.onc.1205998. [DOI] [PubMed] [Google Scholar]
- 45.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D.C: 2006. [Google Scholar]
- 46.Dolan ME, Moschel RC, Pegg AE. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci USA. 1990;87:5368–5372. doi: 10.1073/pnas.87.14.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pegg AE. Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 48.Glassner BJ, Weeda G, Allan JM, Broekhof JL, Carls NH, Donker I, Engelward BP, Hampson RJ, Hersmus R, Hickman MJ, Roth RB, Warren HB, Wu MM, Hoeijmakers JH, Samson LD. DNA repair methyltransferase (Mgmt) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis. 1999;14:339–347. doi: 10.1093/mutage/14.3.339. [DOI] [PubMed] [Google Scholar]
- 49.Tsuzuki T, Sakumi K, Shiraishi A, Kawate H, Igarashi H, Iwakuma T, Tominaga Y, Zhang S, Shimizu S, Ishikawa T, et al. Targeted disruption of the DNA repair methyltransferase gene renders mice hypersensitive to alkylating agent. Carcinogenesis. 1996;17:1215–1220. doi: 10.1093/carcin/17.6.1215. [DOI] [PubMed] [Google Scholar]
- 50.Kaina B, Ziouta A, Ochs K, Coquerelle T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: facts and models. Mutat Res. 1997;381:227–241. doi: 10.1016/s0027-5107(97)00187-5. [DOI] [PubMed] [Google Scholar]
- 51.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Kawate H, Sakumi K, Tsuzuki T, Nakatsuru Y, Ishikawa T, Takahashi S, Takano H, Noda T, Sekiguchi M. Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc Natl Acad Sci U S A. 1998;95:5116–5120. doi: 10.1073/pnas.95.9.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bignami M, O’Driscoll M, Aquilina G, Karran P. Unmasking a killer: DNA O(6)-methylguanine and the cytotoxicity of methylating agents. Mutat Res. 2000;462:71–82. doi: 10.1016/s1383-5742(00)00016-8. [DOI] [PubMed] [Google Scholar]
- 54.O’Brien V, Brown R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis. 2006;27:682–692. doi: 10.1093/carcin/bgi298. [DOI] [PubMed] [Google Scholar]
- 55.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair (Amst) 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 56.Aravind L, Koonin EV. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2001;2:RESEARCH0007. doi: 10.1186/gb-2001-2-3-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M, Sundheim O, Bjoras M, Slupphaug G, Seeberg E, Krokan HE. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 58.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 59.Chen BJ, Carroll P, Samson L. The Escherichia coli AlkB protein protects human cells against alkylation-induced toxicity. Journal of Bacteriology. 1994;176:6255–6261. doi: 10.1128/jb.176.20.6255-6261.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koivisto P, Robins P, Lindahl T, Sedgwick B. Demethylation of 3-methylthymine in DNA by bacterial and human DNA dioxygenases. J Biol Chem. 2004;279:40470–40474. doi: 10.1074/jbc.M407960200. [DOI] [PubMed] [Google Scholar]
- 61.Ringvoll J, Nordstrand LM, Vagbo CB, Talstad V, Reite K, Aas PA, Lauritzen KH, Liabakk NB, Bjork A, Doughty RW, Falnes PO, Krokan HE, Klungland A. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 2006;25:2189–2198. doi: 10.1038/sj.emboj.7601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bjelland S, Bjoras M, Seeberg E. Excision of 3-methylguanine from alkylated DNA by 3-methyladenine DNA glycosylase I of Escherichia coli. Nucleic Acids Research. 1993;21:2045–2049. doi: 10.1093/nar/21.9.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glassner BJ, Rasmussen LJ, Najarian MT, Posnick LM, Samson LD. Generation of a strong mutator phenotype in yeast by imbalanced base excision repair. Proc Natl Acad Sci USA. 1998;95:9997–10002. doi: 10.1073/pnas.95.17.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendricks CA, Razlog M, Matsuguchi T, Goyal A, Brock AL, Engelward BP. The S. cerevisiae Mag1 3-methyladenine DNA glycosylase modulates susceptibility to homologous recombination. DNA Repair. 2002;1:645–659. doi: 10.1016/s1568-7864(02)00072-1. [DOI] [PubMed] [Google Scholar]
- 65.Posnick LM, Samson LD. Imbalanced base excision repair increases spontaneous mutation and alkylation sensitivity in Escherichia coli. J Bacteriol. 1999;181:6763–6771. doi: 10.1128/jb.181.21.6763-6771.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCarthy TV, Karran P, Lindahl T. Inducible repair of O-alkylated DNA pyrimidines in Escherichia coli. EMBO Journal. 1984;3:545–550. doi: 10.1002/j.1460-2075.1984.tb01844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 68.Wyatt MD, Allan JM, Lau AY, Ellenberger TE, Samson LD. 3-methyladenine DNA glycosylases: structure, function, and biological importance. BioEssays. 1999;21:668–676. doi: 10.1002/(SICI)1521-1878(199908)21:8<668::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 69.Biswas T, Clos LJ, 2nd, SantaLucia J, Jr, Mitra S, Roy R. Binding of Specific DNA Base-pair Mismatches by N-Methylpurine-DNA Glycosylase and Its Implication in Initial Damage Recognition. J Mol Biol. 2002;320:503–513. doi: 10.1016/S0022-2836(02)00519-3. [DOI] [PubMed] [Google Scholar]
- 70.Gros L, Maksimenko AV, Privezentzev CV, Laval J, Saparbaev MK. Hijacking of the human alkyl-N-purine-DNA glycosylase by 3, N-4-ethenocytosine, a lipid peroxidation-induced DNA adduct. J Biol Chem. 2004;279:17723–7730. doi: 10.1074/jbc.M314010200. [DOI] [PubMed] [Google Scholar]
- 71.O’Brien PJ, Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem. 2004;279:9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 72.Berdal KG, Johansen RF, Seeberg E. Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J. 1998;17:363–367. doi: 10.1093/emboj/17.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Brien PJ, Ellenberger T. The Escherichia coli 3-methyladenine-DNA glycosylase AlkA has a remarkably versatile active site. J Biol Chem. 2004;4:26876–26884. doi: 10.1074/jbc.M403860200. [DOI] [PubMed] [Google Scholar]
- 74.Connor EE, Wyatt MD. Active-site clashes prevent the human 3-methyladenine DNA glycosylase from improperly removing bases. Chem Biol. 2002;9:1033–1041. doi: 10.1016/s1074-5521(02)00215-6. [DOI] [PubMed] [Google Scholar]
- 75.Karran P, Hjelmgren T, Lindahl T. Induction of a DNA glycosylase for N-methylated purines is part of the adaptive response to alkylating agents. Nature. 1982;296:770–773. doi: 10.1038/296770a0. [DOI] [PubMed] [Google Scholar]
- 76.Evensen G, Seeberg E. Adaptation to alkylation resistance involves the induction of a DNA glycosylase. Nature. 1982;296:773–775. doi: 10.1038/296773a0. [DOI] [PubMed] [Google Scholar]
- 77.Chen J, Derfler B, Samson L. Saccharomyces cerevisiae 3-methyladenine DNA glycosylase has homology to the AlkA glycosylase of E. coli and is induced in response to DNA alkylation damage. EMBO J. 1990;9:4569–4575. doi: 10.1002/j.1460-2075.1990.tb07910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Memisoglu A, Samson L. Contribution of base excision repair, nucleotide excision repair, and DNA recombination to alkylation resistance of the fission yeast Schizosaccharomyces pombe. J Bacteriol. 2000;182:2104–2112. doi: 10.1128/jb.182.8.2104-2112.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, Hussain SP, Roth RB, Zhou XL, Mechanic LE, Zurer I, Rotter V, Samson LD, Harris CC. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. Journal of Clinical Investigation. 2003;112:1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Calleja F, Jansen JG, Vrieling H, Laval F, van Zeeland AA. Modulation of the toxic and mutagenic effects induced by methyl methanesulfonate in Chinese hamster ovary cells by overexpression of the rat N-alkylpurine-DNA glycosylase. Mutat Res. 1999;425:185–194. doi: 10.1016/s0027-5107(99)00034-2. [DOI] [PubMed] [Google Scholar]
- 81.Coquerelle T, Dosch J, Kaina B. Overexpression of N-methylpurine-DNA glycosylase in Chinese hamster ovary cells renders them more sensitive to the production of chromosomal aberrations by methylating agents--a case of imbalanced DNA repair. Mutat Res. 1995;336:9–17. doi: 10.1016/0921-8777(94)00035-5. [DOI] [PubMed] [Google Scholar]
- 82.Ibeanu G, Hartenstein B, Dunn WC, Chang LY, Hofmann E, Coquerelle T, Mitra S, Kaina B. Overexpression of human DNA repair protein N-methylpurine-DNA glycosylase results in the increased removal of N-methylpurines in DNA without a concomitant increase in resistance to alkylating agents in Chinese hamster ovary cells. Carcinogenesis. 1992;13:1989–1995. doi: 10.1093/carcin/13.11.1989. [DOI] [PubMed] [Google Scholar]
- 83.Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson L. Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. EMBO J. 1996;15:945–952. [PMC free article] [PubMed] [Google Scholar]
- 84.Engelward BP, Weeda G, Wyatt MD, Broekhof JLM, De Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JHJ, Samson LD. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc Natl Acad Sci USA. 1997;94:13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Elder RH, Jansen JG, Weeks RJ, Willington MA, Deans B, Watson AJ, Mynett KJ, Bailey JA, Cooper DP, Rafferty JA, Heeran MC, Wijnhoven SW, van Zeeland AA, Margison GP. Alkylpurine-DNA-N-glycosylase knockout mice show increased susceptibility to induction of mutations by methyl methanesulfonate. Mol Cell Biol. 1998;18:5828–5837. doi: 10.1128/mcb.18.10.5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roth RB, Samson LD. 3-Methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance. Cancer Res. 2002;62:656–660. [PubMed] [Google Scholar]
- 87.Paik J, Duncan T, Lindahl T, Sedgwick B. Sensitization of human carcinoma cells to alkylating agents by small interfering RNA suppression of 3-alkyladenine-DNA glycosylase. Cancer Res. 2005;65:10472–10477. doi: 10.1158/0008-5472.CAN-05-1495. [DOI] [PubMed] [Google Scholar]
- 88.Rinne ML, He Y, Pachkowski BF, Nakamura J, Kelley MR. N-methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing non-toxic 7-methylguanine adducts. Nucleic Acids Res. 2005;33:2859–2867. doi: 10.1093/nar/gki601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Connor EE, Wilson JJ, Wyatt MD. Effects of substrate specificity on initiating the base excision repair of N-methylpurines by variant human 3-methyladenine DNA glycosylases. Chem Res Toxicol. 2005;18:87–94. doi: 10.1021/tx049822q. [DOI] [PubMed] [Google Scholar]
- 90.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 91.Xiao W, Chow BL, Hanna M, Doetsch PW. Deletion of the MAG1 DNA glycosylase gene suppresses alkylation-induced killing and mutagenesis in yeast cells lacking AP endonucleases. Mutat Res. 2001;487:137–147. doi: 10.1016/s0921-8777(01)00113-6. [DOI] [PubMed] [Google Scholar]
- 92.Xiao W, Samson L. In vivo evidence for endogenous DNA alkylation damage as a source of spontaneous mutation in eukaryotic cells. Proc Natl Acad Sci U S A. 1993;90:2117–2121. doi: 10.1073/pnas.90.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Monti P, Campomenosi P, Ciribilli Y, Iannone R, Inga A, Shah D, Scott G, Burns PA, Menichini P, Abbondandolo A, Gold B, Fronza G. Influences of base excision repair defects on the lethality and mutagenicity induced by Me-lex, a sequence-selective N3-adenine methylating agent. J Biol Chem. 2002;277:28663–28668. doi: 10.1074/jbc.M203384200. [DOI] [PubMed] [Google Scholar]
- 94.Kelly JD, Inga A, Chen FX, Dande P, Shah D, Monti P, Aprile A, Burns PA, Scott G, Abbondandolo A, Gold B, Fronza G. Relationship between DNA methylation and mutational patterns induced by a sequence selective minor groove methylating agent. J Biol Chem. 1999;274:18327–18334. doi: 10.1074/jbc.274.26.18327. [DOI] [PubMed] [Google Scholar]
- 95.Horton JK, Prasad R, Hou E, Wilson SH. Protection against methylation-induced cytotoxicity by DNA polymerase β-dependent long patch base excision repair. J Biol Chem. 2000;275:2211–2218. doi: 10.1074/jbc.275.3.2211. [DOI] [PubMed] [Google Scholar]
- 96.Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL. Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clin Cancer Res. 1999;5:2908–2917. [PubMed] [Google Scholar]
- 97.Taverna P, Liu L, Hwang HS, Hanson AJ, Kinsella TJ, Gerson SL. Methoxyamine potentiates DNA single strand breaks and double strand breaks induced by temozolomide in colon cancer cells. Mutat Res. 2001;485:269–281. doi: 10.1016/s0921-8777(01)00076-3. [DOI] [PubMed] [Google Scholar]
- 98.Wu X, Wang Z. Relationships between yeast Rad27 and Apn1 in response to apurinic/apyrimidinic (AP) sites in DNA. Nucleic Acids Res. 1999;27:956–962. doi: 10.1093/nar/27.4.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matsuzaki Y, Adachi N, Koyama H. Vertebrate cells lacking FEN-1 endonuclease are viable but hypersensitive to methylating agents and H2O2. Nucleic Acids Res. 2002;30:3273–3277. doi: 10.1093/nar/gkf440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Osheroff WP, Jung HK, Beard WA, Wilson SH, Kunkel TA. The fidelity of DNA polymerase β during distributive and processive DNA synthesis. J Biol Chem. 1999;274:3642–3650. doi: 10.1074/jbc.274.6.3642. [DOI] [PubMed] [Google Scholar]
- 101.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 102.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 103.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 104.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 105.Fortini P, Pascucci B, Belisario F, Dogliotti E. DNA polymerase β is required for efficient DNA strand break repair induced by methyl methanesulfonate but not by hydrogen peroxide. Nucleic Acids Res. 2000;28:3040–3046. doi: 10.1093/nar/28.16.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair. 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 107.Ochs K, Sobol RW, Wilson SH, Kaina B. Cells deficient in DNA polymerase β are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res. 1999;59:1544–1551. [PubMed] [Google Scholar]
- 108.Sobol RW, Kartalou M, Almeida KH, Joyce DF, Engelward BP, Horton JK, Prasad R, Samson LD, Wilson SH. Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses. J Biol Chem. 2003;278:39951–39959. doi: 10.1074/jbc.M306592200. [DOI] [PubMed] [Google Scholar]
- 109.Lehmann AR, Willis AE, Broughton BC, James MR, Steingrimsdottir H, Harcourt SA, Arlett CF, Lindahl T. Relation between the human fibroblast strain 46BR and cell lines representative of Bloom’s syndrome. Cancer Res. 1988;48:6343–6347. [PubMed] [Google Scholar]
- 110.Henderson LM, Arlett CF, Harcourt SA, Lehmann AR, Broughton BC. Cells from an immunodeficient patient (46BR) with a defect in DNA ligation are hypomutable but hypersensitive to the induction of sister chromatid exchanges. Proc Natl Acad Sci U S A. 1985;82:2044–2048. doi: 10.1073/pnas.82.7.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ho EL, Satoh MS. Repair of single-strand DNA interruptions by redundant pathways and its implication in cellular sensitivity to DNA-damaging agents. Nucleic Acids Res. 2003;31:7032–7040. doi: 10.1093/nar/gkg892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, Cleaver JE, Pedersen RA. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 113.Thompson LH, Brookman KW, Dillehay LE, Carrano AV, Mazrimas JA, Mooney CL, Minkler JL. A CHO-cell strain having hypersensitivity to mutagens, a defect in DNA strand-break repair, and an extraordinary baseline frequency of sister-chromatid exchange. Mutat Res. 1982;95:427–440. doi: 10.1016/0027-5107(82)90276-7. [DOI] [PubMed] [Google Scholar]
- 114.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 115.Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- 116.Babich MA, Day RS., 3rd Potentiation of cytotoxicity by 3-aminobenzamide in DNA repair-deficient human tumor cell lines following exposure to methylating agents or anti-neoplastic drugs. Carcinogenesis. 1988;9:541–546. doi: 10.1093/carcin/9.4.541. [DOI] [PubMed] [Google Scholar]
- 117.Cleaver JE. Stimulation of repair replication by 3-aminobenzamide in human fibroblasts with ligase I deficiency. Carcinogenesis. 1996;17:1–3. doi: 10.1093/carcin/17.1.1. [DOI] [PubMed] [Google Scholar]
- 118.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J Biol Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 119.Burkart V, Wang ZQ, Radons J, Heller B, Herceg Z, Stingl L, Wagner EF, Kolb H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic β-cell destruction and diabetes development induced by streptozocin. Nature Medicine. 1999;5:314–319. doi: 10.1038/6535. [DOI] [PubMed] [Google Scholar]
- 120.Masutani M, Suzuki H, Kamada N, Watanabe M, Ueda O, Nozaki T, Jishage K, Watanabe T, Sugimoto T, Nakagama H, Ochiya T, Sugimura T. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96:2301–2304. doi: 10.1073/pnas.96.5.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, Verma A, Wang ZQ, Snyder SH. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cardinal JW, Margison GP, Mynett KJ, Yates AP, Cameron DP, Elder RH. Increased susceptibility to streptozotocin-induced β-cell apoptosis and delayed autoimmune diabetes in alkylpurine-DNA-N-glycosylase-deficient mice. Mol Cell Biol. 2001;21:5605–5613. doi: 10.1128/MCB.21.16.5605-5613.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 124.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 125.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 126.Wang W, Sitaram A, Scicchitano DA. 3-Methyladenine and 7-methylguanine exhibit no preferential removal from the transcribed strand of the dihydrofolate reductase gene in Chinese hamster ovary B11 cells. Biochemistry. 1995;34:1798–1804. doi: 10.1021/bi00005a037. [DOI] [PubMed] [Google Scholar]
- 127.Scicchitano DA, Hanawalt PC. Repair of N-methylpurines in specific DNA sequences in Chinese hamster ovary cells: absence of strand specificity in the dihydrofolate reductase gene. Proc Natl Acad Sci USA. 1989;86:3050–3054. doi: 10.1073/pnas.86.9.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Plosky B, Samson LD, Engelward BP, Gold B, Schlaen B, Millas T, Magnotti M, Schor J, Scicchitano DA. Base excision repair and nucleotide excision repair contribute to the removal of N-methylpurines from active genes. DNA Repair. 2002;1:683–696. doi: 10.1016/s1568-7864(02)00075-7. [DOI] [PubMed] [Google Scholar]
- 129.Xiao W, Chow BL. Synergism between yeast nucleotide and base excision repair pathways in the protection against DNA methylation damage. Curr Genet. 1998;33:92–99. doi: 10.1007/s002940050313. [DOI] [PubMed] [Google Scholar]
- 130.McDonald JP, Frank EG, Plosky BS, Rogozin IB, Masutani C, Hanaoka F, Woodgate R, Gearhart PJ. 129-derived strains of mice are deficient in DNA polymerase iota and have normal immunoglobulin hypermutation. J Exp Med. 2003;198:635–643. doi: 10.1084/jem.20030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bebenek K, Tissier A, Frank EG, McDonald JP, Prasad R, Wilson SH, Woodgate R, Kunkel TA. 5′-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science. 2001;291:2156–2159. doi: 10.1126/science.1058386. [DOI] [PubMed] [Google Scholar]
- 132.Haracska L, Prakash L, Prakash S. A mechanism for the exclusion of low-fidelity human Y-family DNA polymerases from base excision repair. Genes Dev. 2003;17:2777–2785. doi: 10.1101/gad.1146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Prasad R, Bebenek K, Hou E, Shock DD, Beard WA, Woodgate R, Kunkel TA, Wilson SH. Localization of the deoxyribose phosphate lyase active site in human DNA polymerase iota by controlled proteolysis. J Biol Chem. 2003;278:29649–29654. doi: 10.1074/jbc.M305399200. [DOI] [PubMed] [Google Scholar]
- 134.Zhao B, Xie Z, Shen H, Wang Z. Role of DNA polymerase eta in the bypass of abasic sites in yeast cells. Nucleic Acids Res. 2004;32:3984–3994. doi: 10.1093/nar/gkh710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Haracska L, Prakash S, Prakash L. Replication past O(6)-methylguanine by yeast and human DNA polymerase eta. Mol Cell Biol. 2000;20:8001–8007. doi: 10.1128/mcb.20.21.8001-8007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Takenaka K, Ogi T, Okada T, Sonoda E, Guo C, Friedberg EC, Takeda S. Involvement of vertebrate Polkappa in translesion DNA synthesis across DNA monoalkylation damage. J Biol Chem. 2006;281:2000–2004. doi: 10.1074/jbc.M506153200. [DOI] [PubMed] [Google Scholar]
- 137.Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci U S A. 2006;103:8971–8976. doi: 10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nowosielska A, Smith SA, Engelward BP, Marinus MG. Homologous recombination prevents methylation-induced toxicity in Escherichia coli. Nucleic Acids Res. 2006;34:2258–2268. doi: 10.1093/nar/gkl222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 140.Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res. 2001;477:131–153. doi: 10.1016/s0027-5107(01)00115-4. [DOI] [PubMed] [Google Scholar]
- 141.Fuller LF, Painter RB. A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutat Res. 1988;193:109–121. doi: 10.1016/0167-8817(88)90041-7. [DOI] [PubMed] [Google Scholar]
- 142.Jones NJ, Cox R, Thacker J. Isolation and cross-sensitivity of X-ray-sensitive mutants of V79-4 hamster cells. Mutat Res. 1987;183:279–286. doi: 10.1016/0167-8817(87)90011-3. [DOI] [PubMed] [Google Scholar]
- 143.Lin Z, Kong H, Nei M, Ma H. Origins and evolution of the recA/RAD51 gene family: Evidence for ancient gene duplication and endosymbiotic gene transfer. Proc Natl Acad Sci U S A. 2006;103:10328–10333. doi: 10.1073/pnas.0604232103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pittman DL, Cobb J, Schimenti KJ, Wilson LA, Cooper DM, Brignull E, Handel MA, Schimenti JC. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell. 1998;1:697–705. doi: 10.1016/s1097-2765(00)80069-6. [DOI] [PubMed] [Google Scholar]
- 145.Yoshida K, Kondoh G, Matsuda Y, Habu T, Nishimune Y, Morita T. The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol Cell. 1998;1:707–718. doi: 10.1016/s1097-2765(00)80070-2. [DOI] [PubMed] [Google Scholar]
- 146.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci U S A. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 150.Hinz JM, Tebbs RS, Wilson PF, Nham PB, Salazar EP, Nagasawa H, Urbin SS, Bedford JS, Thompson LH. Repression of mutagenesis by Rad51D-mediated homologous recombination. Nucleic Acids Res. 2006;34:1358–1368. doi: 10.1093/nar/gkl020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Smiraldo PG, Gruver AM, Osborn JC, Pittman DL. Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res. 2005;65:2089–2096. doi: 10.1158/0008-5472.CAN-04-2079. [DOI] [PubMed] [Google Scholar]
- 152.Tsaryk R, Fabian K, Thacker J, Kaina B. Xrcc2 deficiency sensitizes cells to apoptosis by MNNG and the alkylating anticancer drugs temozolomide, fotemustine and mafosfamide. Cancer Lett. 2006;239:305–313. doi: 10.1016/j.canlet.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 153.Cejka P, Mojas N, Gillet L, Schar P, Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Curr Biol. 2005;15:1395–1400. doi: 10.1016/j.cub.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 154.Milne GT, Jin S, Shannon KB, Weaver DT. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zdzienicka MZ, Tran Q, van der Schans GP, Simons JW. Characterization of an X-ray-hypersensitive mutant of V79 Chinese hamster cells. Mutat Res. 1988;194:239–249. doi: 10.1016/0167-8817(88)90025-9. [DOI] [PubMed] [Google Scholar]
- 156.Game JC. The Saccharomyces repair genes at the end of the century. Mutat Res. 2000;451:277–293. doi: 10.1016/s0027-5107(00)00055-5. [DOI] [PubMed] [Google Scholar]
- 157.Debiak M, Nikolova T, Kaina B. Loss of ATM sensitizes against O6-methylguanine triggered apoptosis, SCEs and chromosomal aberrations. DNA Repair (Amst) 2004;3:359–368. doi: 10.1016/j.dnarep.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 158.Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 159.Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 160.Hawn MT, Umar A, Carethers JM, Marra G, Kunkel TA, Boland CR, Koi M. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55:3721–3725. [PubMed] [Google Scholar]
- 161.Cejka P, Stojic L, Mojas N, Russell AM, Heinimann K, Cannavo E, di Pietro M, Marra G, Jiricny J. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003;22:2245–2254. doi: 10.1093/emboj/cdg216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Stojic L, Cejka P, Jiricny J. High doses of SN1 type methylating agents activate DNA damage signaling cascades that are largely independent of mismatch repair. Cell Cycle. 2005;4:473–477. doi: 10.4161/cc.4.3.1528. [DOI] [PubMed] [Google Scholar]
- 163.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Worrillow LJ, Allan JM. Deregulation of homologous recombination DNA repair in alkylating agent-treated stem cell clones: a possible role in the aetiology of chemotherapy-induced leukaemia. Oncogene. 2006;25:1709–1720. doi: 10.1038/sj.onc.1209208. [DOI] [PubMed] [Google Scholar]
- 166.Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 167.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 168.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 169.Fry RC, Begley TJ, Samson LD. Genome-wide responses to DNA-damaging agents. Annu Rev Microbiol. 2005;59:357–377. doi: 10.1146/annurev.micro.59.031805.133658. [DOI] [PubMed] [Google Scholar]
- 170.Begley TJ, Rosenbach AS, Ideker T, Samson LD. Damage recovery pathways in Saccharomyces cerevisiae revealed by genomic phenotyping and interactome mapping. Mol Cancer Res. 2002;1:103–112. [PubMed] [Google Scholar]
- 171.Said MR, Begley TJ, Oppenheim AV, Lauffenburger DA, Samson LD. Global network analysis of phenotypic effects: protein networks and toxicity modulation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2004;101:18006–18011. doi: 10.1073/pnas.0405996101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci U S A. 2002;99:16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Allan JM, Travis LB. Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer. 2005;5:943–955. doi: 10.1038/nrc1749. [DOI] [PubMed] [Google Scholar]