

Abstract
Soluble epoxide hydrolase (sEH) is a therapeutic target for treating hypertension and inflammation. 1,3-Disubstituted ureas functionalized with an ether group are potent sEH inhibitors. However, their relatively low metabolic stability leads to poor pharmacokinetic properties. To improve their bioavailability, we investigated the effect of incorporating various polar groups on the ether function on the inhibition potencies, physical properties, in vitro metabolic stability, and pharmacokinetic properties. The structure-activity relationship (SAR) studies showed that a hydrophobic linker between the urea group and the ether function is necessary to keep their potency. In addition, urea-ether inhibitors having a polar group such as diethylene glycol or morpholine significantly improved their physical properties and metabolic stability without any loss of inhibitory potency. Furthermore, improved pharmacokinetic properties in murine and canine models were obtained with the resulting inhibitors. These findings will facilitate the usage of sEH inhibitors in animal models of hypertension and inflammation.
Introduction
Cytochrome P450 epoxygenases oxidize polyunsaturated endogenous fatty acids, such as arachidonic acid1-5 to generate the corresponding epoxides (epoxyeicosatrienoic acids or EETs). These latter compounds have been reported as a new class of lipid mediators regulating blood pressure6-11 and inflammation.12-17 In addition, the EETs further have vascular protective effects such as suppression of reactive oxygen species following hypoxia-reoxygenation18 or enhancement of a fibrinolytic pathway.19 However, the metabolism of EETs to dihydroxyeicosatrienoic acids (DHETs) by the soluble epoxide hydrolase (sEH) often leads to reductions in these biological activities.6 Thus, stabilizing the in vivo concentration of EETs through sEH inhibition represents a novel therapeutic avenue to treat hypertension, inflammation, and other cardiovascular disorders. This hypothesis is supported by numerous in vivo experiments in animal models. For example, the blood pressure of spontaneously hypertensive or angiotensin II induced hypertensive rats treated with sEH inhibitors is dramatically reduced.7-9,11 Also, tobacco smoke-induced lung inflammation12 or lipopolysaccharide- (LPS) induced acute inflammation13 is attenuated by treating with sEH inhibitors. All of these studies support the hypothesis that anti-hypertensive and the cardio protective effects are mediated by EETs, and are dependent on the extent of epoxide hydrolysis by sEH.20,21
1,3-Disubstituted ureas and related compounds are very potent inhibitors of sEH and these compounds efficiently induce a reduction in epoxide hydrolysis in several cellular and animal models.8,9,22 However, poor physical properties, especially limited solubility in either water or organic solvents of some of these urea inhibitors likely result in poor in vivo availability and difficulty in formulation.23 We previously reported that a polar functional group located on specific positions of one of the alkyl chains of the urea inhibitors improves water solubility and generally decreases melting points without decreasing inhibitory potency.24 Further, a carboxylic acid group present on the thirteenth atom, or a polar group such as ester, sulfonamide, alcohol, ether, carbamate, or ketone located on the fifth/sixth atom from the urea group was effective for producing soluble inhibitors in either water or oil while retaining inhibition potency.23-25 Such derivatives that have improved solubility and low melting point, have enhanced pharmacokinetic properties in mice compared to the lipophilic inhibitor,26,27 indicating that compounds having good physical properties result in a better inhibitors for in vivo study. Although significantly improved pharmacokinetic properties was obtained from these inhibitors, relatively low absorptions and short half-lives were still observed.26,27 These inhibitors may not have adequate pharmacokinetic properties to be effective as pharmaceuticals. Toward obtaining compounds that have the better absorbance and the longer in vivo half-lives, we have previously explored the effect of ureas substituted with adamantyl group at position 1 and piperidine group at position 3 of the urea, 28 and separately replaced the adamantane group with various groups.29 In the present study we investigated the effect of functionalizing 1,3-disubstituted ureas with various polar ether groups on physical properties, metabolic stability, and pharmacokinetic properties.
Chemistry
Scheme 1 outlines the syntheses of 1-adamantan-1-yl-3-(hydroxyalkyl)ureas and their aliphatic alkyl ether analogs. Reaction of 1-adamantyl isocyanate with a hydroxyalkylamine (2-hydroxyethylamine for compound 1, 3-hydroxypropylamine for compound 3, 4-hydroxybutylamine for compound 5, 5-hydroxypentylamine for compound 7, and 6-hydroxyhexylamine for compound 9) in N,N-dimethylformamide (DMF) provided 1-adamantan-1-yl-3-(hydroxyalkyl)ureas in 95-100% yields.24,25 Each hydroxyl group of compounds 1, 3, 5, 7, and 9 was alkylated with an alkyl bromide (1-bromoheptane for compound 2, 1-bromohexane for compound 4, 1-bromopentane for compound 6, 1-bromobutane for compound 8, and 3-bromopropane for compound 10) to afford the corresponding ether compounds in a range of 30-55% yields.
Scheme 1.

Syntheses of 1-adamantyl-3-hydroxyalkylureas (1, 3, 5, 7, and 9) and the corresponding ether derivatives (2, 4, 6, 8, and 10): (a) 2-hydroxyethylamine (for compound 1), 3-hydroxypropylamine (for compound 3), 4-hydroxybutylamine (compound 5), 5-hydroxypentylamine (for compound 7), or 6-hydroxyhexylamine (for compound 9), DMF, rt, (b) 1-bromoheptane (for compound 2), 1-bromohexane (for compound 4), 1-bromopentane (for compound 6), 1-bromobutane (for compound 8), or 1-bromopropane (for compound 10), NaH, DMF, rt.
The syntheses of alkyl ethers with a methyl branch (11 and 12) and ethylene glycol derivatives of compound 7 (15, 18, and 19) are described in Scheme 2 (A) and (B). As outlined in Scheme 2 (A), compound 7 was brominated with triphenylphosphine and carbon tetrabromide in DMF to produce 1-adamantan-1-yl-3-(5-bromopentyl)urea (I) in 87% yield, followed by etherification with the corresponding alcohol (2-hexanol for 11, 2-methylpentanol for 12, and triethylene glycol monomethyl ether for 18) in the presence of sodium hydride in DMF in a range of 25-48% yields. Bromination of diethylene glycol monoethyl ether (for 15) or 3-hydroxypropylmorpholine (for 19) using triphenylphosphine and carbon tetrabromide in THF yielded the corresponding bromides II (87-95%), which were reacted with compound 7 in the presence of sodium hydride in DMF to afford compounds 15 and 19 in 52-65% yields, respectively.
Scheme 2.

Syntheses of 1-adamantyl-3-substituted alkyl-ureas: (a) Ph3P, CBr4, DMF, rt, (b) 2-hexanol (for compound 11), 2-methyl-1-pentanol (for compound 12), or triethylene glycol monomethyl ether (for compound 18), NaH, DMF, rt, (c) Ph3P, CBr4, THF, rt, (d) compound 7, NaH, DMF, rt.
Compounds 13, 14, 16, and 17 were prepared by the procedures depicted in Scheme 3 Monoalkylation of 1,3-dihydroxypropane (for compound 13) or 1,4-dihydroxybutane (for compound 14) with 1-bromopropane in the presence of sodium hydride in DMF gave the corresponding mono-alcohols 3-propoxy-1-propanol and 4-propoxy-1-butanol, respectively, in approximately 18% yield. Reaction of these alcohols with I in the presence of sodium hydride in DMF produced compounds 13 and 14 in 25-32% yields. Monoalkylation of di-(2-bromoethyl) ether with 2,2,2-trifluoroethanol (for compound 16) or 4-ethylphenol (for compound 17) using sodium hydride as a base in DMF afforded the corresponding 2-(2,2,2-trifluoroethoxyethoxy)ethyl bromide and 2-(4-ethylphenoxyethoxy)ethyl bromide in 60% yield. Each intermediate was reacted with compound 7 in the presence of sodium hydride in DMF to afford compounds 16 and 17, respectively, in 45% yield.
Scheme 3.

Syntheses of 1-adamantyl-3-substituted alkyl-ureas: (a) 1,2-ethandiol (for compound 13) or 1,3-propandiol (for compound 14), 1-propanol, NaH, DMF, rt, (b) I, NaH, DMF, rt, (c) 2,2,2-trifluoroethanol (for compound 16) or 4-ethylphenol (for compound 17), NaH, DMF, rt, (d) compound 7, NaH, DMF, rt.
Scheme 4 shows the syntheses of methanesulfonamide ureas (20 and 21) and two corresponding amide derivatives (22 and 23) of compound 15. As shown in Scheme 4 (A), reaction of 2-(2-aminoethoxy)ethyl alcohol (for compound 20) and 5-amino-1-pentanol (for compound 21) with methanesulfonic anhydride in acetonitrile gave 2-(2-hydroxyethoxy)ethylmethanesulfonamide and 5-hydroxypentylmethanesulfonamide, respectively, in 66% yield, which were reacted with I in the presence of sodium hydride in DMF to afford compounds 20 and 21 in 40-55% yield. As described in (B), alkylation of 7-bromoheptanoic acid ethyl ester with diethylene glycol monoethyl ether in the presence of sodium hydride in DMF afforded substituted heptanoate in 40% yield, followed by hydrolysis. Coupling of the resulting acid with 1-adamantylamine using 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide hydrochloride (EDCI) in the presence of N,N-4-dimethylaminopyridine (DMAP) in dichloromethane provided compound 22 in 97% yield. For the preparation of the other amide analog 23 with the nitrogen on the “right side” of the carbonyl, the amino group of 5-amino-1-pentanol was protected in the reaction with di-(tert-butyl) dicarbonate in dioxane to give the corresponding protected intermediate in 97% yield, followed by alkylation of the hydroxyl group with 2-(ethoxyethoxy)ethyl bromide (II) in the presence of a catalytic amount of sodium iodide and sodium hydride in DMF (28%). Then, the amine was deprotected with 4N HCl in dioxane to afford amine salt, which was coupled with 1-adamantylacetic acid using EDCI and DMAP in dichloromethane to yield compound 23 (97%).
Scheme 4.

Syntheses of 1-adamantyl-3-substituted alkyl-ureas with a sulfonamide (20 and 21) and of amide derivatives (22 and 23) of compound 15: (a) 2-aminoethoxyethanol (for compound 20) or 5-amino-1-pentanol (for compound 21), methanesulfonic anhydride, acetonitrile, rt, (b) I, NaH, DMF, rt, (c) diethylene glycol monoethyl ether, NaH, DMF, rt, (d) 1N NaOH, ethanol, rt, (e) 1-adamantylamine, EDCI, DMAP, dichloromethane, rt, (f) di-(tert-butyl) dicarbonate, dioxane, rt, (g) 2-(ethoxyethoxy)ethyl bromide (prepared as described in Scheme 2 (B); II), NaH, DMF, rt, (h) (1) 4N HCl in dioxane, rt, (2) 1-adamantylacetic acid, triethylamine, EDCI, DMAP, dichloromethane, rt.
Scheme 5 outlines syntheses of analogs with a substituted cyclohexyl (24-26) or phenyl (27-29) group as a linker between the urea group and the oxygen atom in an ether group. Reaction of 1-adamantyl isocyanate with trans-4-aminocyclohexanol in DMF gave 1-adamantan-1-yl-3-(4-hydroxycyclohexyl)urea (IV) in 95% yield, followed by O-alkylation of IV with 1-bromopentane (for compound 24), 2-(ethoxyethoxy)ethyl bromide (for compound 25), or 3-morpholinopropyl bromide (for compound 26) in the presence of sodium hydride in DMF to provide compounds 24, 25, and 26, respectively, in 25-40% yields. Reaction of 1-adamantyl isocyanate with 3- or 4-aminophenol gave intermediates in 95% yield, which were alkylated with 2-(ethoxyethoxy)ethyl bromide (for compounds 27 and 28) or 3-morpholinopropyl bromide (for compound 29) to afford compounds 27-29 in a range of 80-90% yields.
Scheme 5.

Syntheses of 1-adamantyl-3-substituted cyclohexyl- or phenyl-ureas: (a) trans-4-aminocyclohexanol hydrochloride, triethylamine, DMF, rt, (b) 1-bromopentane (for compound 24), 2-(ethoxyethoxy)ethyl bromide (II; for compound 25), or 3-morpholinopropyl bromide (II; for compound 26), (c) 3-aminophenol (for compound 27) or 4-aminophenol (for compounds 28 and 29), DMF, rt, (d) -(ethoxyethoxy)ethyl bromide (II), NaH, DMF, rt.
Results and Discussion
In order to synthesize soluble inhibitors with improved pharmacokinetic properties, a variety of urea derivatives functionalized with various polar groups were explored in this study. We previously showed that an alcohol or ether function present at least 5atoms away from the urea carbonyl is very useful to make very potent soluble inhibitors.24 This observation was extended to include many polar groups as such secondary pharmacophore. Because ease and cost-effectiveness in synthesizing compounds are critical to facilitate extensive SAR studies, in this study an ether function was selected as the secondary functionality, which can be easily prepared in two reaction steps in a high yield, while five or six reactions are required for the preparation of the corresponding alcohol function, eventually resulting in a lower total reaction yield compared to that obtained in the preparation of the ether.24 First, five ether derivatives were synthesized to determine the appropriate location for the incorporation of the ether or other hydrogen binding pharmacophore. As shown in Table 1, compared to 1-adamantyl-3-decylurea (IC50 =9.4 nM),24 compounds 4, 6, 8, and 10 showed potent inhibition on the target enzyme indicating that at least three methylene carbons between the primary urea and the secondary ether pharmacophores are necessary to produce potent inhibitory activity. In compounds with a shorter carbon chain length (2) or free alcohol functionality (1, 3, 5, 7, and 9), reduced inhibition potencies were observed, which is a similar result to that obtained previously with an ester functionality. It should be noted that when the free alcohol is sufficiently far from the central pharmacophore as in 9, the reduction in potency is minor. Thus, compounds such as 9 could be used to give enhanced water solubility or for the synthesis of prodrugs and soft drugs. Based on this SAR result and cost-effectiveness, compound 8 with a pentyl linker between the two pharmacophores was selected as a lead structure for further modifications in this study. When water solubility of compound 8 was examined, a 4-fold better solubility was obtained compared to a non-functionalized lipophilic inhibitor. However, its solubility is still limited (< 2 μg/mL) and similar to that obtained in the ester compound.24 Moreover, only 25% of compound 8 remained when it was incubated with human liver microsomes for 60 min suggesting that 75% was metabolized by microsomal enzymes such as P450s. These results suggest that compound 8 needs to be modified for making compounds with not only increased solubility but also improved metabolic stability, which can possibly result in inhibitors possessing enhanced pharmacokinetic properties.
Table 1.
Inhibition of human sEH by 1-adamantyl-3-hydroxyalkylureas or the corresponding ether derivatives.
| No. | 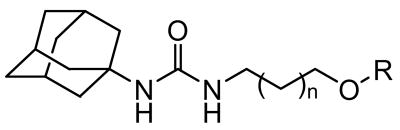 |
IC50 (nM)a Human sEH |
|
|---|---|---|---|
| n | R | ||
| 1 | 0 | H | 482 |
| 2 | 0 | n-heptyl | 29 |
| 3 | 1 | H | 746 |
| 4 | 1 | n-hexyl | 5.6 |
| 5 | 2 | H | 531 |
| 6 | 2 | n-pentyl | 4.4 |
| 7 | 3 | H | 84 |
| 8 | 3 | n-butyl | 4.1 |
| 9 | 4 | H | 12 |
| 10 | 4 | n-propyl | 3.7 |
Human sEH (1 nM) was incubated with inhibitors for 5 min in 25 mM Bis-Tris/HCl buffer (200 μL; pH 7.0) at 30°C before fluorescent substrate (CMNPC) introduction ([S] = 5 μM), results are triplicate averages. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of two-fold or greater are significant.33
As listed in Table 2, various ether compounds modified with a variety of functionalities such as a branched alkyl chain (11 and 12), polar ether groups (13-18), a morpholine (19), or a sulfonamide (20 and 21) were synthesized to investigate the effect of functionalization of compound 8 on inhibition potency on the target enzyme, physical properties, and in vitro metabolic stability. Incorporation of a methyl branch on the carbon alpha (11) or beta (12) to the ether oxygen atom of compound 8 decreased both inhibition potency and metabolic stability, while slightly enhanced physical properties were exhibited. Two derivatives with a propoxy group (13 and 14) showed increases in inhibition and physical properties compared to compound 8. However, a 3-fold reduction in stability was observed in the compounds (13 and 14), implying that a polar propyloxy group or a branch chain incorporated in compound 8 does not effectively produce improved inhibitors in either metabolic stability or inhibition potency. Interestingly, a 80-fold better water solubility than that of compound 8 was observed when an ethoxyethoxy group was introduced in the place of the propoxy group of compounds 13 and 14 (15). Furthermore, a significant enhancement of in vitro metabolic stability was exhibited without a loss of inhibition potency, suggesting that the diethylene glycol group of compound 15 would be very useful for yielding inhibitors with improved pharmacokinetic properties. It was anticipated that compound 15 would be rapidly metabolized by ω hydroxylation or by ω-1 hydroxylation alpha to the heteroatom. It was hypothesized that this metabolism would be blocked by a terminal trifluoromethyl substituent in 16. To our surprise compound 16 showed decreased stability in the S9 incubation, although dramatically improved water solubility was obtained with 16 without a decrease in the inhibition. An aryl ether derivative (17) also did not have significant improvement in either solubility or stability, while inhibition potency was maintained. Thus, neither a branch chain (11 and 12) nor a trifluoromethyl (16) group incorporated in the molecules provided a significant increase in metabolic stability. As expected, compound 17 containing a p-ethylphenyl group did not improve metabolic stability. On the other hand, comparing compounds 13 and 14 to 15, the increased polarity of compound 15 seemed to be effective for enhancing stability as well as other physical properties.
Table 2.
Inhibition of human sEH, physical properties, and in vitro metabolic stability of 1-adamantyl-3-substituted alkyl-urea derivatives.
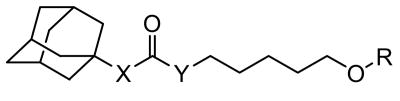 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | X | Y | R | IC50 (nM)a Human sEH |
Mpb (°C) |
Solubility in waterc (μg/mL) |
Stability (%)d | |
| microsomes | S9 | |||||||
| 8 | NH | NH |
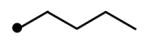
|
4.1 | 71 | 1.6 | 25 | NDe |
| 11 | NH | NH |
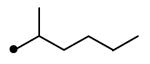
|
11 | 45 | 2.3 | 12 | ND |
| 12 | NH | NH |
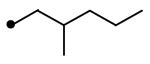
|
10 | 49 | 2.0 | 12 | ND |
| 13 | NH | NH |
|
2.5 | oil | 26 | 6.8 | ND |
| 14 | NH | NH |
|
2.3 | oil | 15 | 7.5 | ND |
| 15 | NH | NH |
|
14 | 78 | 120 | 30 | 11 |
| 16 | NH | NH |
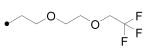
|
1.4 | 34 | 234 | 39 | 1.1 |
| 17 | NH | NH |
|
1.7 | 71 | 0.38 | 21 | ND |
| 18 | NH | NH |
|
23 | oil | 3709 | 54 | 14 |
| 19 | NH | NH |
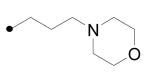
|
9.0 | oil | 897 | 89 | 34 |
| 20 | NH | NH |
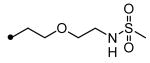
|
58 | oil | 1082 | 42 | 12 |
| 21 | NH | NH |
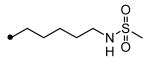
|
11 | 93 | 75 | 59 | <LDf |
| 22 | NH | CH2 |
|
247 | oil | ND | 32 | 5 |
| 23 | CH2 | NH |
|
10 | oil | ND | 5 | ND |
Human sEH (1 nM) was incubated with inhibitors for 5 min in 25 mM Bis-Tris/HCl buffer (200 μL; pH 7.0) at 30°C before fluorescent substrate (CMNPC) introduction ([S] = 5 μM), results are averages of three separate measurements. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of two-fold or greater are significant.33
Melting point.
Solubility in sodium phosphate buffer (0.1 M, pH 7.0) at 25 ± 1.5°C.
Percentage (%) of test compounds (8, 11-22, and 23) remaining after 1h incubation with enzymes in human liver microsomes or S9 fractions. Enzymes (0.125 mg for microsomes or 0.056 mg for S9 fractions) were incubated with inhibitors ([S] = 1.0 μM) for 0 and 60 min with data shown for 60 min in sodium phosphate buffer (0.1 M, pH 7.4) at 37°C.
Non-determined
Limit of detection
Based on the results above, compound 18 was prepared. It showed a dramatic improvement in physical properties and similar stability to that of compound 15 while approximately a 5-fold drop in inhibition was obtained. This indicates that the polar ethylene glycol tail of compound 18 would be very useful for producing soluble compounds in either water or organic solvents although decreased inhibition was observed in this series. On the other hand, a morpholino derivative (19) exhibited more desirable physical properties compared to compound 15 while retaining reasonable inhibition. Moreover, a 3-fold improved stability in the microsomal or S9 incubation was gained in the polar morpholine compound (19), indicating that a morpholino group is a very effective functionality for producing enhanced sEH inhibitors not only with regard to physical properties but also with regard to metabolic stability. When the morpholino group of compound 19 was replaced by a methanesulfonamide function at least a 2-fold decrease in stability was observed (20 and 21). Also, unfortunately, a considerable drop in inhibition potency was shown with the sulfonamide (20), which is 12-fold less active than compound 15. Although the other sulfonamide derivative (21) exhibited a reasonable inhibition on the enzyme, reductions in both solubility and stability were observed in this case, indicating that a sulfonamide group attached to the ether derivatives is not useful for either inhibiting the target enzyme effectively or stabilizing parent compounds against metabolism in this series. The result in Table 2 indicate that significantly enhanced improvements in physical properties and metabolic stability were exhibited with the introduction of a diethylene glycol or a morpholino group as shown in compounds 15 and 19. We have reported that modification of the urea pharmacophore of potent sEH inhibitors to the corresponding amide functionality does not dramatically alter the inhibition potency and that an improvement in physical properties is observed in the amide derivatives.25 This indicates that the physical properties of the ether derivatives (15 or 19) might be further improved with the modification of the urea pharmacophore to the amide function without a drop in inhibition. Thus, when two amide analogues of compound 15 were prepared (22 and 23), compound 22 with the nitrogen atom on the left side of the carbonyl of the amide function had approximately 50-fold decreased inhibition, while a similar stability to that of compound 15 was obtained. The presence of the nitrogen atom on the right side of the amide function did not drop inhibition potency as shown in compound 23, which is consistent with the previous report that the nitrogen on the right side of the carbonyl group of the amide pharmacophore is important for producing potent inhibitors.25 However, a 6-fold decrease in metabolic stability was obtained in the amide compound 23, indicating that an amide pharmacophore might not be effective in this ether series for making stable compounds in vivo while retaining potent inhibition unless the amide functionality could be stabilized.
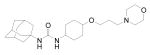
As shown in Table 3, the corresponding cyclohexyl (24-26) and phenyl (27-29) derivatives of compounds 15 and 19 were further synthesized. Interestingly, when the pentyl linker of compounds 8, 15, and 19 was replaced by a cyclohexyl or phenyl group (24-29), 4-8-fold increases in inhibition were obtained, indicating that the increased structural rigidity and/or increased hydrophobicity between the two pharmacophores is effective for producing highly potent inhibitors. This results in compounds binding to the target enzyme very specifically. Comparing compounds 8 and 24, replacement of the pentyl linker (8) with a cyclohexyl (24) resulted in a poor inhibitor in terms of physical properties (higher melting point and lower water solubility), while similar stability was observed. However, a huge improvement in solubility was observed when the hydrophobic alkyl tail of compound 24 was replaced by a polar diethylene glycol (25) or 3-morpholinopropyl (26) group, which has similar solubility to that of the corresponding pentyl derivatives (15 and 19). Moreover, significantly greater stabilities were exhibited in the cyclohexyl derivatives with a polar tail (25 and 26) over the corresponding pentyl compounds (15 and 19), indicating that a cyclohexyl linker between the primary and secondary pharmacophores, and a polar group attached to this linker are important for yielding very potent compounds with improved metabolic stability while retaining reasonable water solubility. Replacement of the cyclohexyl group by a phenyl group in 25 resulted in approximately a 10-fold drop in water solubility (27 and 28), while similar stability was exhibited with compound 28. Interestingly, When the diethylene glycol group was present on the 3-position of the phenyl ring (27), suggesting that para-position on the phenyl ring is metabolically susceptible. A phenyl derivative with a morpholino group (29) also exhibited a 4-fold lower solubility, but no decrease in stability was observed. Overall, the results in Table 3 indicate that a cyclohexyl group as a linker is effective for increasing not only inhibition potency on the human sEH, but metabolic stability without a drop in water solubility when compared to the corresponding pentyl analogs. Furthermore, a phenyl group as a linker between two pharmacophores is also effective for enhancing both inhibition and metabolic stability, although a decrease in solubility was observed. While in this study we concentrated on the human sEH; in previous studies we found over 90% correlation between the inhibition results observed with the human and the murine sEH.21,23-25 Thus, the SAR conclusions drawn here for the human enzyme are probably valid for the rodent enzyme, and the best inhibitors herein are certainly also very potent inhibitor for the rodent sEH. However, there are occasional large differences in potency of certain compounds among model species.
Table 3.
Inhibitions of human and murine sEHs, physical properties, and in vitro metabolic stability of 1-adamantyl-3-substituted cyclohexyl- or phenyl-urea derivatives.

We then investigated the pharmacokinetics of a series of urea-ether derivatives with improved physical properties and/or metabolic stability in mice, rats, and dogs. As shown in Table 4, the pharmacokinetic parameters (area under the curve (AUC) and half-life (t1/2)) of eight derivatives were determined following oral administration at 5 mg/kg body weight for rodents and at 0.3 mg/kg body weight for dogs. A few sEH inhibitors such as compound 15 (also called AEPU (1-adamantan-3-(5-(2-(ethyl-ethoxy)ethoxy)pentyl)urea) or “950”)13 are bioavailable whether administered as a powder or a solution, however many urea sEH inhibitors are poorly bioavailable if they are not in true solution. Thus, all compounds were administered by oral gavage in tristerate solution at body temperature. In a few cases a trace of ethanol was used to make a clear solution.
Table 4.
Pharmacokinetic parameters of 1-adamantyl-3-substituted pentyl (8, 15, 19)-, cyclohexyl (24-26)- or phenyl (28, 29)-urea derivatives in a mouse, a rat, and a dog.
| No. | LogPa (cLogP)b |
Mouse | Rat | Dog | |||
|---|---|---|---|---|---|---|---|
| AUCc | t1/2d | AUCc | t1/2d | AUCc | t1/2d | ||
| 8 | 5.0
(3.1) |
< LDe | < LD | < LD | < LD | 5600 | 130 |
| 15 | 3.6
(1.9) |
< LD | < LD | 3000 | 39 | 8300 | 180 |
| 19 | 2.4
(1.5) |
340 | 15 | 750 | 200 | 2500 | 90 |
| 24 | > 5.0
(3.4) |
1200 | 170 | 560 | 120 | 5100 | 120 |
| 25 | 2.9
(1.7) |
2800 | 160 | 5900 | 150 | 9800 | 90 |
| 26 | 2.2
(1.4) |
10000 | 80 | 2400 | 110 | 27000 | 130 |
| 28 | 3.9
(2.6) |
< LD | < LD | 2900 | 580 | 34000 | 320 |
| 29 | 2.9
(2.3) |
< LD | < LD | < LD | < LD | 8400 | 130 |
Measured log P.
Calculate logP.
Area under the concentration-time curve to terminal time (min.kg.nM.mg-1).
Elimination half-life (min).
Limit of detection.
The AUC is an expression of how much and how long a drug stays in the body, and it is related to the amount of drug absorbed systemically as well as the amount of drug metabolized, sequestered and eliminated; while the t1/2 is more indicative of the rates of degradation, distribution and elimination. The experimental logP of each derivative was determined and compared to the calculated values (clogP). Although a similar trend was observed between the two values, there are significant differences between the calculated and experimental logP. When we compared these data to water solubility data (Tables 2 and 3), the changes in logP are related to their water solubility. In the mouse, little or no apparent absorption was observed with the compounds that have a pentyl group as a linker (8, 15 and 19). Interestingly, a significant increase in the AUC was obtained with the compounds that have a cyclohexyl group as a linker (24 to 26) compared to the corresponding pentyl compounds (8, 15 and 19). Furthermore in the cyclohexyl series (24 to 26), the AUC decreased with an increase in the logP; while the half-lives increased, suggesting that the improved water solubility of the cyclohexyl derivatives is likely useful for enhancing absorption that consequently results in increased AUC. The replacement of the cyclohexyl by a phenyl group results in compounds that were not significantly absorbed in rodents (28 and 29). Overall, it implies that water solubility is an important factor for the absorption of this series of compounds in mice. In the rat, like in the mouse, the cyclohexyl derivatives (24 to 26) gave overall the best absorption and/or greatest metabolic stability. However, independent of the linker structure, compounds having a diethylene glycol group (15, 25 and 28) have better AUC's. Except for 15, these latter compounds have also a longer half-life. Overall, for the series of chemicals tested herein, water solubility is probably not as important for absorption in rats as it is in mice. There appears to be an optimal value of logP, between 3.0 and 4.0, for good absorption.
Finally, we studied these eight derivatives in a canine model (Table 4). All compounds tested were absorbed quite well, and no direct relationships between structural features and pharmacokinetic parameters could be easily drawn. However, overall the presence of a cyclic linker (cyclohexyl 24 to 26 or phenyl 28 and 29) seems to increase the AUC in the dogs. For such linkers, the bioavailability is increased by the presence of polar groups such as diethylene glycol (25 and 28) or morpholino (26 and 29) groups. Overall, across the three species studied, the rigidification of the structure with cyclohexyl or phenyl group gives more bioavailable inhibitors than straight alkyl linkers such as the butyl or pentyl groups. Moreover, the presence of a polar group attached to this linker improves absorption. There are large differences among the three species with regard to the influence of structure on AUC and t1/2. Overall, there is an apparent trend between AUC and the size of the species. However, even though the dose given to the small animals (5 mg/kg) was higher than the one given to the dogs (0.3 mg/kg), the total quantities received were larger for the dogs (6 mg instead of 0.1-1 mg for the rodents). There are many variables involved in scaling from rodent to canine species including total size, hepatic and renal blood flow, gut residence time and many other factors. It must also be cautioned that blood levels correlate well with efficacy with only about half of the marketed pharmaceuticals. Furthermore, the in vivo behavior of the sEH inhibitors is not easy to predict from microsomal and S9. These suggest that further pharmacokinetic studies are needed to understand the mechanisms of absorption, disposition, metabolism, and elimination for this series of compounds, and thus be able to predict their in vivo behaviors.
In conclusion, this work focused on producing inhibitors of human sEH with improved pharmacokinetic properties. This was done by analyzing the effects of structural changes of 1,3-disubstituted ureas with an ether function present at least 3 atoms away from the urea carbonyl on inhibition potency, physical properties (e.g. water solubility and melting point) and metabolic stability. The inhibition studies showed that a hydrophobic group as a linker between the primary urea and the ether function is necessary to yield potent inhibitors. Furthermore, the presence of polar groups on the other side of the ether does not affect the inhibition potency significantly. However, up to 80-fold and 3-fold increases in water solubility and in vitro metabolic stability, respectively, were observed. The best results were obtained in compounds functionalized with ethylene glycol or morpholine groups. Furthermore, the corresponding cyclohexyl or phenyl derivatives showed improved metabolic stability as well as enhanced inhibition potency while retaining reasonable water solubility. The improved water solubility and in vitro metabolic stability were useful for enhancing pharmacokinetic properties in animal models. Overall, across the three species studied, we found the bioavailability is enhanced by the presence of cyclohexyl as a linker between the urea and the ether, and by polar group, such as diethylene glycol or morpholine, on the other side of the ether. In summary, we were able to increase solubility and bioavailability of sEH inhibitors without any loss in potency. These findings are important basis for the design of improved, orally available therapeutic agents for the treatment of hypertension and inflammation.
Experimental
All melting points were determined with a Thomas-Hoover apparatus (A.H. Thomas Co.) and are uncorrected. Mass spectra were measured by LC-MS/MS (Waters 2790) using positive mode electrospray ionization. Elemental analyses (C, H, N/F) were performed by Midwest Microlab, IN; analytical results were within ± 0.4% of the theoretical values for the formula given unless otherwise indicated. 1H-NMR/13C-NMR spectra were recorded on a QE-300 spectrometer, using tetramethylsilane as an internal standard. IR spectra were recorded on a Thermo Nicolet IR100 spectrometer. Synthetic methods are described for representative compounds.
1-Adamantan-1-yl-3-(5-hydroxypentyl)urea (7)
To a solution of adamantyl isocyanate (0.20 g, 1.13 mmol) in DMF (15 mL) was added a solution of 5-amino-1-pentanol (0.17 g, 1.69 mmol) in DMF (15 mL) at 0°C. After stirring for 12 hrs, an aqueous solution of 1N HCl (40 mL) was added into the reaction at 0°C, and the mixture was stirred for 30 min. The solid product crystallized was filtered, and washed with water (40 mL) and ethyl acetate (20 mL). The resulting solid was dried in the vacuum oven to give 7 as a white solid (0.75 g, 100%). 1H NMR δ (CDCl3) 1.37-1.42 (2H, m), 1.46-1.53 (2H, m), 1.57-1.62 (2H, m), 1.66 (6H, brs), 1.96 (6H, brs), 2.06 (3H, brs), 3.13 (2H,q, J = 6.9 Hz), 3.65 (2H, t, J = 6.9 Hz), 4.14 (1H, s), 4.39 (1H, s). LC-MS (ESI) m/z calcd for C16H28N2O2 [M + H]+ 281.22, found [M + H]+ 281.33, mp 220°C, Anal. (C16H28N2O2) C, H, N.
Compounds 1, 3, 5, and 9 were synthesized with the same procedure used for the preparation of 7 using the corresponding aminoalkanol instead of 5-amino-1-pentanol.
1-Adamantan-1-yl-3-(5-butoxypentyl)urea (8)
To a suspension of 60% sodium hydride (21 mg, 0.53 mmol) and 7 (150 mg, 0.53 mmol) in DMF (20 mL) was added drop wise 1-bromobutane (90 mg, 0.64 mmol) at room temperature. After stirring for 12 hrs, water (40 mL) was poured into the reaction mixture, and the product was extracted with ether (40 mL). The organic solution was dried over MgSO4, and concentrated. The residue was purified using silica gel column chromatography (hexane:ethyl acetate = 3:1) to give 8 (80 mg, 45%) as a solid. 1H NMR δ (CDCl3) 0.92 (3H, t, J = 6.9 Hz), 1.35-1.38 (4H, m), 1.53-1.77 (12H, m), 1.946 (6H, brs), 2.06 (3H, brs), 3.10 (2H, q, J = 6.9 Hz), 3.36-3.43 (4H, m), 4.14 (1H, s), 4.26 (1H, s). LC-MS (ESI) m/z calcd for C20H36N2O2 [M + H]+ 337.28, found [M + H]+ 337.32, mp 71°C, Anal. (C20H36N2O2) C, H, N.
Compounds 2, 4, 6, and 10 were synthesized with the same method used for the preparation of 8 using the corresponding 1-bromoalkane instead of 1-bromobutane.
1-Adamantan-1-yl-3-[5-(1-methylpentyloxy)pentyl]urea (11)
To a solution of 7 (1.02 g, 3.64 mmol) in DMF (40 mL) was added portion wise triphenylphosphine (1.05 g, 4.01 mmol) and carbon tetrabromide (2.00 g, 4.01 mmol) at 0°C. After stirring for 12 hrs, the product was extracted with ether (60 mL), and the ether solution was washed with water (60 mL), dried over MgSO4, and concentrated. The residue was purified using silica gel column chromatography (hexane:ethyl acetate = 3:1) to give 1-adamantan-1-yl-3-(5-bromopentyl)urea I (1.08 g, 87%) as a solid. This bromide (0.10 g, 0.29 mmol) was added portion wise to a suspension of 60% sodium hydride (12 mg, 0.29 mmol) and 2-hexanol (41 mg, 0.35 mmol) in DMF (15 mL) at room temperature. After stirring for 12 hrs, water (30 mL) was poured into the reaction mixture, and the product was extracted with ether (40 mL). The organic solution was dried over MgSO4, and concentrated. The residue was purified using silica gel column chromatography (hexane:ethyl acetate = 3:1) to afford 11 (5.8 mg, 52%) as a solid. 1H NMR δ (CDCl3) 0.89 (3H, t, J = 6.9 Hz), 1.12 (3H, d, J = 6.9 Hz), 1.30-1.40 (4H, m), 1.48-1.68 (14H, m), 1.96 (6H, brs), 2.06 (3H, brs), 3.09 (2H, q, J = 6.9 Hz), 3.32-3.35 (2H, m), 3.48-3.50 (1H, m), 4.12 (2H, brs). LC-MS (ESI) m/z calcd for C22H40N2O2 [M + H]+ 365.31, found [M + H]+ 365.31, mp 45-48°C, Anal. (C22H40N2O2) C, H, N.
Compounds 12 and 18 were synthesized with the same procedure used for the preparation of 11 using 2-methyl-1-pentanol for 12 and triethylene glycol monomethyl ether for 18, respectively, instead of 2-hexanol.
1-Adamantan-1-yl-3-{5-[2-(2-ethoxyethoxy)ethoxy]pentyl}urea (15)
To a solution of diethylene glycol monoethyl ether (13.0 g, 95 mmol) in THF (150 mL) was added portion wise triphenylphosphine (27.6 g, 0.11 mol) and carbon tetrabromide (35.0 g, 0.11 mol) at 0°C. After stirring for 12 hrs at room temperature, hexane (100 mL) was added to the reaction mixture. This crude mixture was filtered to get rid of triphenylphospine oxide, and the organic solvent dissolving the product was washed with water (100 mL), dried over MgSO4, and concentrated. The residue was purified using silica gel column chromatography (hexane only and hexane:ethyl acetate = 3:1) to give brominated product II as an oil (16.4 g, 87%). II (10.4 g, 53 mmol) was added drop wise to a suspension of 60% sodium hydride (2.83 g, 70mmol) and compound 7 (9.9 g, 35 mmol) in DMF (70 mL) at 0°C. After stirring for 12 hrs, water (150 mL) was poured into the reaction mixture, and the product was extracted with ether (100 mL × 2). The organic solution was dried over MgSO4, and concentrated. The residue was purified using silica gel column chromatography (hexane:ethyl acetate = 3:1) to afford 15 (52%) as a solid. 1H NMR δ (CDCl3) 1.22 (3H, t, J = 6.9 Hz), 1.37-1.43 (2H, m), 1.46-1.53 (2H, m), 1.56-1.61 (2H, m), 1.67 (6H, brs), 1.97 (6H, brs), 2.07 (3H, brs), 3.11 (2H, q, J = 6.9 Hz), 3.46 (2H, t, J = 6.9 Hz), 3.48-3.67 (10H, m), 4.21 (1H, s), 4.26 (1H, s). 13C-NMR δ (CDCl3): 15.12, 23.36, 29.05, 29.53, 29.68, 36.43, 40.00, 42.51, 50.74, 66.64, 69.78, 69.97, 70.57, 70.60, 71.01, 157.2, IR: 1631 cm-1, 3299 cm-1, 3379 cm-1. LC-MS (ESI) m/z calcd for C22H40N2O4 [M + H]+ 397.30, found [M + H]+ 397.31, mp 78°C, Anal. (C22H40N2O4) C, H, N.
Compound 19 was synthesized with the same procedure used for the preparation of 15 using 3-morpholinopropyl bromide prepared from the corresponding alcohol instead of 2-(ethoxyethoxy)-ethyl bromide.
1-Adamantan-1-yl-3-(5-{2-[2-(2,2,2-trifluoroethoxy)ethoxy]ethoxy}pentyl)urea (16)
To a solution of 60% sodium hydride (1.71 g, 42.8 mmol) and 2,2,2-trifluoroethanol (7.20 g, 71.4 mmol) in DMF (40 mL) was added di-(2-bromoethyl) ether (9.20 g, 35.7 mmol) at room temperature. The reaction mixture was stirred for 3 hrs, and water (50 mL) was poured into the reaction. The product was extracted with ether (30 mL X 2), and the organic layer was washed with water (30 mL × 2), dried over MgSO4, and evaporated to give 2-(2,2,2-trifluoroethoxyethoxy)ethyl bromide in a crude mixture (∼60% yield). Without further purification, this bromide intermediate was directly used for the next reaction. To a suspension of 60% sodium hydride (3.83 g, 95.7 mmol) and 7 (10.0 g, 39.3 mmol) stirred for 20 min in DMF (40 mL) was added this alkylated 2-bromoethyl ether intermediate at room temperature. After stirring overnight, the product was extracted with ether (50 mL X 2), and the organic layer was washed with water (80 mL), dried over MgSO4, and evaporated. The residue was purified using column chromatography eluting with hexane and ethyl acetate (1:1) to provide 16 (0.36 g, 45%) as a solid. This purified product was further recrystallized in hexane, and dried in the vacuum oven for 2 days to provide very pure compound. 1H NMR δ (CDCl3) 1.40 (2H, quint, J = 6.9 Hz), 1.50 (2H, quint, J = 6.9 Hz), 1.55 (2H, quint, J = 6.9 Hz), 1.65 (6H, brs), 1.95 (6H, brs), 2.05 (3H, brs), 3.09 (2H, t, J = 6.9 Hz), 3.45 (2H, t, J = 6.9 Hz), 3.57 (2H, t, J = 6.9 Hz), 3.64 (2H, t, J = 6.9 Hz), 3.68 (2H, t, J = 6.9 Hz), 3.78 (2H, t, J = 6.9 Hz), 3.90 (2H, q, J = 6.9 Hz), 4.23 (1H, s), 4.32 (1H, s). 13C-NMR δ (CDCl3): 23.36, 29.20, 29.38, 29.52, 29.63, 29.92, 36.41, 40.07, 42.49, 50.71, 69.10, 69.97, 70.56, 70.70, 71.19, 71.89, 122.8, 157.3. IR: 1631 cm-1, 2909 cm-1, 3352 cm-1. LC-MS (ESI) m/z calcd for C22H37F3N2O4 [M + H]+ 451.27, found [M + H]+ 451.27, mp 34°C, Anal. (C22H37F3N2O4) C, H, N, F.
Compound 17 was synthesized with the same procedure used for the preparation of 16 using 4-ethylphenol instead of 2,2,2-trifluoroethanol.
N-{5-[5-(3-Adamantan-1-yl-ureido)pentyloxy]pentyl}methanesulfonamide (21)
To a solution of 5-amino-1-pentanol (0.59 g, 5.7 mmol) in acetonitrile (20 mL) was added methanesulfonic anhydride (0.50 g, 2.9 mmol) at room temperature. After stirring for 5 hrs, the solvent was evaporated. To this residue ethyl acetate (30 mL) and water (30 mL) were poured and partitioned. The organic solution dissolving the product was washed with water (30 mL), dried over MgSO4, and concentrated. The residue was purified using silica gel column chromatography (hexane:ethyl acetate = 1:3) to give 5-hydroxypentylmethanesulfonamide (0.34, 1.9 mmol) in 66% yield. To a suspension of 60% NaH (76 mg, 1.9 mmol) in DMF (20 mL) a solution of the above methanesulfonamide intermediate in DMF (2 mL) was added drop wise at room temperature. After stirring for 30 min, I (0.65 g, 1.9 mmol) was added to the reaction mixture, and the reaction was further stirred for 12 hrs at room temperature. Water (50 mL) was poured into the mixture, and the product was extracted with ether (60 mL). The ether solution was dried over MgSO4 and concentrated. The residue was purified by using silica gel column chromatography eluting with hexane and ethyl acetate (1:4) to afford 21 (0.46 g,1.0 mmol) in 55% yield. 1H NMR δ (CDCl3) 1.36-1.53 (6H, m), 1.59-1.66 (12H, m), 1.96 (6H, brs), 2.06 (3H, brs), 2.82 (3H, s), 3.11-3.19 (6H, m), 3.66 (2H, t, J = 6.9 Hz), 4.12 (1H, s), 4.22 (1H, s). LC-MS (ESI) m/z calcd for C22H41N3O4S [M + H]+ 444.28, found [M + H]+ 444.32, mp 93°C, Anal. (C22H41N3O4S) C, H, N.
Compound 20 was prepared with the same method used for the preparation of compound 21 using 2-aminoethoxyethanol instead of 5-amino-1-pentanol.
1-Adamantan-1-yl-3-(4-pentyloxycylclohexyl)urea (24)
A mixture of 1-adamantyl isocyanate (0.30 g, 1.69 mmol), trans-4-aminocyclohexanol hydrochloride (0.38 g, 2.54 mmol), and triethylamine (0.37 mL, 2.54 mmol) in DMF (30 mL) was stirred at room temperature for 12 hrs, and to the reaction mixture was poured an aqueous solution of 1N HCl (40 mL) at 0°C. After 30 min stirring, the solid product crystallized was filtered, and washed with water (50 mL) and ethyl acetate (30 mL). The resulting solid was dried in the vacuum oven at 50°C to give 1-adamantan-1-yl-3-(4-hydroxycyclohexyl)urea IV (0.72 g, 100%) as a white solid. To a suspension of 60% sodium hydride (0.11 g, 2.74 mmol) in DMF (15 mL) was added a solution of IV (0.40 g, 1.37 mmol) in DMF (3 mL) at room temperature. After stirring for 30 min, 1-bromopentane (0.25 g, 1.64 mmol) was added to the reaction at room temperature. The reaction was stirred for 12 hrs, and water (50 mL) was poured into the reaction mixture. The product was extracted with ether (60 mL), and the ether solution was dried over MgSO4, and concentrated. The residue was purified by column chromatography on silica gel eluting hexane and ethyl acetate (1:1) to afford 24 (0.12 g, 25%) as a solid. 1H NMR δ (CDCl3) 0.89 (3H, t, J = 6.9 Hz), 1.15 (2H, q, J = 6.9 Hz), 1.35 (2H, q, J = 6.9 Hz), 1.53-1.61 (6H, m), 1.66 (6H, brs), 1.96 (6H, brs), 1.99-2.11 (7H, m), 3.14-3.19 (1H, m), 3.41 (2H, t, J = 6.9 Hz), 3.50-3.52 (1H, m), 3.91 (2H, s). LC-MS (ESI) m/z calcd for C22H38N2O2 [M + H]+ 363.29, found [M + H]+ 363.31, mp 205°C, Anal. (C22H38N2O2) C, H, N.
Compounds 25 and 26 were synthesized with the same method used for the preparation of compound 24 using 2-(ethoxyethoxy)ethyl bromide and 3-morpholinopropyl bromide, respectively, instead of 1-bromopentane.
Enzyme preparation
Recombinant human sEH was produced in a polyhedron positive baculovirus expression system following cloning and sequencing in this laboratory and was purified by affinity chromatography as previously reported.30-32
IC50 assay conditions
IC50 values were determined as described using a sensitive fluorescent based assay,33 and a brief description of the procedure is as follows: cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyl-oxyran-2-yl) methyl carbonate (CMNPC) was used as a fluorescent substrate. Human sEH (1 nM) was incubated with inhibitors for 5 min in pH 7.0 Bis-Tris/HCl buffer (25 mM) containing 0.1 mg/mL of BSA at 30°C prior to substrate introduction ([S] = 5 μM). Activity was measured by determining the appearance of the 6-methoxy-2-naphthaldehyde with an excitation wavelength of 330 nm and an emission wavelength of 465 nm for 10 minutes. IC50 results are averages of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of two-fold or greater are significant.33
Solubility
Water solubility was determined experimentally in 1.0 mL of sodium phosphate buffer (0.1 M, pH 7.4) as previously described at 25 ± 1.5°C.24,25,27
LogP measurement and calculation
In a 20 mL glass vial, the test compound (4 mg) was dissolved in 5 mL of buffer-saturated octanol. Octanol-saturated sodium phosphate buffer (0.1 M, pH 7.4; 5 mL) was then added to the octanol solution. The vial content was mixed strongly for 24 hrs at 23 ± 1.5°C. After, the phases separation, the concentration of the tested compound dissolved in the octanol and the aqueous layers were measured by LC/MS-MS following the method described previously.24 The logP value was obtained with the following equation: LogP = log ([octanol]/[water]). The results reported in Table 4 are averages of triplicate analyses. The calculate LogP (cLogP) value estimated by Crippen's method was generated by using CS ChemDraw 8.0.
In vitro metabolic stability in human liver microsomes or S9 fractions
Human liver microsomal (0.125 mg) or S9 (0.056 mg) protein was brought to 0.222 mL or 0.178 mL with sodium phosphate buffer (0.1 M, pH 7.4), respectively. The proteins were preincubated for 5 min in open glass tubes immersed in a shaking bath at 37°C. After this preincubation, a solution of test compound (2.5 μL or 2.0 μL of 100 μM for microsome and S9) was added and the reaction was initiated by the addition of a NADPH generating system (25 μL or 20 μL for microsome and S9, respectively; NADP (2 mM), glucose 6-phosphate (57 mM), glucose 6-phosphate dehydrogenase (3.5 units), and magnesium chloride (50 mM) in 1 mL of sodium phosphate buffer (0.1 M, pH 7.4)). The incubation mixture (0.25 mL or 0.20 mL total volume for microsome and S9, respectively) was shaken in a water bath at 37°C for 60 min. A control was prepared by the addition of ethanol (1 mL) immediately after adding the NADPH generating system. The reaction was terminated by the addition of cold ethanol (0.75 mL for microsomes or 0.60 mL for S9), and a 50 μL aliquot of 500 ng/mL 1-adamantyl-3-decylurea was added to the samples. The samples were then vortexed and centrifuged at 6,000 rpm (4,000 g) for 5 min. The extracts were transferred to a new glass tube and dried under nitrogen. The residue was reconstituted in methanol (0.5 mL). Aliquots (5 μL) were analyzed by LC-MS/MS.34,35 The absolute amount of parent compounds remaining after the incubation was converted to a percentage. The results given are averages of triplicate independent analyses.
In vivo pharmacokinetic studies
In vivo experiments were performed following protocols approved by the U.C.D. Animal Use and Care Committee. Two rodent species (mice and rats) and dogs were treated with test compounds orally at 5 mg/kg for rodents and 0.3 mg/kg body weight for dogs, respectively. Compounds were given by oral gavage in 3 ml of tristerate at body temperature for the dogs, in 0.1 mL of oleic oil solution for the mice, and in 1 mL of oleic oil solution for the rats. For the rodents, 10 μL of whole blood were collected at 0, 0.5, 1, 2, 4, 6, 8 and 24 hours. For dogs, 1 mL of blood sample was collected into EDTA K3 blood collecting tubes at 0, 0.25, 0.5, 1, 2, 3, 4, 5, 6, 8, and 24 hours. The samples were centrifuged at 3000 rpm at 4°C for 10 minutes and the plasma samples were collected for instrumental analysis. Blood sample preparation and LC/MS/MS analysis were performed as previously reported.26,27 Pharmacokinetic parameters (AUC, and t1/2) were calculated by fitting the blood concentration-time data to a noncompartmental model with WinNonlin 5.0 (Pharsight, CA). Data are average results obtained from at least three different animals.
Supplementary Material
Syntheses and detailed analytical data for compounds 1-6, 9-10, 12-14, 17-20, 22-23, 25-28, and 29. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported in part by NIEHS Grant R37 ES02710, NIEHS Center for Environmental Health Sciences P30 ES05707, NIH/NIEHS Superfund Basic Research Program P42 ES04699, NIH/NHLBI R01 HL59699-06A1, and UCDMC Translational Technology Research Grant.
Abbreviations
- AEPU
1-adamantan-3-(5-(2-(ethyl-ethoxy)ethoxy)pentyl)urea
- AUC
area under the curve
- DMAP
N,N-4-dimethylaminopyridine
- EDCI
1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide hydrochloride
- EETs
epoxyeicosatrienoic acids
- DHETs
dihydroxyeicosatrienoic acids
- LPS
lipopolysaccharide
- SAR
structure activity relationship
- sEH
soluble epoxide hydrolase
References
- 1.Hammock BD, Grant D, Storms D. Epoxide hydrolase. In: Sipes I, McQueen C, Gandolfi A, editors. Comprehensive toxicology. Pergamon Press; Oxford: 1977. pp. 283–305. [Google Scholar]
- 2.Fretland AJ, Omiecinski CJ. Epoxide hydrolases: biochemistry and molecular biology. Chem Biol Intereract. 2000;129:41–59. doi: 10.1016/s0009-2797(00)00197-6. [DOI] [PubMed] [Google Scholar]
- 3.Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, Capdevila JH. Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. J Biol Chem. 1993;268:6402–6407. [PubMed] [Google Scholar]
- 4.Carroll MA, McGiff JC. A new class of lipid mediators: cytochrome P450 arachidonate metabolites. Thorax. 2000;55:S13–16. doi: 10.1136/thorax.55.suppl_2.S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: Their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation: molecular and functional properties on the arachidonate monooxygenase. J Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 7.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 8.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 10.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 11.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2006;46:975–981. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc Natl Acad Sci USA. 2005;102:2186–2191. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res. 1998;83:932–939. doi: 10.1161/01.res.83.9.932. [DOI] [PubMed] [Google Scholar]
- 15.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 16.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol Sci. 2000;21:125–127. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- 18.Yang B, Graham L, Dikalov S, Mason RP, Falck JR, Liao JK, Zeldin DC. Overexpression of cytochrome P450 CYP2J2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol Pharmacol. 2001;60:310–320. doi: 10.1124/mol.60.2.310. [DOI] [PubMed] [Google Scholar]
- 19.Node K, Ruan X, Dai J, Yang S, Graham L, Zeldin DC, Liao JK. Activation of Gs mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 20.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med. 1997;3:562–567. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci USA. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman JW, Denton DL, Morisseau C, Koger CS, Wheelock CE, Hinton DE, Hammock BD. Evaluation of fish models of soluble epoxide hydrolase inhibition. Environ Health Perspect. 2001;109:61–66. doi: 10.1289/ehp.0110961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Structural refinement of inhibitors of urea based soluble epoxide hydrolase. Biochem Pharmacol. 2002;63:1599–1608. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 24.Kim IH, Morisseau C, Watanabe T, Hammock BD. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J Med Chem. 2004:2110–2122. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 25.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J Med Chem. 2005;48:3621–3629. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe T, Schulz D, Morisseau C, Hammock BD. High-throughput pharmacokinetic method: cassette dosing in mice associated with minuscule serial bleedings and LC-MS-MS analysis. Anal Chim Acta. 2006;559:37–44. doi: 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim IH, Nishi K, Tsai HJ, Bradford T, Koda Y, Watanabe T, Morisseau C, Blanchfield J, Toth I, Hammock BD. Design of bioavailable derivatives of 12-(3-adamantan-1-yl-ureido)dodecanoic acid, a potent inhibitor of the soluble epoxide hydrolase. Bioorg Med Chem. 2007;15:312–323. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PD, Tsai HJ, Do Z, Moresseau C, Hammock BC. Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolse. Bioorg Med Chem Lett. 2006;16:5212–5216. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang SH, Morisseau C, Do Z, Hammock BD. Solid-phase combinatorial approach for the optimization of soluble epoxide hydrolase inhibitors. Bioorg Med Chem. 2006;16:5773–5777. doi: 10.1016/j.bmcl.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant DE, Storms DH, Hammock BD. Molecular cloning and expression of murine liver soluble epoxide hydrolase. J Biol Chem. 1993;268:17628–17633. [PubMed] [Google Scholar]
- 31.Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 32.Wixtrom RN, Silva MH, Hammock BD. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated sepharose gels. Anal Biochem. 1988;169:71–80. doi: 10.1016/0003-2697(88)90256-4. [DOI] [PubMed] [Google Scholar]
- 33.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem. 2005;343:66–75. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe T, Hammock BD. Rapid determination of soluble epoxide hydrolase inhibitors in rat hepatic microsomes by high performance liquid chromatography with electrospray tandem mass spectrometry. Anal Biochem. 2001;299:227–234. doi: 10.1006/abio.2001.5423. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe T, Morisseau C, Newman JW, Hammock BD. In vitro metabolism of the mammalian soluble epoxide hydrolase inhibitor, 1-cyclohexyl-3-dodecyl-urea. Drug Metab Dispos. 2003;31:846–853. doi: 10.1124/dmd.31.7.846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Syntheses and detailed analytical data for compounds 1-6, 9-10, 12-14, 17-20, 22-23, 25-28, and 29. This material is available free of charge via the Internet at http://pubs.acs.org.