

Abstract
Inhibitors of the Group IVA phospholipase A2 (GIVA cPLA2) and GVIA iPLA2 are useful tools for defining the roles of these enzymes in cellular signaling and inflammation. We have developed inhibitors of GVIA iPLA2 based on the 2-oxoamide backbone that are uncharged, containing ester groups. While the most potent inhibitors of GVIA iPLA2 also inhibited GIVA cPLA2, there were three 2-oxoamide compounds that selectively and weakly inhibited GVIA iPLA2. We further show that several potent 2-oxoamide inhibitors of GIVA cPLA2, containing free carboxylic groups (Kokotos et al. J. Med. Chem. 2002, 45, 2891–2893), do not inhibit GVIA iPLA2, and are therefore selective GIVA cPLA2 inhibitors.
Introduction
Phospholipase A2 (PLA2) constitutes a superfamily of enzymes that catalyze the hydrolysis of the fatty acid ester from the sn-2 position of a membrane phospholipid, yielding a free fatty acid and a lysophospholipid. Among the intracellular PLA2s are the cytosolic Group IVA PLA2 (GIVA cPLA2), which is generally considered a pro-inflammatory enzyme, and the calcium-independent Group VIA iPLA2 (GVIA iPLA2), which is typically referred to in the literature as iPLA2. GVIA iPLA2 is actually a group of cytosolic enzymes ranging from 85 to 88 kDa and expressed as several distinct splice variants of the same gene, only two of which have been shown to be catalytically active (Group VIA-1 and VIA-2 iPLA2).1 The role of GVIA iPLA2 in the inflammatory process is unclear, but this enzyme appears to be the primary PLA2 for basal metabolic functions within the cell, reportedly including membrane homeostasis,2–7 insulin receptor signaling5,8 and calcium channel regulation.9–11
The GVIA iPLA2 enzymes all contain a consensus lipase motif, Gly-Thr-Ser*-Thr-Gly, with the catalytic serine confirmed by site-directed mutagenesis.1,12 More recently the homologous Group VIB iPLA2 was confirmed to have an active site catalytic dyad consisting of the conserved Ser and an equally conserved Asp.13 The first identification of the novel catalytic Ser/Asp dyad was for GIVA cPLA2 based on exhaustive mutagenesis and a crystal structure, which confirmed that the catalytic dyad is present in a non-canonical α/β hydrolase and that the mechanism involves an acyl-enzyme intermediate on the serine.14–19 A similar structure, topology, and conserved catalytic dyad were also found in patatin, a distant plant homolog of both GIV and GVI PLA2.20 The growing family of lipid hydrolases ulilizing a catalytic Ser-Asp dyad now includes bacterial ExoU, fungal phospholipase B/Spo1, plant patatins, and the many mammalian enzymes in the GIV PLA2, GVI PLA2, and neuropathy target esterase groupings.21
Arachidonyl trifluoromethyl ketone (ATFK) has been shown to function as a tight binding, reversible inhibitor of both GIVA and GVIA PLA2,22,23 while methyl arachidonyl fluorophosphonate (MAFP) functions as an irreversible inhibitor of both enzymes.24 Variants of the trifluoromethyl ketones show differential potencies for GIVA and GVIA PLA2; oleic acid- and phenyl-containing compounds are more potent than ATFK with GVIA iPLA2 and less potent than ATFK with GIVA cPLA2.25 Similar trends in potency are seen with the fluorophosphonate inhibitors; oleic acid and phenyl derivatives are more potent than MAFP towards GVIA iPLA2.25 Interestingly, the trifluoromethylketone and fluorophosphonate inhibitors all show fast binding to GVIA iPLA2 and slow binding to GIVA cPLA2,22,25,26 suggesting subtle differences in the active sites of GIVA and GVIA PLA2. Bromoenol lactone (BEL) is an irreversible, covalent inhibitor of GVIA iPLA2 but does not inhibit GIVA cPLA2. Because of this, BEL is commonly used to selectively inhibit GVIA iPLA2 in cellular systems.3,5,7,9,22 However, it has been shown that in addition to inhibiting GVIA iPLA2, BEL inhibits numerous cellular enzymes including the magnesium-dependent phosphatidate phosphohydrolase 1.27
We have recently reported that 2-oxoamides containing a free carboxyl group are potent inhibitors of human GIVA cPLA2.28,29 The aim of the present work was to develop inhibitors based on the 2-oxoamide backbone that are selective for GIVA or GVIA PLA2. Based upon the similarity of substrates, classes of common inhibitors, and the homologous Ser-Asp catalytic dyad, it is very likely that the active sites of GIVA and GVIA PLA2 are similar such that inhibitors of GIVA cPLA2 may show cross-reactivity with GVIA iPLA2. There are, however, significant differences in substrate preference, known inhibitor profiles, and the primary sequence between GIVA and GVIA PLA2 that could be exploited in designing selective inhibitors.
Design and Synthesis of 2-Oxoamide Inhibitors
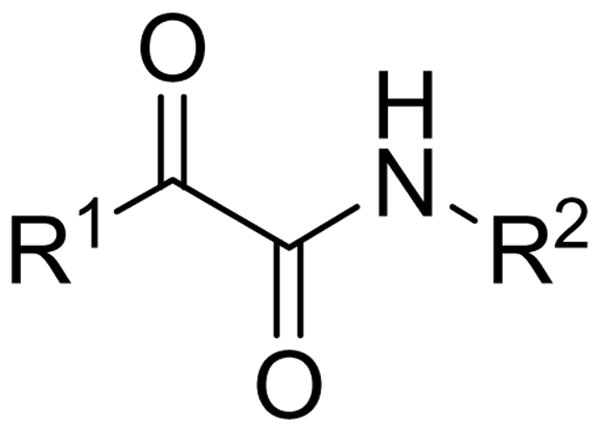
We have developed a strategy for the design of inhibitors of serine-containing lipolytic enzymes, which is based on the principle that the inhibitor should consist of two components: (a) an electrophilic group that is able to react with the active-site serine residue, and (b) a lipophilic segment that contains chemical motifs necessary for both specific interactions and a proper orientation in the substrate binding cleft of the enzyme.30 This strategy has been successfully applied in the development of lipophilic 2-oxoamides,31,32 2-oxoamide and bis-2-oxoamide triacylglycerol analogues,33,34 as well as lipophilic aldehydes35 and trifluoromethyl ketones36 as effective inhibitors of pancreatic and gastric lipases. Accordingly, we have recently developed a novel class of 2-oxoamides that inhibit GIVA cPLA2.28,29 The noted homology of GVIA iPLA2 to GVIB PLA2, patatin and GIVA cPLA2 (lipases known to possess a catalytic Ser-Asp dyad) and the confirmation of its catalytic serine strongly suggest that GVIA iPLA2 would be susceptible to inhibition by 2-oxoamides.12 Thus, we studied a number of 2-oxoamides of the generic structure shown in Scheme 1 in an effort to understand the effect of R1 and R2 groups on GVIA iPLA2 inhibition.
Scheme 1.
2-Oxoamide inhibitors containing either a free carboxyl group or a carboxymethyl ester group and 2-oxoacyl residues based on oleic acid or phenyl groups were synthesized using methods previously developed,29 as depicted in Scheme 2. In Scheme 3, the synthesis of inhibitors based on a γ-amino-α,β-unsaturated acid is shown. It should be noted that the oxidation of the unsaturated 2-hydroxyamides 2c, 6 and 7 was carried out using Dess-Martin periodinane,37 instead of NaOCl/TEMPO, to avoid oxidation of the double bonds.
Scheme 2a.

aReagents and conditions: (a) H2N(CH2)3COOCH3, Et3N, WSCI, HOBt, CH2Cl2; (b) NaOCl, TEMPO, NaBr, NaHCO3, EtOAc/toluene/H2O, 0 ºC; (c) Dess-Martin periodinane, CH2Cl2; (d) 1N NaOH/MeOH; (e) NaOCl, TEMPO, NaBr, NaHCO3, EtOAc/toluene/H2O, 0 ºC, then HCl.
Scheme 3a.

aReagents and conditions (a) NaOCl, TEMPO, NaBr, NaHCO3, EtOAc/toluene/H2O, −5 ºC; (b) Ph3P=CHCOOCH3, THF, reflux; (c) 4 N HCl in THF; (d) CH3(CH2)13CHOHCOOH, Et3N, WSCI, HOBt, CH2Cl2; (e) 1N NaOH/MeOH; (f) Dess-Martin periodinane, CH2Cl2.
Selective Inhibition of GIVA and GVIA PLA2 by 2-Oxoamide Inhibitors
Fourteen 2-oxoamides were tested for inhibition of GVIA iPLA2 in our in vitro assay system27,28 and compared with GIVA cPLA2 inhibition. The data, summarized in Table 1, are represented as XI(50) values. XI(50) is defined as the inhibitor concentration in a two-dimensional micellar surface that produces 50% inhibition. The surface concentration (mole fraction units) is calculated as the moles of inhibitor divided by the total moles of inhibitor, detergent, and phospholipid in the micelle surface. XI(50) is utilized as opposed to the more common IC50 because GIVA and GVIA PLA2 are active at a two-imensional lipid interface containing the substrate phospholipids rather than in three-dimensional solution with soluble, monomeric substrates.22,25,38,42 Because the 2-oxoamide inhibitors also partition to the micelle interface, the relevant concentration of inhibitor for membrane-bound enzymes is the surface concentration (mole fraction) not the bulk concentration (molar units).22,25,28,38,39,42 Of the fourteen compounds listed in Table 1, five show at least partial inhibition of GVIA iPLA2 at the highest concentrations tested.
Table 1.
Structures of 2-Oxoamide Inhibitors and their Effects on GIVA and GVIA PLA2.
| Number | Structure | Inhibition of GVIA iPLA2 | Inhibition of GIVA cPLA2 |
|---|---|---|---|
| 13 |
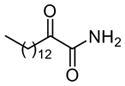
|
NDa,f | NDe |
| 14 |
|
ND | ND |
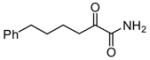
| 15 |
|
LDb,f | NDe |
| 16 |
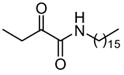
|
LDf | NDe |
| 17 |
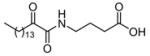
|
ND | XI(50) = 0.017 ± 0.009c,d |
| 18 |
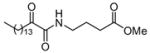
|
LD | ND |
| 5a |
|
ND | ND |
| 4a |
|
ND | ND |
| 5b |
|
ND | ND |
| 4b |
|
ND | ND |
| 12 |
|
ND | XI(50) = 0.011 ± 0.003 |
| 4c |
|
XI(50) = 0.067 ± 0.003 | XI(50) = 0.012 ± 0.014 |
| 11 |
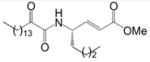
|
XI(50) = 0.032 ± 0.010 | XI(50) = 0.018 ± 0.010 |
| 10 |
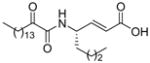
|
ND | XI(50) = 0.003 ± 0.001 |
ND: negligible inhibition (0–25%) at highest dose. Unless otherwise indicated the highest dose tested was 0.091 mole fraction.
LD: limited inhibition (25–50%) at highest dose.
Data taken from Ref. 28.
XI(50) is the surface concentration of inhibitor at which there is 50% inhibition.
0.01 mole fraction.
0.02 mole fraction.
Among the primary 2-oxoamides 13 (AX001)29 and 14 (AX015),29 neither exhibits significant inhibition of GIVA or GVIA PLA2. The secondary 2-oxoamides, 15 (AX002)29 and 16 (AX009),29 with long carbon chains either at the R1 or at the R2 position present limited inhibition of GVIA iPLA2, but Four 2-oxoamides containing a substituted phenyl chain at the no detectable inhibition of GIVA cPLA2. R1 position (4a,b, 5a,b) (AX035-AX038) did not inhibit GVIA iPLA2. This is somewhat unexpected given previous reports of the selectivity of phenyl-containing fluoroketones or fluorophosphonates. None of the phenyl-containing 2-oxoamides inhibits GIVA cPLA2.
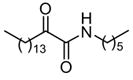
The 2-oxoamides containing a free carboxyl group 17 (AX006),29 12 (AX040), and 10 (AX074) inhibit GIVA cPLA2 but do not inhibit GVIA iPLA2. In fact, in all cases these compounds enhance GVIA iPLA2 enzymatic activity. The increased GVIA iPLA2 activity may be due to increased negative charge at the micelle surface due to addition of inhibitors with a free carboxyl group. Unlike the inhibitors of GIVA cPLA2, the inhibitors of GVIA iPLA2 18 (AX010),29 4c (AX041), and 11 (AX073) are uncharged. The effect of charge is highlighted when comparing 17 to 18, where 18 possesses a carboxymethyl ester in place of the free carboxyl found in 17. Compound 18 exhibits limited inhibition of GVIA iPLA2 but does not significantly inhibit GIVA cPLA2. Compound 17 does not significantly inhibit GVIA iPLA2 at concentrations up to 0.091 mole fraction but is a potent inhibitor of GIVA cPLA2 with an XI(50) value of 0.017 mole fraction.28 Compound 4c is an inhibitor of GVIA iPLA2 with an XI(50) value of 0.067 mole fraction. Interestingly, it also inhibits GIVA cPLA2 with an XI(50) value of 0.012 mole fraction. Compound 12, the charged variant of 4c, does not inhibit GVIA iPLA2 but is an inhibitor of GIVA cPLA2 with an XI(50) value of 0.011 mole fraction. Consistent results were seen with compounds 11 and 10. These compounds are also variants that contain either a carboxymethyl ester (11) or a free carboxyl (10). Compound 10 is the most potent 2-oxoamide inhibitor of GIVA cPLA2 reported to date with an XI(50) of 0.003 mole fraction. By observing the trend of inhibition of GVIA iPLA2 by 18, 4c, and 11, it appears that an unsaturated chain at R1 or R2 is preferable to a saturated one. This is consistent with the presence of unsaturated fatty acids at the sn-2 position of many phospholipids. The inhibition dose-response curve for 18 appears to plateau at the higher mole fractions tested. The in vitro assay contains detergent and phospholipid that should readily form mixed micelles with 18, which has a similar hydrophobicity (ClogP) to many other compounds that behave normally. Most other lower potency 2-oxoamide inhibitors possess a linear dose-response. Compound 18 is unique as a lower potency inhibitor with a logarithmic dose-response.
A known reference inhibitor (non-covalent and readily reversible) for GIVA cPLA2 is not commercially available, but a patented inhibitor of GIVA cPLA2, pyrrophenone, is described in the literature40,41. Comprehensive analysis of pyrrophenone demonstrated that it inhibits GIVA cPLA2 with an XI(50) of 0.002 mole fraction under a variety of assay conditions.42 This level of potency is similar to the most potent GIVA cPLA2 2-oxoamide inhibitors, (4S)-4-[(2-oxododecanoyl)amino]octanoic acid (AX007)29 and 10 (this work). Pyrrophenone was reported to have no effect on the activity of GVIA iPLA2.42 A known reference inhibitor (non-covalent and readily reversible) for GVIA iPLA2 is palmitoyl trifuoromethyl ketone (PATK). Previous tests of this compound in our lab have confirmed the XI(50) of PATK for GVIA iPLA2 is 0.0075 mole fraction.22 A further study tested an expanded panel of hydrophobic trifluoromethyl ketones and found that most are slow, tight-binding inhibitors of GIVA cPLA2 and fast, reversible inhibitors of GVIA iPLA2, so the inhibition of the two enzymes by these compounds are not readily comparable.25
Mechanism of GVIA PLA2 Inhibition by 2-Oxoamide Inhibitors
We tested 18 and 11 to determine if these inhibitors showed either time-dependent or irreversible inhibition of GVIA iPLA2. GVIA iPLA2 (25 ng) was preincubated with either 18 or 11 (5 μM) for 0, 5, 15 or 30 minutes and then assayed in the standard GVIA iPLA2 assay mix containing 5 μM inhibitor. The final concentration of the inhibitors in the assay mix was 0.01 mole fraction, and the samples were incubated for 30 minutes at 40°C. Both 18 and 11 showed no increased potency with prolonged incubation, demonstrating a fast-binding and reversible mode of inhibition (Figure 2A). We next preincubated 25 ng of GVIA iPLA2 with 10 μM 18 or 11 for 10 minutes before diluting the enzyme 1:50 into the standard GVIA iPLA2 assay mix lacking inhibitor, and incubating for 30 minutes at 40 °C. The final inhibitor concentration in these assays was 0.0004 mole fraction, well below surface concentrations that either 18 or 11 inhibit the enzyme. GVIA iPLA2 showed full activity in this system, demonstrating that both 18 and 11 are freely reversible inhibitors (Figure 2B).
Figure 2.

Immediate and reversible inhibition of GVIA iPLA2 by 18 and 11. A. Time-dependent binding of 18 and 11 was tested by pre-incubating no inhibitor (black bar), 5 μM 18 (light bars), or 5 μM 11 (dark bars) with GVIA iPLA2 prior to adding to mixed micelles consisting of 100 μM DPPC and 400 μM Triton X-100 containing 0.01 mole fraction inhibitor. B. Reversibility of 18 and 11 was tested by preincubating no inhibitor, 10 μM 18, or 10 μM 11 with GVIA iPLA2 for 10 minutes prior to diluting 1:50 into mixed micelles consisting of 100 μM DPPC and 400 μM Triton X-100 and assaying for activity.
Inhibition of PGE2 Production by 2-Oxoamide Inhibitors
We tested several 2-oxoamides in the long-term lipopolysaccharide (LPS) stimulation pathway in the murine RAW 264.7 macrophage-like cell line.43,44 This pathway requires GIVA cPLA2 activity for maximal extracellular release of many eicosanoid compounds, including the prostaglandin PGE2.45 Compound 18, which does not significantly inhibit GIVA cPLA2 in vitro, also did not inhibit PGE2 release from the RAW cells (data not shown). In the low μM range, 4c and 11 reduced PGE2 release by roughly 40% (Figure 3). Based on previous work, this is the fraction of PGE2 release attributable to GIVA cPLA2.44,45 At 1 μM and 5 μM concentrations, small activations were often seen, suggesting minor stimulation of the cells from membrane perturbing compounds.
Figure 3.

Inhibition of PGE2 production in RAW 264.7 cells by 2-oxoamides containing a methyl ester. Increasing concentrations of 4c (○) or 11 (▼) were added to cells for 30 minutes prior to stimulation with 100 ng/ml LPS for 24 hours. Media was harvested and assayed for PGE2 production as described in the experimental section.
In conclusion, based on the 2-oxoamide backbone structure we have developed inhibitors that selectively inhibit GIVA cPLA2 or inhibit both GIVA and GVIA PLA2. The selective 2-oxoamide inhibitors of GIVA cPLA2 were found to be charged, containing a free carboxyl group. Interestingly some non-charged 2-oxoamides showed dual specificity in inhibiting both GIVA cPLA2 and GVIA iPLA2. Inhibitors selective for GIVA cPLA2 or dual specificity inhibitors reduced PGE2 levels in cellular assays that test for inhibition of GIVA cPLA2. Several 2-oxoamide compounds that significantly inhibit GVIA iPLA2 are promising leads for selective inhibitors of GVIA iPLA2 that would significantly improve investigations into the role of GVIA iPLA2 in cellular systems. As we have previously demonstrated for 2-oxoamide inhibitors of GIVA cPLA2, the inhibitors of GVIA iPLA2 are also fast-binding and freely reversible. Such selective inhibitors of GIVA and GVIA enzymes will be a significant asset in examining the role of these enzymes in cellular signaling and inflammation.
Experimental Section
Synthesis of 2-Oxoamide Inhibitors
Melting points were determined on a Buchi 530 apparatus and are uncorrected. Specific rotations were measured at 25 °C on a Perkin-Elmer 343 polarimeter using a 10 cm cell. NMR spectra were recorded on a Varian Mercury (200 MHz) spectrometer. Fast atom bombardment (FAB) mass spectra were recorded using a VG analytical ZAB-SE instrument. Electron spray ionization (ESI) mass spectra were recorded on a Finnigan, Surveyor MSQ Plus spectrometer. TLC plates (silica gel 60 F254) and silica gel 60 (70–230 or 230–400 mesh) for column chromatography were purchased from Merck.
Coupling of 2-hydroxy acids with amino components
To a stirred solution of 2-hydroxy acid (2.0 mmol) and hydrochloride amino component (2.0 mmol) in CH2Cl2 (20 mL), Et3N (0.61 mL, 4.4 mmol) and subsequently 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide (WSCI) (0.42 g, 2.2 mmol) and 1-hydroxybenzotriazole (HOBt) (0.27 g, 2.0 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and overnight at room temperature. The solvent was evaporated under reduced pressure and EtOAc (20 mL) was added. The organic layer was washed consecutively with brine, 1N HCl, brine, 5% NaHCO3, and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by column-chromatography using CHCl3 as eluent.
4-(2-Hydroxy-5-phenyl-pentanoylamino)-butyric acid methyl ester (2a)
yield 82%; white solid; m.p. 34–35 °C; 1H NMR: δ 7.24-7.11 (5H, m, C6H5), 6.82 (1H, m, NHCO), 4.06 (1H, m, CH), 3.62 (3H, s, CH3O), 3.53 (1H, d, J = 5.2 Hz, OH), 3.26 (2H, m, CH2NH), 2.59 (2H, t, J = 7.8 Hz, CH2C6H5), 2.30 (2H, t, J = 6.8 Hz, CH2COO), 1.82-1.70 (6H, m, 3×CH2); 13C NMR: δ 174.2, 173.8 142.0, 128.3, 128.2, 125.7, 71.7, 51.7, 38.3, 35.5, 34.3, 31.3, 26.8, 24.6; MS (ESI): m/z (%): 316 (100) [M + Na]+. Anal. (C16H23NO4) C, H, N.
4-(2-Hydroxy-6-phenyl-hexanoylamino)-butyric acid methyl ester (2b)
yield 85%; white solid; m.p. 50–51 °C; 1H NMR: δ 7.31-7.15 (5H, m, C6H5), 6.76 (1H, m, NHCO), 4.08 (1H, m, CH), 3.68 (3H, s, CH3O), 3.32 (2H, m, CH2NH), 3.10 (1H, d, J = 4.8 Hz, OH), 2.62 (2H, t, J = 7.8 Hz, CH2C6H5), 2.36 (2H, t, J = 7.4 Hz, CH2COO), 1.91-1.49 (8H, m, 4×CH2); 13C NMR: δ 174.0, 142.3, 128.3, 128.2, 125.7, 72.0, 51.7, 38.4, 35.7, 34.7, 31.4, 31.1, 24.6; MS (ESI): m/z (%): 330 (88) [M + Na]+, 308 (100) [M + H]+. Anal. (C17H25NO4) C, H, N.
4-(2-Hydroxy-nonadec-10-enoylamino)-butyric acid methyl ester (2c)
yield 82%; white solid; m.p. 55–57 °C; 1H NMR: δ 6.80 (1H, m, NHCO), 5.33 (2H, m, CH=CH), 4.07 (1H, m, CH), 3.67 (3H, s, CH3O), 3.30 (2H, m, CH2NH), 2.37 (2H, t, J = 7.2 Hz, CH2COO), 1.98 (4H, m, 2×CH2CH=CH), 1.85 (2H, m, CH2CH2NH), 1.26 (24H, br s, 12×CH2), 0.87 (3H, t, J = 6.6 Hz, CH3); 13C NMR: δ 174.2, 173.8, 129.9, 129.7, 72.1, 51.7, 38.4, 34.8, 31.8, 31.3, 29.7, 29.5, 29.4, 29.3, 29.2, 27.2, 25.0, 24.6, 22.6, 14.1. Anal. (C24H45NO4) C, H, N.
4-(2-Hydroxy-hexadecanoylamino)-oct-2-enoic acid methyl ester (9)
The oxidation of compound 4 follows method A. The Wittig reaction of the resulting N-protected α-aminoaldehyde with a stabilized ylide and the general method for the removal of the Boc group was carried out as described previously.29 The coupling reaction to yield compound 9 is as described above. The overall yield 52%; white solid; m.p 40–42 °C; 1H NMR: δ 6.85 (1H, dd, J1 = 5.2 Hz, J2 = 15.4 Hz, CHCH =CH), 6.60 (1H, d, J = 9.2 Hz, NHCO), 5.87 (1H, d, J = 15.4 Hz, CH=CHCOOCH3), 4.62 (1H, m, CH), 4.14 (1H, m, CH), 3.73 (3H, s, COOCH3), 2.77 (1H, m, OH), 1.98-1.01 (32H, m, 16xCH2), 0.86 (6H, t, J = 7 Hz, 2×CH3); 13C NMR: δ 173.3, 166.7, 148.0, 120.5, 72.3, 51.6, 49.6, 37.0, 34.9, 34.0, 31.9, 29.7, 29.5, 29.3, 27.7, 25.0, 24.9, 22.7, 22.3, 14.1, 13.8; MS (ESI): m/z (%): 448 (100) [M + Na]+. Anal. (C25H47NO4) C, H, N.
Oxidation of 2-hydroxy-amides
Method A
To a solution of 2-hydroxy-amide (5.00 mmol) in a mixture of toluene-EtOAc 1:1 (30 mL), a solution of NaBr (0.54 g, 5.25 mmol) in water (2.5 mL) was added followed by TEMPO (11 mg, 0.050 mmol). To the resulting biphasic system, which was cooled at −5 °C, an aqueous solution of 0.35 M NaOCl (15.7 mL, 5.50 mmol) containing NaHCO3 (1.26 g, 15 mmol) was added dropwise under vigorous stirring, at −5 °C over a period of 1 h. After the mixture had been stirred for a further 15 min at 0 °C, EtOAc (30 mL) and H2O (10 mL) were added. The aqueous layer was separated and washed with EtOAc (20 mL). The combined organic layers were washed consecutively with 5% aqueous citric acid (30 mL) containing KI (0.18 g), 10% aqueous Na2S2O3 (30 mL), and brine and dried over Na2SO4. The solvents were evaporated under reduced pressure and the residue was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C), 1:9].
4-(2-Oxo-5-phenyl-pentanoylamino)butyric acid methyl ester (4a)
yield 67%; white solid; m.p. 30–31 °C; 1H NMR: δ 7.19-7.15 (6H, m, C6H5, NHCO), 3.67 (3H, s, CH3O), 3.35 (2H, m, CH2NH), 2.94 (2H, t, J = 7.4 Hz, CH2COCO), 2.65 (2H, t, J = 7.8 Hz, CH2C6H5), 2.36 (2H, t, J = 7.0 Hz, CH2COO), 1.91 (4H, m, 2×CH2); 13C NMR: δ 198.7, 173.2, 160.0, 141.1, 128.3, 128.2, 125.8, 51.6, 38.5, 35.9, 34.8, 31.1, 24.6, 24.1; MS (ESI): m/z (%): 314 (63) [M + Na]+. Anal. (C16H21NO4) C, H, N.
4-(2-Oxo-6-phenyl-hexanoylamino)butyric acid methyl ester (4b)
yield 75%; white solid; m.p. 52–54 °C; 1H NMR: δ 7.29-7.16 (6H, m, C6H5, NHCO), 3.69 (3H, s, CH3O), 3.37 (2H, m, CH2NH), 2.95 (2H, t, J = 7.0 Hz, CH2COCO), 2.64 (2H, t, J = 7.0 Hz, CH2C6H5), 2.38 (2H, t, J = 7.0 Hz, CH2COO), 1.89-1.66 (6H, m, 3×CH2); 13C NMR: δ 198.8, 173.2, 160.1, 141.9, 128.21, 128.15, 125.6, 51.6, 38.5, 36.4, 35.4, 31.1, 30.6, 24.2, 22.6; MS (ESI): m/z (%): 328 (75) [M + Na]+. Anal. (C17H23NO4) C, H, N.
4-(2-Oxo-5-phenyl-pentanoylamino)butyric acid (5a)
The procedure is the same as that followed in method A described above, with the difference that in this case the aqueous layer was acidified before the work-up, and then extracted with EtOAc, and the combined organic layers were washed with 5% aqueous citric acid containing KI, and 10% aqueous Na2S2O3 (30 mL). The residue was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C)]. Yield 48%; white solid; m.p. 65–67 °C; 1H NMR: δ 7.25-7.11 (6H, m, C6H5, NHCOCO), 3.33 (2H, m, CH2NH), 2.86 (2H, t, J = 7.4 Hz, CH2COCO), 2.60 (2H, m, CH2), 2.36 (2H, m, CH2), 1.86 (4H, m, 2×CH2); 13C NMR: δ 198.8, 178.5, 160.3, 141.2, 128.41, 128.37, 126.0, 38.5, 36.1, 34.9, 31.2, 24.7, 24.0; MS (ESI) : m/z (%): 276 (100) [M – H]−. Anal. (C15H19NO4) C, H, N.
4-(2-Oxo-6-phenyl-hexanoylamino)-butyric acid (5b)
The procedure is the same as that followed for 5a. Yield 47%; white solid; m.p. 60–62 °C; 1H NMR: δ 7.27-7.15 (6H, m, C6H5, NHCOCO), 3.35 (2H, m, CH2NH), 2.94 (2H, t, J = 7.4 Hz, CH2COCO), 2.60 (2H, m, CH2), 2.38 (2H, m, CH2), 1.86 (2H, m, CH2), 1.64 (4H, m, 2×CH2); 13C NMR: δ 198.8, 178.8, 160.3, 142.0, 128.33, 128.27, 125.7, 38.6, 36.5, 35.5, 31.4, 30.7, 24.2, 22.6; MS (FAB) : m/z (%): 292 (100) [M + H]+. Anal. (C16H21NO4) C, H, N.
Oxidation of 2-hydroxyamides
Method B
To a solution of 2-hydroxyamide (1 mmol) in dry CH2Cl2 (20 mL) Dess-Martin periodinane was added (0.64 g, 1.5 mmol) and the mixture was stirred for 2 h at room temperature. The organic solution was washed with 10% aqueous NaHCO3, dried over Na2SO4 and the organic solvent was evaporated under reduced pressure. The residue was purified by recrystallization [EtOAc/petroleum ether (bp 40–60 °C)].
4-(2-Oxononadec-10-enoylamino)butyric acid methyl ester (4c)
yield 82%; oily solid; 1H NMR: δ 7.13 (1H, m, NHCOCO), 5.33 (2H, m, CH=CH), 3.67 (3H, s, CH3O), 3.33 (2H, m, CH2NH), 2.91 (2H, t, J = 7.2 Hz, CH2COCO), 2.38 (2H, t, J = 7.4 Hz, CH2COO), 1.98 (4H, m, 2×CH2CH=CH), 1.88 (2H, m, CH2CH2NH), 1.59 (2H, m, CH2CH2COCO), 1.26 (20H, br s, 10×CH2), 0.87 (3H, t, J = 6.6 Hz, CH3); 13C NMR: δ 199.2, 173.3, 160.3, 129.9, 129.7, 51.7, 38.0, 36.7, 31.8, 31.3, 29.7, 29.6, 29.5, 29.3, 29.2, 29.0, 28.98, 27.2, 27.1, 24.3, 23.1, 22.6, 14.1; MS (FAB): m/z (%): 410 (100) [M + H]+. Anal. (C24H43NO4) C, H, N.
4-(2-Oxohexadecanoylamino)oct-2-enoic acid methyl ester (11)
yield 81%, white solid; m.p. 48–50 °C; [α]D −19.7 (c 0.95 CHCl3); 1H NMR: δ 6.93 (1H, d, J = 8 Hz, NHCOCO), 6.85 (1H, dd, J1 = 6 Hz, J2 = 16 Hz, CHCH=CH), 5.87 (1H, d, J = 16 Hz, CH=CHCOOCH3), 4.58 (1H, m, CH), 3.73 (3H, s, COOCH3), 2.91 (2H, t, J = 7 Hz, CH2COCO), 1.61 (4H, m, 2×CH2), 1.30 (26H, m, 13×CH2), 0.88 (6H, t, J = 7 Hz, 2×CH3); 13C NMR: δ 199.3, 166.7, 159.8, 146.9, 121.4, 51.9, 50.4, 37.0, 34.1, 32.1, 29.9, 29.8, 29.6, 29.5, 29.3, 27.9, 23.4, 22.9, 22.5, 14.3, 14.0; MS (ESI): m/z (%): 446 (85) [M + Na]+. Anal. (C25H45NO4) C, H, N.
4-(2-Oxohexadecanoylamino)oct-2-enoic acid (10)
The procedure is the same as that followed in method B with the difference that the organic layer was not washed with 10% aqueous NaHCO3. Yield 69%, white solid; m.p. 65–67 °C; [α]D −7.7 (c 0.84 CHCl3); 1H NMR: δ 7.0 (1H, m, NHCOCO), 6.82 (1H, dd, J1 = 6 Hz, J2 = 16 Hz, CHCH=CH), 5.87 (1H, d, J = 16 Hz, CH=CHCOOCH3), 4.62 (1H, m, CH), 2.91 (2H, t, J = 7 Hz, CH2COCO), 1.61 (4H, m, 2×CH2), 1.44-1.25 (26H, m, 13×CH2), 0.88 (6H, t, J = 7 Hz, 2×CH3); 13C NMR: δ 199.0, 170.8, 159.6, 149.0, 120.8, 50.2, 36.7, 33.7, 31.9, 29.6, 29.4, 29.3, 29.0, 27.7, 23.1, 22.7, 22.3, 14.1, 13.8; MS (ESI): m/z (%): 408 (100) [M – H]−. Anal. (C24H43NO4) C, H, N.
Saponification of methyl esters
To a stirred solution of methyl ester (2.00 mmol) in a mixture of dioxane-H2O (9:1, 20 mL), 1N NaOH (2.2 mL, 2.2 mmol) was added and the mixture was stirred for 12 h at room temperature. The organic solvent was evaporated under reduced pressure and H2O (10 mL) was added. The aqueous layer was washed with EtOAc, acidified with 1N HCl, and extracted with EtOAc (3 × 12 mL). The combined organic layers were washed with brine, dried over Na2SO4, and evaporated under reduced pressure. The residue was purified after recrystallization [EtOAc-petroleum ether (bp 40–60 °C)].
4-(2-Hydroxy-5-phenylpentanoylamino)butyric acid (3a)
yield 79%; white solid; m.p. 63–65 °C; 1H NMR: δ 7.26-7.12 (6H, m, C6H5, NHCO), 4.09 (1H, m, CH), 3.27 (2H, m, CH2NH), 2.59 (2H, t, J = 6.6 Hz, CH2C6H5), 2.31 (2H, t, J = 6.6 Hz, CH2COOH), 1.78 (6H, m, 3×CH2); 13C NMR: δ 177.3, 175.5, 142.0, 128.3, 125.8, 71.8, 38.4, 35.5, 34.1, 31.3, 26.8, 24.3. Anal. (C15H21NO4) C, H, N.
4-(2-Hydroxy-6-phenylhexanoylamino)butyric acid (3b)
yield 86%; white solid; m.p. 78–80 °C; 1H NMR: δ 7.30-7.13 (6H, m, C6H5, NHCO), 4.11 (1H, m, CH), 3.30 (2H, m, CH2NH), 2.60 (2H, t, J = 7.8 Hz, CH2C6H5), 2.35 (2H, t, J = 6.6 Hz, CH2COOH), 1.81-1.47 (8H, m, 4×CH2); 13C NMR: δ 177.4, 175.5, 142.4, 128.3, 128.2, 125.7, 71.9, 38.4, 35.7, 34.3, 31.4, 31.1, 24.7, 24.4; MS (ESI): m/z (%): 316 (54) [M + Na]+, 294 (100) [M + H]+. Anal. (C16H23NO4) C, H, N.
4-(2-Hydroxyhexadecanoylamino)oct-2-enoic acid (8)
yield 62%; white solid; m.p 46–48 °C; 1H NMR: δ 6.92 (1H, m, NHCO), 6.76 (1H, dd, J1 = 6 Hz, J2 = 16 Hz, CHCH =CH), 5.87 (1H, d, J = 16 Hz, CH=CHCOOH), 4.64 (1H, m, CH), 4.20 (1H, m, CH), 3.42 (1H, br, OH), 1.95-1.25 (32H, m, 16xCH2), 0.88 (6H, t, J = 7 Hz, 2xCH3); 13C NMR: δ 172.3, 170.5, 150.0, 120.5, 72.6, 49.9, 35.1, 34.2, 32.1, 29.9, 29.6, 28.0, 25.3, 22.9, 22.7, 22.5, 14.3, 14.1; MS (ESI): m/z (%): 434 (100) [M + Na]+. Anal. (C24H45NO4) C, H, N.
Inhibitor 12 was prepared by similar procedures.
4-(2-Oxononadec-10-enoylamino)butyric acid (12)
yield 69%; white solid; m.p. 57–59 °C; 1H NMR: δ 10.05 (1H, br, COOH), 7.23 (1H, m, NHCOCO), 5.33 (2H, m, CH=CH), 3.38 (2H, m, CH2NH), 2.90 (2H, t, J = 7.2 Hz, CH2COCO), 2.41 (2H, t, J = 6.8 Hz, CH2COOH), 1.98 (4H, m, 2×CH2CH=CH), 1.89 (2H, m, CH2CH2NH), 1.58 (2H, m, CH2CH2COCO), 1.26 (20H, br s, 10×CH2), 0.87 (3H, t, J = 6.6 Hz, CH3); 13C NMR: δ 199.1, 178.4, 160.4, 129.9, 129.7, 38.5, 36.7, 32.7, 31.8, 31.2, 29.7, 29.6, 29.5, 29.3, 29.2, 29.02, 28.96, 27.1, 24.1, 23.1, 22.6, 14.1; MS (ESI): m/z (%): 418 (95) [M + Na]+. Anal. (C23H41NO4) C, H, N.
Inhibitors 13–18 were prepared as described previously.28,29
Expression and Purification of Recombinant Group VIA PLA2
Protein was produced in Sf9 insect cells using a recombinant baculovirus. The virus had been constructed using the cDNA coding for human Group VIA-2 iPLA2, kindly provided by Dr. Brian Kennedy at Merck-Frost, modified with a six residue histidine tag added three amino acids from the amino terminus using PCR with oligonucleotides 5′-ATGCAGTTCCACCATCACCATCACCATTTTGGAGCGCTGGTCAATACC-3′ and 5′-CCTCAGGGTGAGAGCAGCAGCTG-3′. Gateway cloning ends were added to the histidine-tagged Group VIA-2 cDNA followed by insertion into pDONOR201 (Invitrogen) to produce a Gateway entry clone. The gene construct was then transferred to pDEST8 using Gateway cloning technology and used to make recombinant baculovirus using the Bac-to-Bac system (Invitrogen).
A suspension culture of Sf9 insect cells at a density of 1.1 to 1.5 million cells per mL was infected with the recombinant baculovirus with an MOI of approximately 0.1. Infections were carried out for 72 hours and the cells were harvested by centrifugation at 3,000 × g for 10 minutes and stored at −80 °C. The frozen cell pellets from 200 mL of suspension culture Sf9 cells were resuspended in 25 mL resuspension buffer (25 mM Tris pH 8.0, 150 mM NaCl, 10 mM DTT, 5 mM EDTA, 2 mM ATP, 0.2% methyl-β-cyclodextrin (Sigma-Aldrich) and 1X protease inhibitor cocktail). The cells were lysed by repeated sonication, and the lysate was allowed to sit on ice for 10 minutes and then clarified by centrifugation at 15,000 × g for 30 minutes at 4 °C. The resulting pellet was resuspended in solubilization buffer (25 mM Tris pH 8.0, 150 mM NaCl, 10 mM β-mercaptoethanol, 2 mM ATP, 1 M urea and 1X protease inhibitor cocktail) by 20 passes of a Dounce homogenizer with the tight pestle. The resuspended pellet was then stirred at 4 °C for one hour followed by centrifugation at 15,000 × g for 30 minutes at 4 °C to remove insoluble material. At this point, 2.5 mL of Fast-flow Ni-NTA resin per 200 mL cell pellet was mixed with the soluble protein fraction and allowed to incubate at 4 °C for 30 minutes for batch binding. The protein/resin slurry was poured into a column and allowed to settle. The column was washed with 15 column volumes of Ni-wash buffer (25 mM NaHPO4 pH 7.4, 250 mM NaCl, 2 mM ATP, 0.2% dodecyl maltoside (Anatrace) and 1X protease inhibitor cocktail) and eluted with 10 column volumes of Ni-elution buffer (25 mM NaHPO4 pH 7.4, 100 mM NaCl, 50 mM urea, 2 mM ATP and 200 mM imidazole, 30% v/v glycerol). Eluate was collected as 1.5 mL fractions into tubes containing 15 μL 500 mM DTT (5 mM DTT final). Fractions containing protein were pooled, measured for activity and protein concentration, and stored as 200 μL aliquots at −80 °C.
Group VIA iPLA2 Activity Assays
The standard Group VIA iPLA2 activity assay utilizes DPPC/Trition X-100 mixed micelles at a ratio of 1:4 as previously described.46,47 A stock solution of lipid was generated by drying down 50 nmoles of dipalmitoyl phosphatidylcholine (DPPC) mixed with 1 × 105 cpm of 1-palmitoyl, 2-[1-14C]-palmitoyl PC per assay tube under a stream of nitrogen gas. The dried lipids were solubilized in 50 μL of 10X assay buffer (100 mM HEPES pH 7.5, 50 mM EDTA, 20 mM DTT, 10 mM ATP, 4 mM Triton X-100) per assay tube by repeated vortexing and heating to 40 °C. The resulting 10X substrate mixture was combined with 100 mM HEPES pH 7.5 to give a final volume of 500 μL upon addition of enzyme and inhibitor. Inhibitors were dissolved in DMSO to a stock concentration of 5 mM and diluted with DMSO prior to addition of 5 μL to the reaction tube, yielding a final DMSO concentration of 1%. The final substrate concentration in this mixed-micelle assay is 100 μM DPPC and 400 μM Triton X-100. Purified enzyme (190 ng) was added to start the reaction followed by incubation for 30 minutes at 40 °C. The reaction was quenched, extracted and analyzed using the modified Dole assay.48
Group IVA cPLA2 Activity Assays
The GIVA cPLA2 assays have been described previously.28,29,46 Pure, native, human GIVA cPLA2 was a generous gift from Dr. Ruth Kramer of Lilly Research Laboratories. Briefly, the final assay conditions were 10 ng GIVA cPLA2 in 100 mM HEPES (pH 7.5), 80 μM CaCl2, 0.1 mg/mL fatty acid free bovine serum albumin, 2 mM DTT, 97 μM 1-palmitoyl-2-[14C]-arachidonoyl phosphatidylcholine (100,000 cpm), 3 μM phosphatidylinositol 4,5-bisphosphate, and 400 μM Triton X-100 in 500 μL. The reaction contained 1% DMSO with varying amounts of inhibitors added as described above. The assays were incubated at 40 °C for 30 min. Reactions were quenched, extracted and analyzed using the modified Dole assay as above.48
Cell Culture and PGE2 Assay
The RAW 264.7 macrophage-like cell line was maintained at 37 °C in a humidified 5% CO2 atmosphere. Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal calf serum (HyClone Labs, Provo Utah), 100 units/mL penicillin and 100 μg/mL streptomycin (Invitrogen, Carlsbad California). Prior to stimulation, cells were plated at a density of 5 × 105 cells per well in standard 12 well tissue culture plates and were allowed to adhere for 24 hours. They were then washed with serum-free medium and allowed to adjust for 18 hours. Cells were then exposed to 100 ng/mL LPS (Sigma L4130 from E. coli 0111:B4) for 24 hours. Following stimulation, the media was removed and the cells were scraped into 1 mL PBS and counted. Deuterated PGE2 internal standard (10 ng) was added to the media of each sample, and the media was cleared of cellular debris by centrifugation (3000 × g, 10 min). Methanol and acetic acid were added to the cleared supernate to a final concentration of 10% and 2% respectively. Prostaglandins were extracted using 60 mg/3 mL Strata-X columns (Phenomenex). The columns were preconditioned with 2 mL methanol followed by 2 mL water. The sample was loaded and the column washed with 2 mL 0.5% methanol. The sample was eluted from the column with 1 mL 100% methanol.
Inhibitors, when included, were dissolved in DMSO and diluted into serum-free medium prior to addition to cells. The DMSO concentration was kept below 0.5% v/v in all studies. All inhibitors were added 30 minutes prior to stimulation.
PGE2 released by the cells was quantitated by the following LCMS procedure. The chromatography was performed on a Grace-Vydac reverse phase C18 column (2.1 mm X 250 mm) run with a gradient beginning with 100% Buffer A (63:37:0.02 water:acetonitrile:formic acid) and ending with 100% Buffer B (50:50 acetonitrile:isopropanol). PGE2 was detected on an ABI 4000 Qtrap mass spectrometer in MRM mode with the electrospray ion source operating in negative ion mode using the following settings: curtain gas = 10, spray voltage = −4.5kV, source temperature = 525 °C, source gas 1 = 60, source gas 2 = 60, declustering potential = −50V. The PGE2 was detected via CID with a precursor ion of 351 and a product ion of 189 amu and a collision energy = −27V and Q2 collision gas = high.
Supplementary Material
Elemental analysis data. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

Dose-response curves for 2-oxoamide inhibitors of GVIA iPLA2. The activity of human GVIA iPLA2 was tested on mixed-micelles containing 100 μM DPPC and 400 μM Triton X-100. The surface concentration of 18 (●), 4c (○), and 11 (▼) was increased as shown. A logarithmic or linear fit function was used to calculate the XI(50) values shown in Table 1.
Acknowledgments
We thank Dr. Brian Kennedy at Merck-Frost for a generous gift of the cDNA coding for human Group VIA-2 iPLA2 and Dr. Ruth Kramer at Lilly Research Laboratories for a generous gift of pure native human GIVA cPLA2. We would like to thank Dr. Karin Lucas for her input and valuable discussions while preparing this manuscript. This work was supported by NIH Grants GM 20501 and GM 064611 (E.A.D.) and by AnalgesiX/UC Biotechnology Grant # B1002-10303. The Regents of the University of California, G.K., and E.A.D. hold equity in AnalgesiX.
References
- 1.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem. 1998;273:207–214. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 2.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci US A. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 4.Balsinde J, Dennis EA. Function and inhibition of intracellular calcium-independent phospholipase A2. J Biol Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- 5.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 6.Birbes H, Drevet S, Pageaux JF, Lagarde M, Laugier C. Involvement of calcium-independent phospholipase A2 in uterine stromal cell phospholipid remodelling. Eur J Biochem. 2000;267:7118–7127. doi: 10.1046/j.1432-1327.2000.01814.x. [DOI] [PubMed] [Google Scholar]
- 7.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A(2) Lipids. 2001;36:689–700. doi: 10.1007/s11745-001-0774-9. [DOI] [PubMed] [Google Scholar]
- 8.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, Turk J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2β) indicate a signaling rather than a housekeeping role for iPLA2β. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 9.Guo Z, Su W, Ma Z, Smith GM, Gong MC. Ca2+-independent phospholipase A2 is required for agonist-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2003;278:1856–1863. doi: 10.1074/jbc.M211075200. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2)β, and not iPLA2γ, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J Biol Chem. 2002;277:32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 11.Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum Ca2+-independent phospholipase A(2) in oxidant-induced renal cell death. Am J Physiol Renal Physiol. 2002;283:F492–F498. doi: 10.1152/ajprenal.00022.2002. [DOI] [PubMed] [Google Scholar]
- 12.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka H, Minakami R, Kanaya H, Sumimoto H. Catalytic residues of group VIB calcium-independent phospholipase A2 (iPLA2γ) Biochem Biophys Res Commun. 2004;320:1284–1290. doi: 10.1016/j.bbrc.2004.05.225. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds LJ, Hughes LL, Louis AI, Kramer RM, Dennis EA. Metal ion and salt effects on the phospholipase A2, lysophospholipase, and transacylase activities of human cytosolic phospholipase A2. Biochim Biophys Acta. 1993;1167:272–280. doi: 10.1016/0005-2760(93)90229-3. [DOI] [PubMed] [Google Scholar]
- 15.Sharp JD, Pickard RT, Chiou XG, Manetta JV, Kovacevic S, Miller JR, Varshavsky AD, Roberts EF, Strifler BA, Brems DN, Kramer RM. Serine 228 is essential for catalytic activities of 85-kDa cytosolic phospholipase A2. J Biol Chem. 1994;269:23250–23254. [PubMed] [Google Scholar]
- 16.Huang Z, Payette P, Abdullah K, Cromlish WA, Kennedy BP. Functional identification of the active-site nucleophile of the human 85-kDa cytosolic phospholipase A2. Biochem. 1996;35:3712–3721. doi: 10.1021/bi952541k. [DOI] [PubMed] [Google Scholar]
- 17.Pickard RT, Chiou XG, Strifler BA, DeFelippis MR, Hyslop PA, Tebbe AL, Yee YK, Reynolds LJ, Dennis EA, Kramer RM, Sharp JD. Identification of essential residues for the catalytic function of 85-kDa cytosolic phospholipase A2. J Biol Chem. 1996;271:19225–19231. doi: 10.1074/jbc.271.32.19225. [DOI] [PubMed] [Google Scholar]
- 18.Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999;97:349–360. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- 19.Dessen A. Structure and mechanism of human cytosolic phospholipase A2. Biochim Biophys Acta. 2000;1488:40–47. doi: 10.1016/s1388-1981(00)00108-6. [DOI] [PubMed] [Google Scholar]
- 20.Rydel TJ, Williams JM, Krieger E, Moshiri F, Stallings WC, Brown SM, Pershings JC, Purcell JP, Alibhai MF. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry. 2003;42:6696–6708. doi: 10.1021/bi027156r. [DOI] [PubMed] [Google Scholar]
- 21.Phillips RM, Six DA, Dennis EA, Ghosh P. In vivo phospholipase activity of the Pseudomonas aeruginosa cytotoxin ExoU and protection of mammalian cells with phospholipase A2 inhibitors. J Biol Chem. 2003;278:41326–41332. doi: 10.1074/jbc.M302472200. [DOI] [PubMed] [Google Scholar]
- 22.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 23.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, Tremblay NM, Huang Z, Weech PK, Gelb MH. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 24.Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca2+-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim Biophys Acta. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- 25.Ghomashchi F, Loo R, Balsinde J, Bartoli F, Apitz-Castro R, Clark JD, Dennis EA, Gelb MH. Trifluoromethyl ketones and methyl fluorophosphonates as inhibitors of group IV and VI phospholipases A2: structure-function studies with vesicle, micelle, and membrane assays. Biochim Biophys Acta. 1999;1420:45–56. doi: 10.1016/s0005-2736(99)00056-5. [DOI] [PubMed] [Google Scholar]
- 26.Balsinde J, Balboa MA, Insel PA, Dennis EA. Regulation and inhibition of phospholipase A(2) Annu Rev Pharmacol Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- 27.Balsinde J, Dennis EA. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D(1) macrophages. J Biol Chem. 1996;271:31937–31941. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- 28.Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. Novel 2-oxoamide inhibitors of human Group IVA phospholipase A2. J Med Chem. 2002;45:2891–2893. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]
- 29.Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. Inhibition of Group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells and in vivo. J Med Chem. 2004;47:3615–3628. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 30.Kokotos G. Inhibition of digestive lipases by 2-oxo amide triacylglycerol analogues. J Mol Catal B-Enzym. 2003;22:255–269. [Google Scholar]
- 31.Chiou A, Verger R, Kokotos G. Synthetic routes and lipase-inhibiting activity of long chain α-keto amides. Lipids. 2001;36:535–542. doi: 10.1007/s11745-001-0754-0. [DOI] [PubMed] [Google Scholar]
- 32.Chiou A, Markidis T, Constantinou-Kokotou V, Verger R, Kokotos G. Synthesis and study of a lipophilic α-keto amide inhibitor of pancreatic lipase. Org Lett. 2000;2:347–350. doi: 10.1021/ol991295s. [DOI] [PubMed] [Google Scholar]
- 33.Kotsovolou S, Chiou A, Verger R, Kokotos G. Bis-2-oxo amide triacylglycerol analogues: a novel class of potent human gastric lipase inhibitors. J Org Chem. 2001;66:962–967. doi: 10.1021/jo005705y. [DOI] [PubMed] [Google Scholar]
- 34.Kokotos G, Verger R, Chiou A. Synthesis of 2-oxo amide triacylglycerol analogues and study of their inhibition effect on pancreatic and gastric lipases. Chemistry - A European Journal. 2000;6:4211–4217. doi: 10.1002/1521-3765(20001117)6:22<4211::aid-chem4211>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 35.Kotsovolou S, Verger R, Kokotos G. Synthesis of lipophilic aldehydes and study of their inhibition effect on human digestive lipases. Org Lett. 2002;4:2625–2628. doi: 10.1021/ol026039l. [DOI] [PubMed] [Google Scholar]
- 36.Kokotos G, Kotsovolou S, Verger R. Novel trifluoromethyl ketones as potent gastric lipase inhibitors. ChemBioChem. 2003;4:90–95. doi: 10.1002/cbic.200390019. [DOI] [PubMed] [Google Scholar]
- 37.Dess DB, Martin JC. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J Am Chem Soc. 1991;113:7277–7287. [Google Scholar]
- 38.Deems RA. Interfacial enzyme kinetics at the phospholipid/water interface: practical considerations. Anal Biochem. 2000;287:1–16. doi: 10.1006/abio.2000.4766. [DOI] [PubMed] [Google Scholar]
- 39.Homan R, Hamelehle KL. Influence of membrane partitioning on inhibitors of membrane-bound enzymes. J Pharm Sci. 2001;90:1859–1867. doi: 10.1002/jps.1135. [DOI] [PubMed] [Google Scholar]
- 40.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M. Pyrrolidine inhibitors of human cytosolic phospholipase A(2) J Med Chem. 2000;43:1041–1044. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 41.Ono T, Yamada K, Chikazawa Y, Ueno M, Nakamoto S, Okuno T, Seno K. Characterization of a novel inhibitor of cytosolic phospholipase A2α, pyrrophenone. Biochem J. 2002;363:727–735. doi: 10.1042/0264-6021:3630727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghomashchi F, Stewart A, Hefner Y, Ramanadham S, Turk J, Leslie CC, Gelb MH. A pyrrolidine-based specific inhibitor of cytosolic phospholipase A(2)α blocks arachidonic acid release in a variety of mammalian cells. Biochim Biophys Acta. 2001;1513:160–166. doi: 10.1016/s0005-2736(01)00349-2. [DOI] [PubMed] [Google Scholar]
- 43.Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Ableson leukemia virus. Cell. 1978;15:261–267. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- 44.Shinohara H, Balboa MA, Johnson CA, Balsinde J, Dennis EA. Regulation of delayed prostaglandin production in activated P388D1 macrophages by group IV cytosolic and group V secretory phospholipase A2s. J Biol Chem. 1999;274:12263–12268. doi: 10.1074/jbc.274.18.12263. [DOI] [PubMed] [Google Scholar]
- 45.Gijon MA, Leslie CC. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J Leukoc Biol. 1999;65:330–336. doi: 10.1002/jlb.65.3.330. [DOI] [PubMed] [Google Scholar]
- 46.Yang HC, Mosior M, Johnson CA, Chen Y, Dennis EA. Group-specific assays that distinguish between the four major types of mammalian phospholipase A2. Anal Biochem. 1999;269:278–288. doi: 10.1006/abio.1999.4053. [DOI] [PubMed] [Google Scholar]
- 47.Lucas KK, Svensson CI, Hua XY, Yaksh TL, Dennis EA. Spinal phospholipase A2 in inflammatory hyperalgesia: role of Group IVA cPLA2. Br J Pharmacol. 2005;144:940–952. doi: 10.1038/sj.bjp.0706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reynolds LJ, Washburn WN, Deems RA, Dennis EA. Assay strategies and methods for phospholipases. Methods in Enzymology. 1991;197:3–23. doi: 10.1016/0076-6879(91)97129-m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data. This material is available free of charge via the Internet at http://pubs.acs.org.