

Abstract
Members of the syntaxin protein family participate in the docking–fusion step of several intracellular vesicular transport events. Tlg1p has been identified as a nonessential protein required for efficient endocytosis as well as the maintenance of normal levels of trans-Golgi network proteins. In this study we independently describe Tlg1p as an essential protein required for cell viability. Depletion of Tlg1p in vivo causes a defect in the transport of the vacuolar protein carboxypeptidase Y through the early Golgi. Temperature-sensitive (ts) mutants of Tlg1p also accumulate the endoplasmic reticulum/cis-Golgi form of carboxypeptidase Y at the nonpermissive temperature (38°C) and exhibit underglycosylation of secreted invertase. Overexpression of Tlg1p complements the growth defect of vti1-11 at the nonpermissive temperature, whereas incomplete complementation was observed with vti1-1, further suggesting a role for Tlg1p in the Golgi apparatus. Overexpression of Sed5p decreases the viability of tlg1 ts mutants compared with wild-type cells, suggesting that tlg1 ts mutants are more susceptible to elevated levels of Sed5p. Tlg1p is able to bind His6-tagged Sec17p (yeast α-SNAP) in a dose-dependent manner and enters into a SNARE complex with Vti1p, Tlg2p, and Vps45p. Morphological analyses by electron microscopy reveal that cells depleted of Tlg1p or tlg1 ts mutants incubated at the restrictive temperature accumulate 40- to 50-nm vesicles and experience fragmentation of the vacuole.
INTRODUCTION
Proteins destined for the secretory pathway are initially targeted to the endoplasmic reticulum (ER) and transported through the Golgi and then to their subsequent destinations. This transport process is mediated through a series of membrane-bound vesicles that bud from a donor membrane and fuse with a target membrane (Rothman and Wieland, 1996; Schekman and Orci, 1996; Bock and Scheller, 1997; Pelham, 1997; Rowe et al., 1998).
Combined biochemical and genetic studies in yeast as well as in mammalian cells have identified a significant number of components of the molecular machinery that mediates transport. In mammalian cells, cytosolic transport factors such as the N-ethylmaleimide-sensitive factor (NSF) and its three soluble NSF attachment proteins (α-, β-, γ-SNAPs) have been isolated through in vitro intra-Golgi transport assays (Clary and Rothman, 1990; Whiteheart et al., 1993). In Saccharomyces cerevisiae, Sec17p (the functional homologue of α-SNAP), and Sec18p (NSF homologue) function at several transport steps of the secretory and endocytotic pathways (Graham and Emr, 1991; Whiteheart and Kubalek, 1995; Nichols et al., 1997). Transport vesicles contain integral membrane proteins called SNAREs (SNAP receptors) that contribute to the specificity of docking and participate in the subsequent fusion with the acceptor membranes (for review see Kee et al., 1995; Rothman and Sollner, 1997; Schimmoller et al., 1997).
The SNARE complex was first characterized in neurons when an integral membrane protein, syntaxin 1, in association with a synaptosomal protein of 25 kDa (SNAP-25) and vesicle-associated membrane protein (VAMP) were coimmunoprecipitated as a protein complex with α-SNAP and NSF (Sollner et al., 1993; Pevsner and Scheller, 1994; Bock and Scheller, 1997). Similar complexes have been examined extensively in yeast and mammalian cells. Some well-characterized SNARE complexes in yeast include those involved in ER to Golgi and Golgi to cell surface transport events (Sogaard et al., 1994) (for review, see Rothman, 1994) as well as the new SNARE complex found to be involved in the retrograde transport from the Golgi back to the ER (Lewis and Pelham, 1996; Lewis et al., 1997). The syntaxin-like Sed5p, located on the cis-Golgi, interacts with the v-SNAREs Bos1p, Sec22p, and Bet1p on ER-derived vesicles. These complexes were isolated using Sec18p ts mutant cells at 37°C by antibodies against Sed5p (Sogaard et al., 1994). Recent evidence has indicated that Sec17p and Sec18p are required only for the symmetric priming of both vesicle-derived (v)- and target-derived (t)-SNAREs and thus are a prerequisite for docking in homotypic membrane fusion of vacuoles (Mayer et al., 1996; Mayer and Wickner, 1997; Nichols et al., 1997). Nonetheless, sec18-1 cells grown at the restrictive temperature display a condition in which the concentration of SNARE complexes within the cell increases (Sogaard et al., 1994; Sapperstein et al., 1996; Lupashin and Waters, 1997; Holthuis et al., 1998a). The ER to Golgi SNARE complex has also been isolated from mammalian cells, and similar sets of interacting proteins have been identified (Nagahama et al., 1996; Subramaniam et al., 1996; Hay et al., 1997). The SNARE complex involved in the Golgi to cell surface transport event contains several proteins, including the syntaxin-like Sso1p and Sso2p, VAMP-like Snc1p and Snc2p, and the SNAP-25 like Sec9p (Protopopov et al., 1993; Brennwald et al., 1994). The SNARE complex involved in retrograde Golgi to ER transport includes Ufe1p, Sec22p, Sec20p, and Tip20 (Lewis et al., 1997). Aberrations affecting retrograde transport to the ER lead to the rapid impairment of anterograde transport, making it difficult to distinguish by phenotypic analyses whether a protein is involved in forward or retrograde transport. This tight association of anterograde and retrograde transport suggests that factors needed for forward transport have to be recycled (Lewis and Pelham, 1996; Hay et al., 1997). Recent experiments by Lupashin and Waters (1997) elucidated a model for the regulation of v/t-SNARE complex assembly by Ypt1p and Sly1p that act as throttles and dampers respectively of the fusion event at this particular step (Rothman and Sollner, 1997). Thus far, little is known about the molecular mechanism involved in transport within the Golgi complex, and it is not known whether similar SNARE complexes are involved (Schekman and Mellman, 1997).
During the preparation of this report, Holthuis et al. (1998a) identified Tlg1p as an endosomal protein and Tlg2p as a trans-Golgi network (TGN) protein and examined some of their functions. In their study, Tlg1p and Tlg2p were not essential for growth and were postulated to be involved in endocytosis and maintenance of the normal levels of TGN proteins (Holthuis et al., 1998a). Tlg2p has also been described by others as a t-SNARE involved in early endosome biogenesis and endocytosis (Abeliovich et al., 1998; Seron et al., 1998). Our present study shows that Tlg1p is required for normal growth in three different yeast strains, whereas deletion of VAM3, TLG2, and VTI1 in two of the strains tested gave results similar to the published data (Fischer von Mollard et al., 1997; Lupashin et al., 1997; Holthuis et al., 1998a). Transport of carboxypeptidase Y (CPY) between the ER and the Golgi is blocked in Δtlg1 cells as well as in tlg1 temperature-sensitive (ts) mutants at the restrictive temperature. The secreted form of invertase is underglycosylated in these cells with some intracellular accumulation of the core-glycosylated invertase. Large-scale immunoprecipitation of Tlg1p identified several associated proteins, including Sec17p, Vti1p, Tlg2p, and Vps45p. Immunoprecipitation of extracts of cells that favor the v/t-SNARE complex assembly (sec18-1 at 37°C) with either anti-Vti1p or Tlg2p antibodies reciprocally coprecipitates both proteins as well as Tlg1p and Sec17p. These data indicate that these proteins are part of the same SNARE complex and are possibly regulated by the Sec1p family member Vps45p.
MATERIALS AND METHODS
Strains and Media
DH5α and GM119 bacterial strains were used to propagate all plasmids. Yeast strains were grown in either YEPD (2% bactopeptone, 1% yeast extract, and 2% glucose) or standard minimal glucose medium with the appropriate supplements. To induce the expression from the GAL1 promoter, glucose was replaced by 2% raffinose and 1–2% galactose. The strains and genotypes are described in Table 1.
Table 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| Yeast | ||
| RSY255 | MATα ura3-52, leu2-3, 112 | R. Schekman |
| W303-D | MATa/α leu2-3,-112 ade2-1 ura3-1 his3-11,-15 trp1-1 can1-100 | U. Surana |
| JC20 | MATa Δtlg1::LEU2, GAL1-TLG1 ade2-1 ura3-1 his3-11,-15 leu2-3,-112 trp1-1 can1-100 | This study |
| JC21 | MATa Δtlg1::LEU2, GAL1-c-mycTLG1 ade2-1 ura3-1 his3-11,-15 leu-2-3,-112 trp1-1 can1-1-100 | This study |
| JC22 | same as W303-D with Δtlg1::LEU2/TLG1 | This study |
| ANY115 | MATa bet1-1 his4-619 ura3-52 | S. Ferro-Novick |
| SEY6210-sed5-1 | MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 sed5-1 | H. Pelham |
| RSY324 | MATa sec22-1 ura3-52 | S. Ferro-Novick |
| SEY6210 | MATa ura3-53 his3-Δ200 leu2-3,-112 trp1-Δ901 suc2-Δ9 lys2-801 | S. Emr |
| SEY6211 | MATα ura3-52 his2-Δ200 leu2-3,-112 trp1-Δ901 ade2-101 suc2-Δ9 lys2-801 | S. Emr |
| Y97 | RSY255 x SEY 6210 | This study |
| Y98 | MATa/α ade2-1, ade3Δ his3-11,-15 leu2-3,-112 trp1-Δ1 ura3-1 can1-100 | M. Cai |
| Y100 | SEY6210 X SEY6211 | This laboratory |
| FvMY7 | MATa ura3-52 his3-Δ200 leu2-3,-112 trp1-Δ901 suc2-Δ9 lys2-801 vti1-1 | T. Stevens |
| FvMY21 | MATa ura3-52 his3-Δ200 leu2-3,-112 trp1-Δ901 suc2-Δ9 lys2-801 vti1-11 | T. Stevens |
| RSY271 | MATa sec18-1 | R. Schekman |
| E. coli | ||
| GM119 | F′ supE44 lacY1 galK2 galT22 metB1 dam-3 dcm-6 tsx-78 λ- | ATCC |
| DH5α | F′ endA1 hsdR17 supE44 thil λ-recA gyrA96 relA1 (φ80 dlacZΔM15) | Hanahan |
Plasmids and Strains
Pfu polymerase (Stratagene, La Jolla, CA) was used to amplify the 2.1-kb yeast genomic sequence containing TLG1 from a genomic DNA of W303-D yeast strain using primers derived from database sequence information (primer 1, 5′-CACGTAACGGATCCTCTGGTTGTTGTAC-3′; primer 2, 5′-TTGTCAAGCTTTAACGCTGGTGAAACTCC-3′, incorporating BamHI and HindIII sites, respectively) and cloned into pUC18 (New England Biolabs, Beverly, MA) to generate pUCTLG1. A 537-bp fragment of the TLG1 DNA coding region between StyI and AccI was removed from pUCTLG1 and replaced with 1.6 kb of LEU2 sequence. To delete the TLG1 in the genomic locus, Δtlg1::LEU2 construct was amplified and linearized with HindIII and SmaI, and the digested DNA was transformed into the diploid yeast strain W303-D and later into Y97, Y98, and Y100 yeast diploid strains. Leu+ transformants were checked by Southern blotting as described by Maniatis et al. (1989), and correct integrants were later sporulated and dissected. Of 30 tetrads dissected, only Leu− cells grew and segregated in a 2:0 pattern in W303-D, Y97, and Y98 diploids but not in Y100 diploid.
The c-myc-tagged version of Tlg1p was generated by PCR using primer 2 (5′-AAAGAATTCATGGAACAAAAACTAATTTCTGAA-GAAGATCTAAACAACAGTGAAGATCCGTTTCAAC-3′) and primer 4 (5′-ATCAAGTCGACAGATCTAAGTCCTATACAACAATCGTCGT-A3′); wild-type TLG1 was amplified using primer 5 (5′-TTCTTGGATCCATGAACAACAGTGAAGATCCGTTTCAAC-3′) and primer 4. A TLG1 form lacking the transmembrane anchor was amplified by PCR using primers 4 and 6 (5′-GTGCAGTCGACAGATCTAGCAATGAATGCCAAAACTAATAAAAC-3′). Pfu polymerase was used to amplify TLG1, myc-TLG1, and TLG1 lacking the transmembrane anchor and the DNA fragments were cloned into pYES2 (Invitrogen, San Diego, CA) to generate pYmTLG1, pYTLG1, and pYTLG1M, respectively.
The DNA insert of pUCTLG1 was excised and cloned into YCplac22 (CEN4,TRP1) (Gietz and Sugino, 1988) and pRS426 (2μ, URA3) or pRS424 (2μ, TRP1) (Sikorski and Hieter, 1989). A modified PCR-based procedure (Lupashin et al., 1997) was used to construct ts tlg1. The DNA fragment containing TLG1 was amplified in four separate reaction tubes with Taq polymerase (Amersham, Buckinghamshire, UK) using skewed dNTP concentrations such that one of the dNTPs was at 10% of the normal concentration using primers 1 and 4. The resulting mutagenized TLG1 PCR products were pooled, ligated to YCplac22 at BamHI and SalI sites, and transformed into Escherichia coli DH5α. The minilibrary was transformed into JC20 (Δtlg1::LEU2, GAL1-TLG1) cells, which were kept viable by the presence of GAL1::TLG1 plasmid. Approximately 10,000 transformants were selected on glucose base–selective plates and were incubated at 23°C. Once grown, transformants were replica plated on selective plates with one set incubated at 38°C and the other maintained at 23°C. Temperature-conditional mutants that grew at 23°C but not at 38°C were isolated.
A 1.7-kb DNA fragment spanning the VTI1 coding frame was amplified using Pfu polymerase and cloned into pUC18. The coding DNA region of VTI1 between BclI and BglII was removed and replaced with either LEU2 or HIS3 to create Δvti1. A 2-kb DNA fragment containing TLG2 was amplified using Pfu polymerase and cloned into pUC18 and pRS424 to generate pUCTLG2 and pRTLG2, respectively. The DNA coding region of TLG2 between BclI and BglII was replaced with LEU2 to create a Δtlg2 plasmid. These constructs were linearized at the multiple cloning region and transformed into W303-D and Y100 as described previously (Ito et al., 1983).
The SEC17 of RSB-775 (gift of Dr. R. Schekman, University of California, Berkeley, CA) was cleaved with BspHI and blunted with T4 DNA polymerase as described in the manufacturer’s instructions. The fragment was later cut with HindIII, and the truncated SEC17 DNA fragment was cloned into pQE30 (Qiagen, Hilden, Germany) at the SmaI and HindIII sites. This construct removed 21 amino acids at the N terminus of Sec17p. Recombinant His6-tagged Sec17p was expressed and purified using nickel-nitrilotriacetic acid agarose as described in the manufacturer’s instructions. Purified GST, GST-retinoblastoma protein, and His6-tagged Sec17p proteins (5 μg) bound to beads were mixed with 100 μg of total yeast extract from cells overexpressing Tlg1p and incubated overnight at 4°C as described by Sogaard et al. (1994). The beads were washed with 20–30 ml of buffer E (20 mM HEPES-KOH, pH 7.3, 100 mM KCl, 1 mM DTT, 2 mM EDTA, pH 8.0, 0.5 mM ATP, pH 7.0, 1 mM PMSF, and 0.5% Triton X-100), mixed with an equal volume of 2× gel loading buffer, and then analyzed by SDS-PAGE. The proteins were transferred on a nitrocellulose membrane and probed for Tlg1p.
Antibodies
Sec17p (residues 203–282), Tlg1p (residues 1–208), Vti1p (residues 1–105), and Tlg2p (residues 100–276) were fused in-frame to the appropriate GST vector (Pharmacia, Piscataway, NJ), expressed in E. coli, purified, and used to immunize rabbits (Harlow and Lane, 1988). The resulting serum was precleaned with a GST column, and specific antibodies were affinity purified on specific columns. Western analyses were performed as described by Maniatis et al. (1989). Antibodies specific for α-1,6- and α-1,3-mannose linkages were the generous gifts of Dr. T. Aust (University of Basel, Basel, Switzerland) and Dr. R. Schekman.
Immunofluorescence
W303 cells were either transformed with SM3M-414-expressing triple myc-tagged Mnt1p (Jungmann and Munro, 1998) or a myc-tagged Tlg1p (JC21). Transformants were fixed and mounted on slides as described previously by Kilmartin and Adams (1984). Appropriate affinity-purified antibodies and 9E10 were incubated on the fixed cells for 2 h at 30°C. FITC-conjugated secondary anti-mouse antibodies and Cy3-conjugated secondary anti-rabbit antibodies (Amersham) were used for visualization. Dual images were taken with an MRC-600 confocal laser scanning microscope (Bio-Rad, Hercules, CA) using appropriate wavelengths to avoid bleed-through.
Metabolic Labeling and Immunoprecipitation of CPY and Invertase
CPY immunoprecipitation was performed as described (Gaynor et al., 1994) with slight modifications. After the addition of Tween 20-immunoprecipitation (IP)/BSA buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 150 mM NaCl, 0.5% Tween 20, and 10 mg/ml BSA) to the lysed cells, 50 μl of precleaned agarose (Sigma, St. Louis, MO) were added and mixed for 30 min at 4°C. After a brief centrifugation, the supernatant was incubated overnight at 4°C with affinity-purified anti-CPY antibodies (6 μg) with gentle shaking. Protein A-Sepharose CL-4B (7 mg) was later added to the suspension and incubated for a further 2 h at 4°C. The beads were collected by centrifugation, washed twice in Tween 20-urea buffer (100 mM Tris-HCl, pH 7.5, 200 mM NaCl, 2 M urea, and 0.5% Tween 20), once in Tween 20-IP buffer, and once in Tris-buffered saline (50 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, and 150 mM NaCl). Gel loading buffer (50 μl of 2×) was added, the sample was heated at 100°C for 3 min, and the supernatant was loaded onto a 7.5% SDS-PAGE gel.
The invertase transport assay was performed as described (Banfield et al., 1995) with modifications. The wild-type and the indicated mutants were transformed with a CEN-based plasmid expressing the secreted form of invertase tagged with a myc epitope under the control of the PHO5 promoter (a gift from S. Munro, Medical Research Council, Cambridge, United Kingdom). They were grown at 23°C to an OD600 of 0.4–0.5 or shifted to 38°C for 1 h. After centrifugation, cells were spheroplasted with the addition of a 0.3 mg/ml Zymolyase 100T-lyticase mix in a final sorbitol concentration of 1 M. The spheroplasts were further incubated at either 23 or 38°C for 1 h before being isolated by centrifugation at 3000 × g for 5 min and lysed in 1 ml of synthetic complete media. Proteins from the lysed spheroplasts (I) and the spheroplasting media (E) were precipitated with trichloroacetic acid (TCA) to a final concentration of 10%, left on ice for 20 min, and centrifuged. After washing with ice-cold acetone, the pellets were resuspended in sample buffer, loaded onto 0.7% SDS-PAGE, and probed with anti-c-myc monoclonal antibodies. Primary antibodies were detected with an HRP-conjugated anti-mouse immunoglobulin G secondary antibody followed by Pierce (Rockford, IL) Super Signal chemiluminescence kit. To detect outer chain mannosyl residues, the c-myc-tagged invertase was first immunoprecipitated with c-myc monoclonal antibodies bound to protein A+G-agarose (Calbiochem, La Jolla, CA). The immunoprecipitates were heated at 90°C for 15 min to inactivate the antibodies and centrifuged to remove the agarose. The supernatants were diluted 10-fold with Tween 20-IP/BSA buffer and reimmunoprecipitated with anti-α-1,6-mannose linkage-specific or anti-α-1,3-mannose linkage-specific antisera as described by Franzusoff and Schekman (1989).
Cell Fractionation, Membrane Extractions, and Immunoblotting
The procedure as outlined by Whitters et al., (1994) was followed with some modifications. RSY255 cells were grown to an OD600 nm of 0.5 in YEPD medium at 30°C. The cells were washed with PBS containing 10 mM NaN3 twice and converted to spheroplasts by resuspending in the spheroplast buffer (50 mM KPO4, pH 7.5, 1.4 M sorbitol, 50 mM β-mercaptoethanol, 10 mM NaN3, and 2.5 μg/OD600 lyticase). Spheroplast conversion was observed under a light microscope by adding a drop of culture and a drop of 0.2% SDS in water. Spheroplasts were pelleted by a 5-min spin at 500 × g and resuspended in 1.5 ml of lysis buffer (10 mM HEPES, pH 7.3, 0.3 M sorbitol, 0.1 mM EGTA, 1 mM PMSF, 1 μg/ml protease inhibitor mixture, and 100 μg/ml BSA). An aliquot (500 μl) of the lysate was reserved as the whole-cell extract. The remaining cell lysate was first centrifuged for 3 min at 500 × g to remove intact cells and other large debris, yielding the low-speed pellet. The resulting supernatant was spun at 13,000 × g for 20 min at 4°C to generate the S13 (supernatant) and P13 (pellet) fractions. The S13 fraction was centrifuged for 1 h at 100,000 × g in a TL100 ultracentrifuge (Beckman Instruments, Palo Alto, CA) to generate the high-speed supernatant (S100) and the high-speed pellet (P100) fractions. An aliquot of proteins (50 μg) was separated by SDS-PAGE and analyzed by Western blotting using appropriate antibodies. Membrane extracts were prepared by glass bead lysis as described by Ossig et al. (1991).
Subcellular Fractionations
Spheroplasts were prepared from RSY255 cells that express the c-myc-tagged Mnt1p (SM3M-414) as described (Becherer et al., 1996; Jungmann and Munro, 1998). The S13 fraction (resuspended in ∼2 ml of lysis buffer) was loaded on top of the gradient and centrifuged in a Beckman SW41 rotor at 170,000 × g for 18 h at 4°C as outlined by Becherer et al. (1996). Fifteen fractions were collected starting from the top of the tube, and the proteins were precipitated by the addition of TCA to 10%. The fractions were analyzed by immunoblot with various antibodies.
Electron Microscopy
This was performed as described by Numata et al. (1993). After being fixed in 3% (wt/vol) glutaraldehyde for 1 h at room temperature, cells were collected by centrifugation and washed twice in water. The cells were postfixed in 2% fresh potassium permanganate solution for 2 h at room temperature, washed twice in water, and dehydrated using a graded series of ethanol (25% ethanol for 5 min, 50% ethanol for 10 min, 75% for 10 min, 95% for 10 min, and 100% for 10 min) and twice with 100% acetone. Cells were embedded in low-viscosity Spurr resin for 24 h. Sections were stained with uranyl acetate and lead citrate and observed at 60 kV with a JEOL (Tokyo, Japan) 1200 EX transmission electron microscope.
Immunoprecipitation
The procedure described by Sogaard et al. (1994) was followed with some modifications. Twenty milligrams of yeast spheroplast detergent extract were diluted 10-fold with buffer E (20 mM HEPES-KOH, pH 7.3, 100 mM KCl, 1 mM DTT, 2 mM EDTA, 0.5 mM ATP, pH 7.0, 1 mM PMSF, and 0.5% Triton X-100) and rotated overnight at 4°C with 200 μg of affinity-purified antibodies coupled to cyanogen bromide–activated Sepharose beads. The beads were washed four times with 15 ml of buffer E in a disposable column (Pierce) and eluted with 1.5 ml of 0.1 M glycine, pH 2.6. Postimmunoprecipitation supernatants were precipitated with TCA (added to a final concentration of 10%), centrifuged for 30 min on ice, and washed twice in ice-cold acetone. The pellets were resuspended in sample buffer, and 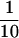 to
to 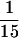 of the samples was examined. Immunodetection of Western blots was performed using affinity-purified antibodies.
of the samples was examined. Immunodetection of Western blots was performed using affinity-purified antibodies.
For large-scale immunoprecipitation of the Tlg1p-complex(es) shown in Figure 9B, 400 μg of affinity-purified anti-Tlg1p antibodies were cross-linked to protein A-Sepharose CL-4B with dimethyl pimelimidate as described by Brew et al. (1975). Yeast detergent (100 mg) extracts from indicated strains were diluted with buffer E, preadsorbed with protein A-Sepharose CL-4B for 2 h at 4°C, and centrifuged at 3000 rpm for 10 min. The supernatant was incubated overnight at 4°C with beads coupled with anti-Tlg1p antibodies. The mixture was then transferred to a disposable column, and the beads were washed with 50 ml of ice-cold buffer E. Proteins were eluted three times each with 200 μl of 0.1 M glycine, pH 2.6, and the combined eluant was neutralized with 20 μl of 1 M Tris-HCl, pH 9.4. The eluate was concentrated in a vacuum dryer and analyzed by 15% SDS-PAGE. The gel was stained in Coomassie blue dye. Individual proteins were excised and digested in situ with either trypsin or lysyl endopeptidase. The resulting peptides were fractionated by HPLC and sequenced by Edman microsequencing techniques at the Institute of Molecular and Cell Biology Microchemistry Facility.
Figure 9.

Characterization of Tlg1p-containing SNARE complex. Spheroplasts of sec18-1 cells were incubated at either 25 or 37°C for 1 h and lysed with Triton X-100, and extracts were prepared as described (see MATERIALS AND METHODS). (A) Twenty milligrams of the extract from the indicated cells were incubated with anti-Tlg1p antibodies (200 μg), which had been covalently coupled to cyanogen bromide–activated Sepharose CL4B (400 μg). The supernatants and eluted immunoprecipitates were TCA precipitated, and 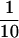 of the samples was analyzed by immunoblot for Tlg1p and Sec17p. Sec17p was not coimmunoprecipitated by anti-Tlg1p antibodies when extracts from wild-type cells were used (lanes 1 and 4). However, Sec17p was coimmunoprecipitated by anti-Tlg1p antibodies when sec18-1 cell extracts were used (lanes 2 and 3), particularly when spheroplasts were preincubated at 37°C (lane 3). (B) Large-scale immunoprecipitation of the Tlg1p–SNARE complex from sec18-1 cells. Proteins immunoprecipitated with anti-GST antibodies (lane 1) or anti-Tlg1p antibodies (lane 2) were resolved on a 12% SDS-PAGE gel and stained with Coomassie brilliant blue. The molecular mass markers are shown in lane 3. The heavy chain of the rabbit antibody is detectable despite the chemical cross-linking. All of the specific protein bands denoted by different letters were individually excised and sequenced for identity (refer to Table 3).
of the samples was analyzed by immunoblot for Tlg1p and Sec17p. Sec17p was not coimmunoprecipitated by anti-Tlg1p antibodies when extracts from wild-type cells were used (lanes 1 and 4). However, Sec17p was coimmunoprecipitated by anti-Tlg1p antibodies when sec18-1 cell extracts were used (lanes 2 and 3), particularly when spheroplasts were preincubated at 37°C (lane 3). (B) Large-scale immunoprecipitation of the Tlg1p–SNARE complex from sec18-1 cells. Proteins immunoprecipitated with anti-GST antibodies (lane 1) or anti-Tlg1p antibodies (lane 2) were resolved on a 12% SDS-PAGE gel and stained with Coomassie brilliant blue. The molecular mass markers are shown in lane 3. The heavy chain of the rabbit antibody is detectable despite the chemical cross-linking. All of the specific protein bands denoted by different letters were individually excised and sequenced for identity (refer to Table 3).
RESULTS
Isolation and Cloning of a Novel SNARE Protein
Members of the syntaxin family play a critical role in the vesicle docking and fusion of diverse transport events (Bock and Scheller, 1997; Schimmoller et al., 1997; Weimbs et al., 1997; Weber et al., 1998). Homology searches of the yeast genome with known syntaxins identified a novel gene (ORF YDR468c) of 676-nucleotide pairs potentially coding for a 225–amino acid protein with an estimated molecular mass of 25.8 kDa. A study on the ORF YDR468c was recently published by Holthuis et al. (1998a), and the protein was named Tlg1p. The coiled-coil domain of Tlg1p shows homology to other syntaxins, namely syntaxin-6 (28% identity and 58% similarity) and the SNAP-25 homologue Vam7p (21% identity and 51% similarity) (Wada and Anraku, 1992; Bock et al., 1996; Klumperman et al., 1998). In mammalian syntaxins, this conserved domain acts as a site for the binding of α-SNAP, VAMP, and SNAP-25 to rat syntaxin 1A (Chapman et al., 1994; Kee et al., 1995).
Tlg1p Is Essential for Cell Viability or Normal Cell Growth in Three Different Yeast Strains
To determine whether the product of TLG1 is functional and required for protein transport, the TLG1 ORF was replaced with a functional 1.6 kb of the LEU2 gene (Figure 1A). The LEU2 is flanked with stop codons in all three reading frames to prevent the formation of a hybrid protein (Berber et al., 1991). This deletion removed TLG1 sequences between 43 bp downstream of the ATG and 90 bp upstream of the original stop codon. The DNA fragment containing the disrupted TLG1 was integrated at the native TLG1 locus of the yeast diploid W303-D. Correct integrants were confirmed by Southern blotting, sporulated, and dissected (our unpublished results). Of the 30 tetrads dissected, only two live leu− colonies per tetrad were obtained (Figure 1B). Examination by a dissection microscope revealed that Δtlg1::LEU2 spores were able to germinate but ceased dividing at the four- to six-cell stage (our unpublished results). These results suggest that Tlg1p is essential for cell growth as depletion of this protein causes rapid growth arrest. However, Holthuis et al. (1998a) have shown that deletion of Tlg1p in SEY6211/SEY6210 and K699 rendered these strains ts but otherwise superficially normal. This inconsistency prompted us to further investigate the true character of cells lacking Tlg1p. Three other diploid leu− yeast strains (Y97, Y98, and Y100; see MATERIALS AND METHODS and Table 1) were generated and transformed with the same disrupting TLG1 DNA construct. Dissection of correct integrants of these different strains revealed two distinct phenotypes (Figure 1C). Δtlg1 cells of the Y100 (SEY6211/SEY6210) background grew at slower rates compared with wild-type cells (Figure 1C, left panel). Δtlg1 cells of Y100 were growth affected when they were incubated at 37°C as described by Holthuis and colleagues (our unpublished results). In Y98 and Y97 cells, a deletion of TLG1 gave results similar to that of the W303-D strain; however, longer incubation of the dissection plates at 30°C for 5–6 d did show some very small Δtlg1 cells (Figure 1C, middle and right panels). Under light microscopy, stationary phase Δtlg1 cells of Y98 and Y97 have large granules when compared with similarly aged wild-type cells (our unpublished results). Upon further streaking, Δtlg1 haploid cells of Y98 and Y97 did propagate but at a much slower rate compared with wild-type cells, suggesting that Tlg1p is required for normal cell propagation. The doubling time of Δtlg1 cells from Y98 and Y97 yeast backgrounds grown in rich glucose media at 23°C were ∼2.5 times longer than that of wild-type cells (doubling time of 90 min in wild-type cells vs. 225 min in Δtlg1 cells in YEPD).
Figure 1.

TLG1 is essential for cell growth. (A) Disruption strategy to create a null mutation of TLG1 in the yeast genome. The arrow represents the direction of transcription. (B) Dissection result of heterozygous W303 yeast diploids containing Δtlg1::LEU2. Correct integrants verified by Southern blotting were incubated on a sporulation plate for 3 d at 30°C. Asci were glusulase treated, and tetrads were segregated on a dissection microscope. Surviving progeny were wild-type for TLG1. (C) Dissection of Y100 (SEY6210 × SEY6211)-, Y98 (MATa/α ade2-1, ade3Δ his3-11,-15 leu2-3,-112 trp1-Δ1 ura3-1 can1-100)-, and Y97 (RSY255 × SEY6210)-based asci in which two of the spores from each tetrad contain Δtlg1::LEU2. The middle and right panels of C were dissection plates incubated at 30°C for 5 d. All other plates were incubated for 3 d at 30°C. Δtlg1 cells grew poorly, as revealed by the tiny colonies, particularly in Y97 and Y98 cells. (D) Reintroduction of an episomal TLG1 under the control of an inducible GAL1 promoter in Δtlg1::LEU2 cells from W303 background restores cell growth in galactose-based media but not in glucose-based media. The growth curves of wild type (open squares), a heterozygous diploid of Δtlg1::LEU2 (open triangles), and Δtlg1::LEU2 cells (closed circles) containing the wild-type TLG1 under the control of the GAL1 promoter (pYTLG1) are shown. Cells were grown in YEP-galactose (D) to induce or in YEP-glucose (E) liquid media to deplete the Tlg1p. Cell growth was measured by optical density at 600 nm at the indicated times.
To confirm that the observed effects were solely due to disruption of the TLG1 locus, the wild-type TLG1, an N-terminal myc-tagged TLG1, as well as a truncated form of TLG1 encoding the cytoplasmic domain alone were amplified by PCR and placed under the control of the GAL1 promoter in the multicopy pYES2 vector. The constructs were transformed into W303Δtlg1, Y97Δtlg1, Y98Δtlg1, and Y100Δtlg1 (diploid yeasts in which one copy of TLG1 was disrupted), and the transformants were grown in galactose to induce the expression of various forms of Tlg1p before being sporulated and dissected. All four spores from each tetrad carrying the full-length Tlg1p or the myc-tagged Tlg1p under the control of GAL1 promoter were viable on galactose plates (our unpublished results), whereas only two of the progeny from each tetrad were able to grow on glucose plates, and these were leu− cells that were wild type for TLG1 (our unpublished results). The Tlg1p cytoplasmic domain was unable to rescue the lethality of Δtlg1::LEU2 in W303 strain cells, suggesting that proper membrane anchorage of Tlg1p is essential for its function. In addition, TLG1 under its own promoter in either the CEN (low-copy number) or 2μ (high-copy number) plasmid was able to suppress the lethality of the Δtlg1 haploid cells of W303 and Y98 strains (Table 2). These data establish that low-copy or multicopy plasmids bearing Tlg1p, with the exception of the transmembrane domain deleted mutant of Tlg1p, were able to complement Δtlg1 cells, and that overexpression of Tlg1p does not affect normal cellular function. The growth kinetics of transformants containing the different constructs suggest that overexpression of Tlg1p does not compromise growth, whereas cells depleted of Tlg1p undergo rapid growth arrest (Figure 1, D and E, respectively).
Table 2.
Overexpression of Tlg1p or its cytoplasmic domain alone does not have any detectable phenotype in wild-type cells, whereas the transmembrane domain of Tlg1p is required for its function.
| Strain | WT | Growth of Δtlg1::LEU2 haploid cells |
|---|---|---|
| Genomic TLG1/CEN | ++++ | ++++ |
| Genomic TLG1/2μ | ++++ | ++++ |
| myc-tagged Tlg1p/2μ | ++++ | +++ |
| Tlg1p/ΔTM/2μ | ++++ | − |
Plasmid constructs were transformed into haploid wild-type W303 as well as diploid W303Δtlg1 and Y98Δtlg1 cells in which one copy of the TLG1 was disrupted. Transformants of W303Δtlg1 and Y98Δtlg1 cells were sporulated and dissected. Spore progeny were grown on selective plates with galactose and raffinose as sole carbon sources to induce the expression of various Tlg1p proteins. +, degree of growth of cells on plates; −, no apparent growth after 3 d at 30°C.
Tlg1p Is Essential for Protein Transport in the Early Secretory Pathway
The phenotype of Δtlg1 cells was severe in three of four yeast strains. We concentrated our further study in the W303 background, because in this background a deletion of Tlg1p caused lethality. To determine the stage of the secretory pathway in which Tlg1p is required, both wild-type and Δtlg1::LEU2 cells (JC20) of W303 progeny were initially grown at 30°C in medium containing 1% galactose and 2% raffinose and then resuspended at 30°C in glucose medium to repress the Tlg1p protein expression. Aliquots were removed at 2-h intervals, and the cells were labeled with [35S]methionine for 15 min and subsequently chased for 30 min at 30°C. The fate of CPY was monitored by immunoprecipitation of cell extracts and analyzed by SDS-PAGE and autoradiography (Figure 2).
Figure 2.

Depletion of plasmid-borne Tlg1p in Δtlg1::LEU2 cells leads to an accumulation of the ER form of CPY (p1CPY). The wild-type and Δtlg1::LEU2 cells containing the GAL1-TLG1 plasmid (JC20) were grown in 1% galactose at 30°C, pelleted, washed, and incubated in 2% glucose medium at 30°C for the indicated periods. Cells (3 OD600nm) were pulse labeled with [35S]methionine for 15 min and chased for a further 30 min at 30°C. CPY was isolated from cell extracts by immunoprecipitation with 6 μg of anti-CPY antibodies and resolved by 8% SDS-PAGE. The different forms of CPY are indicated on the left. p1CPY, p2CPY, and mCPY represent the ER/early Golgi, late Golgi, and mature vacuolar forms of CPY, respectively. After 2–4 h of incubation in glucose, processing of p1CPY to p2CPY and mCPY is inhibited.
CPY exits the ER and is transported to the ER/early Golgi as a 67-kDa p1 form which is modified in the Golgi to generate the 69-kDa p2 form upon arrival at the late Golgi compartment (Stevens et al., 1982). p2CPY is then transported to the vacuole and cleaved by the Pep4p, to give rise to a 61-kDa mature form of CPY (Hemmings et al., 1981). Two hours after cells were transferred to glucose medium, most of the CPY was detected as the p1 precursor form in Δtlg1 cells, and this defect was enhanced 4 h after JC20 were transferred to the glucose medium. However, in wild-type cells, most of the CPY had matured to the 61-kDa with some p2CPY still remaining (Figure 2). Most of the invertase secreted by JC20 cells incubated in the glucose medium for 4 h or longer migrated faster in SDS-PAGE than the hyperglycosylated forms secreted from wild-type cells, suggesting that invertase was underglycosylated (our unpublished results). These results indicate that Tlg1p is required for the transport of CPY in the early secretory pathway and depletion of this protein causes a severe transport block in the ER to Golgi and/or intra-Golgi transport.
Golgi Function Is Affected in tlg1 Temperature-sensitive Mutants
To further analyze the role of Tlg1p in protein transport, ts alleles of TLG1 were constructed by PCR-mediated mutagenesis (Fromant et al., 1995; Lupashin et al., 1997). Five tlg1 ts mutants were obtained in the W303 background, all of which have a much-reduced growth when incubated at 38°C compared with 23°C (Figure 3). These ts mutants contain similar amounts of Tlg1p after prolonged incubation at 23°C, as detected by Western blot analysis of total yeast lysates, indicating that the stability of the mutant proteins was not compromised (our unpublished results).
Figure 3.

Temperature-sensitive tlg1 mutants are defective for growth at 38°C. Δtlg1::LEU2 cells in W303 haploids containing either TLG1 or its mutant alelles under the control of its endogenous promoter were streaked on a glucose-based selective plate without trptophan and incubated for 3 d at either 23 or 38°C. The orientation of the respective cells is indicated on the left.
To examine the effect on three of the ts mutations on the intracellular transport of CPY, Δtlg1 cells bearing either TLG1 or tlg1 ts alleles on CEN plasmids were grown at 23°C, and half of the cultures were shifted to 38°C for 30 min. These cells were pulse labeled with [35S]methionine for 15 min and chased for 60 min, and immunoprecipitated CPY was analyzed by SDS-PAGE and autoradiography (Figure 4A). After 60 min of chase at 38°C, most of the CPY in cells harboring the wild-type TLG1 had matured. Under similar experimental conditions, all tlg1 ts cells accumulated predominantly the p1 form of CPY, with the majority of CPY not matured after 60 min of chase (Figure 4A). The presence of some p2CPY was detected in tlg1-1 and tlg1-15 mutants at 38°C but not in tlg1-24 cells. At 23°C, all ts alleles had transported their CPY quite efficiently, and most of the CPY had matured after a 60-min chase period (Figure 4A). The presence of some p2CPY in tlg1-1 and tlg1-15 mutant cells at 38°C after 60 min chase may be indicative of the slight leakiness in these mutants at 38°C, because a dilution of Tlg1p in Δtlg1 cells accumulated the p1CPY and complete inhibition of growth (Figure 2). These results suggest that Tlg1p may function between ER to Golgi and/or cis- to medial- intra-Golgi transport but does not exclude its possible function in intraGolgi retrograde events.
Figure 4.

Transport of CPY in the early secretory pathway is affected at the restrictive temperature for tlg1 ts mutants. (A) Accumulation of p1CPY at 38°C in Δtlg1 cells bearing various tlg1 ts mutant alelles. Cells were initially grown in yeast standard minimal glucose medium (without Trp, Leu, and Met) at 23°C, after which half of each sample was shifted to 38°C for 60 min. The cultures were pulse labeled with [35S]methionine for 15 min and chased for 60 min at either 23 or 38°C. CPY was immunoprecipitated from cell lysates and analyzed by SDS-PAGE. (B) The ER/cis-Golgi form of CPY is poorly glycosylated by α-1,6-mannose-specific enzymes in tlg1 ts cells at 38°C. tlg1 ts mutants were grown at 38°C, labeled for 15 min with [35S]methionine, and chased for 20 min at 38°C. The immunoprecipitated CPY was heated at 90°C for 15 min, and the supernatants were reimmunoprecipitated with either anti-CPY or anti-α-1,6-mannose-specific antibodies. The second immunoprecipitates were analyzed by SDS-PAGE and autoradiography. The different forms of CPY are indicated on the left. p1CPY, p2CPY, and mCPY represent the ER/early Golgi, late Golgi, and mature vacuolar forms of CPY, respectively.
To examine whether post-ER glycosyl modifications had occurred, we eluted the CPY immunoprecipitate of TLG1, tlg1-1, and tlg1-15 mutants by heating the samples at 90°C for 15 min and diluted the supernatant 10-fold with IP buffer containing 1 mg/ml BSA. The samples were divided into two fractions and reimmunoprecipitated with antisera against the α-1,6-mannose linkages or antisera against CPY. α-1,6-Mannose linkages are added at the cis- and medial-Golgi (Franzusoff and Schekman, 1989; Graham et al., 1994). The p2CPY and mCPY were immunoprecipitated with antisera against α-1,6-mannose (Figure 4B) as efficiently as those samples that were immunoprecipitated with anti-CPY antiserum. However, the p1CPY form from all samples was poorly detected, suggesting that the p1CPY was either not modified or poorly modified by α-1,6-mannose. Indeed in similar CPY immunoprecipitation experiments on vti1-24 cells that were incubated at 38°C, Lupashin et al. (1997) obtained a similar result. Vti1p is an intra-Golgi retrograde v-SNARE protein, and vti1-24 mutant cells accumulate p1CPY at the restrictive temperature (Lupashin et al., 1997).
To further test the effect of tlg1 ts mutations on protein secretion, we analyzed the transport of the yeast periplasmic enzyme invertase in TLG1, tlg1-15, and tlg1-24 cells. These cells were transformed with a CEN plasmid expressing the secreted form of invertase tagged with c-myc epitope. The c-myc-tagged invertase is controlled under the PHO5 promoter; hence no induction in low-glucose medium is required. The cells were grown at 23°C, and half of the culture were shifted to 38°C for 1 h. Cells were spheroplasted, which were further incubated at 23 or 38°C for 1 h, resulting in the intracellular (I) and extracellular (E) fractions. Total cell extract (I) and the culture medium (E) were electrophoresed on SDS-PAGE gels and probed with anti-c-myc antibodies (Figure 5A). It was found that invertase was secreted from tlg1-15 and tlg1-24 mutants at both the permissive and restrictive temperatures (Figure 5A, lanes 5–12) similar to wild-type cells (Figure 5, lanes 1–4). However, invertase secreted by both mutants at the restrictive temperature migrated faster in SDS-PAGE than the hyperglycosylated forms secreted by the wild-type cells or by mutants at 23°C, suggesting that invertase was underglycosylated (Figure 5A, lanes 8 and 12). Furthermore, tlg1-24 mutant incubated at 38°C accumulated the core glycosylated as well as the hypoglycosylated form of invertase intracellularly, indicating a lesion of invertase transport in the early secretory pathway (Figure 5A, lanes 11 and 12).
Figure 5.

Secretion of the underglycosylated form and intracellular accumulation of core-glycosylated forms of invertase in tlg1 cells at 38°C. (A) Autoradiograph of invertase immunoprecipitations from Δtlg1 strains bearing the TLG1, tlg1-15 ts, or tlg1-24 ts mutant alelles. Cells expressing myc-tagged invertase were grown at 23 or 38°C for 1 h and converted to spheroplasts. The spheroplasts were further incubated for 1 h at 23 or 38°C, and the resulting spheroplast (I, intracellular fraction) and the culture media (E, extracellular fraction) were analyzed by immunoblot to detect myc-tagged invertase. Note the intracellular accumulation of the core glycosylated invertase and the underglycosylation of the secreted invertase at the 38°C in tlg1-24 cells. (B) The secreted hypoglycosylated invertase is modified by α-1,6- and α-1,3-mannose-specific linkages. The c-myc-tagged invertase was immunoprecipitated from the extracellular (E) fractions of spheroplasts that were incubated at 38°C. Immunoprecipitates were heated to 90°C for 15 min, and the supernatants were diluted in IP buffer. Samples were reimmunoprecipitated with anti-invertase, anti-α-1,6-mannose-specific antiserum, and anti-α-1,3-mannose-specific antiserum. Immunoprecipitates were analyzed on 7% SDS-PAGE and autoradiographed.
To examine post-ER glycosyl modifications of invertase in TLG1, tlg1-15, and tlg1-24 cells, we immunoprecipitated the extracellular fractions with anti-c-myc antibody bound to protein A+G-agarose, divided the supernatants into three fractions, and reimmunoprecipitated with polyclonal antisera against invertase, anti-α-1,6-mannose, and anti-α-1,3-mannose linkages, which are added in the cis- and medial-Golgi or medial- to trans-Golgi, respectively (Franzusoff and Schekman, 1989; Graham and Krasnov, 1995). The mature hyperglycosylated forms of invertase secreted at 38°C by wild-type cells (Figure 5B, lane 1–3) as well as the underglycosylated forms of invertase secreted at 38°C by tlg1-15 (Figure 5B, lanes 4–6) and tlg1-24 (Figure 5B, lanes 7–9) cells were immunoprecipitated well by all three antibodies. Under similar experimental conditions, the core glycosylated form of invertase was immunoprecipitated only by anti-invertase antibodies but not by anti-α-1,6-mannose and anti-α-1,3-mannose linkage-specific antibodies (our unpublished results). These results suggest that the hypoglycosylated invertase secreted by the mutants at 38°C was modified by both α-1,6-mannose and α-1,3-mannose enzymes (Figure 5B, lane 4–9). Thus, a loss of Tlg1p function compromises traffic of CPY in the Golgi without seriously affecting the transit of invertase to the cell surface. Although invertase is secreted, it is underglycosylated with intracellular accumulation of the core glycosylated form, particularly in tlg1-24 cells. Similar observations have been reported for ts mutants of Vti1p, which interacts with both the cis-Golgi resident Sed5p and the endosomal t-SNARE Pep12p (Becherer et al., 1996).
Loss of Tlg1p Causes Accumulation of Vesicles as Well as Fragmentation of the Vacuole
Electron microscopy analysis of cells that were depleted of Tlg1p for 16 h revealed an accumulation of small 40- to 50-nm vesicles as well as fragmentation of the vacuole, neither of which was observed in wild-type cells (Figure 6, B and A, respectively). Similar phenotypes were also observed in cells that have been depleted of Tlg1p for 4 h (our unpublished results). Electron microscopy data from tlg1-24 at the restrictive temperature revealed the accumulation of vesicles as well as vacuolar fragmentation similar to that of Δtlg1 cells, indicating that Tlg1p may act at multiple steps. The accumulation of vesicles is consistent with the phenotypes of SNARE mutants affecting vesicle docking and membrane fusion.
Figure 6.

Accumulation of vesicles and fragmentation of the vacuole upon depletion of cellular Tlg1p. Wild-type (A) and Tlg1p-depleted cells containing the episomal TLG1 under the GAL1 promoter in Δtlg1::LEU2 cells (B) were incubated in YEP-glucose medium for 16 h at 30°C and then examined by electron microscopy. V, vacuole; N, nucleus. Large white arrows and the region within the box indicate the areas where 50-nm vesicles accumulated. The large dark structures of irregular size shown in left panels of B are fragmented vacuoles. Right panels of B are enlargements of the regions indicated by large arrows in the left panels. Large white arrows in the left panels indicate transport vesicles. Bars, 1 μm for the whole cell and 0.15 μm for enlarged regions shown in the right panels of B.
Tlg1p Is an Integral Golgi Membrane Protein
The primary sequence of Tlg1p, in conjunction with the observation that the hydrophobic domain of Tlg1p is essential for its function, suggests that it is an integral membrane protein. Affinity-purified rabbit antibodies against Tlg1p recognized specifically a 26-kDa protein that is present in extracts of wild-type cells but not in the Δtlg1 or pYES2:c-mycTLG1 (pJC21) cells in which the plasmid-derived myc-tagged Tlg1p had been depleted by incubation in glucose medium (Figure 7A, lanes 1 and 2, respectively). High levels of Tlg1p were detected in Δtlg1 cells, which overexpress the plasmid myc-tagged Tlg1p cDNA (Figure 7A, lane 3).
Figure 7.

Tlg1p is an integral membrane protein that cofractionates mainly with the Golgi fraction. (A) Polyclonal antibodies against Tlg1p specifically recognize Tlg1p in wild-type cells (lane 1), Δtlg1 cells that have been depleted of the myc-tagged Tlg1p regulated by the episomal GAL1 promoter (lane 2), and Δtlg1 cells overexpressing myc-tagged Tlg1p (lane 3). (B) Yeast homogenates were separated into different fractions by differential centrifugation. The resulting low-speed pellet (LSP), 13,000 × g pellet (P13), 100,000 × g supernatant (S100), and 100,000 × g pellet (P100) fractions were analyzed by immunoblot to detect proteins as indicated. Tlg1p has a similar distribution as the Golgi t-SNARE Sed5p. (C) Tlg1p is an integral membrane protein. Yeast membranes were extracted with different buffers as indicated and separated into pellet (P) and supernatant (S) fractions, which were analyzed by immunoblot to detect Tlg1p. (D) The majority of Tlg1p cofractionated with Sed5p. The 13,000 × g supernatant from RSY255 cells was fractionated on a sucrose gradient as described in MATERIALS AND METHODS. Fractions were collected from the top of the gradient and analyzed for the distribution of Vti1p, Mnt1p, Tlg1p, Sed5p, and Pep12p by immunoblotting.
To examine whether Tlg1p is an integral membrane protein, cells were homogenized with glass beads, extracted with buffers of different composition, and centrifuged at 100,000 × g to pellet membrane fractions. Proteins from the supernatant as well as the pellet were analyzed by SDS-PAGE, electroblotted onto nitrocellulose membranes, and probed with Tlg1p antibodies. Figure 7C indicates that untreated cell extracts accumulate Tlg1p in the membrane fraction. The protein could not be extracted from the membrane with 0.15 M Na2CO3, pH 11.5, buffer or 1 M NaCl, conditions known to release peripheral membrane proteins. However, treatment of the membranes with 1% Triton X-100 extracted Tlg1p efficiently, suggesting that Tlg1p is indeed an integral membrane protein.
Cell fractionation by differential centrifugation was used to fractionate yeast lysates to obtain membrane and cytoplasmic fractions. The presence of Tlg1p in each fraction was determined by SDS-PAGE and immunoblot analysis. The integrity of various membrane-enclosed organelle fractions was determined by the localization of characterized markers. Figure 7B indicates that Tlg1p was predominantly partitioned into the 100,000 × g membrane pellet (P100), which contains small vesicles and Golgi membranes, as defined by the enrichment of the Golgi proteins Sed5p and Kex2p. Similar to the cis-Golgi protein Sed5p, some Tlg1p was also present in the P13 pellet.
To further examine the compartmental localization of Tlg1p, the S13 fraction was subjected to equilibrium sedimentation in sucrose gradients as described by Becherer et al. (1996) (Figure 7D). The gradient was fractionated, and aliquots were subjected to immunoblotting. Vti1p, myc-tagged Mnt1p, and Sed5p were used as Golgi markers, whereas Pep12p was used as an endosomal marker. Under these conditions, the majority of Tlg1p cofractionated quite well with Sed5p and Mnt1p and to some extent with Vti1p. The majority of the Tlg1p fraction was well separated from the endosomal Pep12p fraction. Under similar experimental conditions, Lupashin et al. (1997) have shown that the majority of the Sed5p cofractionates with Vti1p. These results suggest that Tlg1p is most likely located in the Golgi. Under standard sucrose fractionation assay, it is difficult to separate the cis- and medial-Golgi markers, because they show very similar profiles. Similarly, Jungmann and Munro (1998) have shown that a cis-Golgi Anp1p cofractionates with similar profiles as that of the medial-Golgi Mnt1p on sucrose gradients.
To determine the intracellular localization of Tlg1p with greater accuracy, epitope-tagged Tlg1p was expressed from a multicopy vector, and the staining pattern was compared with those of endogenous Sed5p by immunofluorescence. The staining patterns of a triple myc-tagged Mnt1p was also compared with that of the endogenous Tlg1p (Figure 8). Although under confocal microscopy, the punctate staining of Tlg1p showed considerable overlap with cis-Golgi Sed5p and medial-Golgi Mnt1p, some of the Tlg1p spots did not coincide with either of the two markers (Figure 8). Jungmann and Munro (1998) have shown that even two cis-Golgi proteins, Anp1p and Sed5p, have some punctate staining that did not correspond, indicating that components of the Golgi are highly dynamic (Lussier et al., 1995; Jungmann and Munro, 1998). These results suggest that the majority of the Tlg1p is located in the Golgi apparatus.
Figure 8.

Golgi association of Tlg1p as revealed by imunofluorescence microscopy. Cells expressing the myc-tagged Tlg1p were double labeled with 9E10 antibody (B) and polyclonal antibodies against endogenous Sed5p (C). Similarly, cells expressing myc-tagged Mnt1p were double labeled with 9E10 (E) and polyclonal antibodies against the endogenous Tlg1p (F). Also shown are the corresponding differential interference contrast images. Bar, 4 μm.
Suppression of Tlg1
Because the majority of Tlg1p is enriched in the Golgi apparatus, we examined whether other Golgi SNAREs were able to complement tlg1 ts mutants. Transformation of multiple copies of myc-tagged Sed5p (Banfield et al., 1995) in tlg1-20 and tlg1-24 at 23°C leads to an ∼10-fold reduction in the number of transformants compared with wild-type cells. Moreover, propagation of the tlg1 ts mutants as well as wild-type transformants in selective medium at the nonrestrictive temperature abrogates the lethal effects of Sed5p overexpression. Overexpression of other SNAREs such as Bet1p, Tlg2p, Vam3p, Gos1p, or Bos1p in tlg1 ts cells did not give any noticable variation in the number of transformants, nor did they rescue the tlg1 mutants at the restrictive temperatures (our unpublished results). Overexpression of Tlg1p or Tlg2p did not rescue the growth defect of sed5-1 cells at 37°C or induce any discernible phenotype (our unpublished results). One possible explanation for the observed effect of Sed5p overexpression on tlg1 ts mutants is that Sed5p and Tlg1p may interact with some common proteins, for example, Vti1p, and thus overexpression of Sed5p may titrate away the protein available for interaction with Tlg1p and exaggerate the ts phenotype of Tlg1p.
Interestingly, overexpression of Tlg1p can moderately complement the growth defect of vti1-11 at 38°C but only weakly in vti1-1 (Fischer von Mollard et al., 1997). This correlates with the accumulation in vti1-11 of the early Golgi p1 form of the CPY at the restrictive temperatures, whereas vti1-1 accumulates p2CPY (Fischer von Mollard et al., 1997). Overexpression of Vti1p was not able to complement tlg1-20 and tlg1-24 ts mutants at 38°C.
Tlg1p Is a Component of a SNARE Complex
We next investigated whether Tlg1p functions as a SNARE. Total cell lysate from wild-type cells overexpressing Tlg1p was mixed with His6-tagged Sec17p, GST-retinoblastoma, or GST alone coupled to either nickel-nitrilotriacetic acid agarose or glutathione-Sepharose beads and incubated under conditions that favor the formation of SNARE complexes. Beads were extensively washed and analyzed by immunoblot for Tlg1p. Tlg1p was specifically retained by immobilized His6-tagged Sec17p in a concentration-dependent manner, but not by GST-retinoblastoma or GST alone (our unpublished results).
Incubation of sec18-1 ts mutant cells at 37°C results in a deficiency of NSF activity, causing a block in transport. This was thought to be due to inability of the docked vesicles to fuse with the acceptor membrane, allowing the concentration of Sec17p as well as SNARE complexes to increase (for review, see Sogaard et al., 1994; Sapperstein et al., 1996). Further evidence from in vitro assays of homotypic vacuolar fusion and microscopic analyses indicated that Sec18p and Sec17p are required only for the priming stage of the v/t-SNAREs before docking (Mayer and Wickner, 1997; Xu et al., 1997). Nevertheless, other reports have shown that sec18-1 cells incubated at the restrictive temperature accumulate Sed5p-SNARE complexes in which Sec17p was still associated (Sapperstein et al., 1996; Rothman and Sollner, 1997). To determine whether the presence of the Tlg1p-SNARE complex was enhanced in sec18-1 cells grown at the restrictive temperature, Tlg1p was immunoprecipitated from different yeast lysates solubilized with 2% Triton X-100. The samples were analyzed by SDS-PAGE and immunoblotted for Tlg1p and Sec17p. Sec17p was found to coimmunoprecipitate with Tlg1p under conditions that favor SNARE complex formation (sec18-1 cells grown at 37°C for 1 h; Figure 9A, lane 3), whereas Sec17p was barely detectable in the immunoprecipitate from wild-type cells grown at 37°C (Figure 9A, lane 1, lower panel). Lesser but significant amounts of Sec17p were still detectable in sec18-1 cells grown at 25°C (Figure 9A, lane 2). Similarly, Tlg1p was also coimmunoprecipitated by antibodies against Sec17p, particularly from the lysate of sec18-1 cells grown at 37°C (our unpublished results).
Purification and Characterization of the Tlg1p SNARE Complex(es)
To characterize Tlg1p SNARE complexes, large-scale immunoprecipitations were performed, using 400 μg of affinity-purified anti-Tlg1p antibodies coupled to protein A-Sepharose beads with 100 mg of spheroplast detergent extract of sec18-1 cells that were preincubated at 37°C. Proteins specifically retained by the beads were separated on an SDS-PAGE gel and stained with Coomassie blue dye. At least five proteins coimmunoprecipitated specifically with Tlg1p were detected and individually excised for amino acid sequencing (Figure 9B). Two tryptic peptides derived from each of the 29-kDa (E) and 45-kDa (C) protein bands revealed that these polypeptides were Vti1p and Tlg2p, respectively (Table 3). Tlg2p was identified by Holthuis et al. (1998a) as a TGN syntaxin that is required for retrograde Golgi trafficking. However, Abeliovich et al. (1998) have suggested that Tlg2p is an endosomal protein. Others have shown that Tlg2p is required for the biogenesis of normal early endosomes and is involved in endocytosis, because a deletion of TLG2 is synthetic lethal with vma2Δ (Seron et al., 1998). Alternatively, Tlg2p could function in several transport events. Two peptides sequenced from the 26-kDa band (F) correspond to Tlg1p, whereas a tryptic peptide from the 32-kDa band (D) corresponds to Sec17p. We have identified the 58-kDa protein band (B) as Vps45p, a peripheral membrane protein of the Sec1p protein family (Cowles et al., 1994; Piper et al., 1994). Vps45p is a multiregulator in that it is able to form a complex with Vps21p, Vac1p, and Pep12p in a SEC18-dependent manner and function directly in the docking and fusion of Golgi-derived transport vesicles with the prevacuolar endosome (Burd et al., 1997; Cowles et al., 1997). Vps45p is thus required for Pep12p/Vac1p function and may mediate a conformational change in one or both of these proteins required for receptor activity. (Burd et al., 1997). Furthermore, Vps45p is able to complement the lethality of Apm3p, a gene coding for the yeast AP-3 μ chain (Stepp et al., 1997). A peptide sequence from the 76-kDa band was identical to a stretch of amino acids in the major coat protein of the double-stranded RNA virus of S. cerevisiae (Icho and Wickner, 1989), which may reflect an interaction of Tlg1p with this molecule or a nonspecific association. We were unable to obtain amino acid sequences for other protein bands.
Table 3.
The identities of components of the Tlg1p SNARE complex components
| Size (kDa) | Protein band | Peptide sequences obtained | Identity (residues) |
|---|---|---|---|
| 76 | A | KIKLPVTIDDT | Major coat protein (156–166) |
| 58 | B | KNIWEISEIEQ | Vps45p (357–368) |
| 45 | C | REGLDIEDYSK | Tlg2p (218–229) |
| REMQDLVVDQGTI | Tlg2p (268–281) | ||
| 32 | D | ELNLAGDSFLKR | Sec17p (52–62) |
| 29 | E | QTLFQADSYVDK | Vti1p (167–178) |
| RLPQSLVDS | Vti1p (88–97) | ||
| 26 | F | DVEETIVDLDRS | Tlg1p (47–58) |
| HNTAGDDDQEEE | Tlg1p (29–40) |
Individual protein bands shown in Figure 9B were excised from the SDS-PAGE gels and in situ digested with either trypsin or lysyl endopeptidase, and the resulting peptides were fractionated, purified by HPLC, and sequenced by Edman microsequencing. The identity of the proteins were determined using the peptide sequences to search the yeast Saccharomyces database. The size of the proteins and the GenBank accession numbers are shown.
Tlg1p, Vti1p, and Tlg2p Can Be Found in the Same SNARE Complex
To determine whether Vti1p and Tlg2p are part of the same Tlg1p-SNARE complex, we made specific polyclonal antibodies against Vti1p and Tlg2p. Affinity-purified antibodies (200 μg) generated against GST alone, GST-Vti1p, or GST-Tlg2p were individually cross-linked to protein A-Sepharose beads with dimethyl pimelimidate. The bound antibodies were incubated overnight with cell extracts of either wild-type or sec18-1 mutant cells grown at 37°C for 1 h. Washed beads were eluted with glycine buffer, and 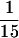 of the sample was analyzed by SDS-PAGE and probed for the presence of Sec22p, Bet1p, Tlg1p, Vti1p, Sec17p, and Tlg2p (Figure 10). Anti-Vti1p antibodies immunoprecipitated Sec17p, Tlg1p, and Tlg2p in both wild-type (Figure 10, lane 2) and sec18-1 (Figure 10, lane 3) cells. The presence of these proteins is enhanced in the sec18-1 cells that potentially accumulate vesicles at the nonpermissive temperature (Figure 10, compare lanes 3 and 2). Conversely, anti-Tlg2p antibodies immunoprecipitated Sec17p, Tlg1p, and Vti1p in a manner similar to that of anti-Vti1p antibodies (Figure 10, lane 4). The Bet1p and Sec22p were not detected in any of these immunoprecipitates. These results indicate that Tlg1p, Vti1p, and Tlg2p can be found in the same SNARE complex.
of the sample was analyzed by SDS-PAGE and probed for the presence of Sec22p, Bet1p, Tlg1p, Vti1p, Sec17p, and Tlg2p (Figure 10). Anti-Vti1p antibodies immunoprecipitated Sec17p, Tlg1p, and Tlg2p in both wild-type (Figure 10, lane 2) and sec18-1 (Figure 10, lane 3) cells. The presence of these proteins is enhanced in the sec18-1 cells that potentially accumulate vesicles at the nonpermissive temperature (Figure 10, compare lanes 3 and 2). Conversely, anti-Tlg2p antibodies immunoprecipitated Sec17p, Tlg1p, and Vti1p in a manner similar to that of anti-Vti1p antibodies (Figure 10, lane 4). The Bet1p and Sec22p were not detected in any of these immunoprecipitates. These results indicate that Tlg1p, Vti1p, and Tlg2p can be found in the same SNARE complex.
Figure 10.

Tlg1p, Vti1p, and Tlg2p are components of the same complex. Spheroplasts of wild-type or sec18-1 yeast cells were preincubated for 1 h at 37°C. Cell extracts were then prepared as described (Sogaard et al., 1994). Extracts (20 mg of protein) from wild-type (lane 2) and sec18-1 mutant cells (lanes 1, 3, and 4) were incubated with anti-Vti1p (lanes 2 and 3), anti-Tlg2p (lane 4), and anti-GST (lane 1) antibodies, which had been covalently coupled to protein A-Sepharose CL4B. Immunoprecipitates were eluted with glycine elution buffer, and 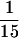 of the sample was analyzed by immunoblot to detect the respective proteins.
of the sample was analyzed by immunoblot to detect the respective proteins.
DISCUSSION
This work describes the independent identification and characterization of Tlg1p. A previous study (Holthuis et al., 1998a,b) had indicated that Tlg1p is not essential in cells of the Y100 (SEY6210 × SEY6211) strain. Consistent with this, our deletion of TLG1 in Y100 gave a similar phenotype. However, deletion of TLG1 in three other strains resulted in a more severe phenotype. In the W303 strain, Tlg1p deletion gave a lethal phenotype. Although Tlg1p is not needed for germination, it is required for normal cell growth. Deletion of TLG1 in Y97 and Y98 yeast strains resulted in a severe defect in normal cell growth. The reasons for the differing phenotypes from TLG1 deletion in these alternative yeast strains are currently unknown. We have focused our present study on the W303 strain, because in this strain TLG1 is essential. Ectopic expression of Tlg1p in either low- or high-copy vectors was able to complement the lethal phenotype of Δtlg1 cells in W303 and the severe growth defect in Y98 backgrounds, confirming that the observed phenotypes were indeed due to the deletion of TLG1. Western analyses further confirmed TLG1 deletion. Under similar conditions, deletion of TLG2, VAM3, or VTI1 in W303 cells resulted in phenotypes as previously published, regardless of the different strains used.
Cells depleted of Tlg1p as well as tlg1 ts mutants incubated at the restrictive temperature accumulate the p1 form of CPY, which is poorly glycosylated by the α-1,6-mannose-specific linkages. Although invertase is secreted at the restrictive temperature by tlg1 ts mutants, nevertheless it is underglycosylated. Some intracellular accumulation of core glycosylated as well as the hypoglycosylated invertase is observed in tlg1-24, suggesting intracellular transport of invertase is also affected to some extent in this strain. This more severe invertase transport defect in tlg1-24 is also detected by the more acute accumulation of the p1CPY compared with tlg1-15. The reason why intracellular transport of CPY is affected more dramatically than invertase in Tlg1p-depleted cells and tlg1 ts mutant cells is not known; however, similar phenotypes were observed in other studies (Lupashin et al., 1997; Yang et al., 1998). Electron microscopy data indicate that cells depleted of Tlg1p accumulate 40- to 50-nm vesicles, a characteristic of SNARE mutants that are defective in docking and membrane fusion as observed by Holthuis et al. (1998a). Because our studies were performed in the W303 yeast background, we were unable to dissect Tlg1p participation in the later part of transport as described by Holthuis et al. (1998a). Other than a role in early Golgi, the Tlg1p functions in the TGN–endosomal pathway, based on the presence of Vps45p and Tlg2p in the Tlg1p-SNARE complex immunoprecipitate. Together, these results suggest that Tlg1p may participate in two pathways: the intra-Golgi transport and the endosome pathway (Holthuis et al., 1998a). Multiple functions for SNAREs have been reported previously. Vam3p, a vacuolar t-SNARE, and the v-SNARE Vti1p are able to interact with multiple partners from different compartments. Vam3p is able to interact with Vti1p and Nyv1p, a vacuolar v-SNARE (Nichols et al., 1997). Vam3p is also required for the docking and/or fusion of autophagosomes with the vacuole and is able to complement the pep12Δ mutant phenotype and vice versa (Darsow et al., 1997). Vti1p is able to interact with multiple t-SNAREs, namely Sed5p, Tlg1p, Tlg2p, and Pep12p. These interactions are required for the efficient recycling of v-SNAREs from different vesicles and compartments (Fischer von Mollard et al., 1997; Lupashin et al., 1997; Holthuis et al., 1998a). Vti1p is also genetically able to complement an sft1-1 mutant (a t-SNARE) at the restrictive temperature (Lupashin et al., 1997). Overexpression of the t-SNARE Sed5p is able to rescue the lethal phenotype of cells lacking Vti1p, and immunoprecipitation analyses have shown that Vti1p interacts with both Pep12p and Sed5p (Fischer von Mollard et al., 1997). Tlg1p may have a similar multifunctional character as exemplified by Vam3p and Vti1p. Besides its necessity for efficient endocytosis and maintenance of normal TGN proteins (Holthuis et al., 1998a), our study suggests that Tlg1p is also required for intra-Golgi traffic.
Genetic evidence also supports a functional role for Tlg1p in intra-Golgi events. Overexpression of Tlg1p is able to complement the defect of vti1-11 mutant cells at 38°C but not vti1-1 cells under similar conditions. The complementation of vti1-11 by Tlg1p is probably due to the fact that vti1-11 vesicular transport is blocked in the early Golgi pathway where Tlg1p also functions, whereas vti1-1 accumulates vesicles at the late Golgi (Fischer von Mollard et al., 1997). Overproduction of Sed5p in any of the tlg1 ts cells leads to a marked reduction in the number of transformants compared with wild-type cells, suggesting that tlg1 ts mutants are sensitive to Sed5p overexpression. This may be explained if overexpression of Sed5p titrates out proteins that interact with both Sed5p and Tlg1p and thus reduces the targets for interaction with Vti1p. This decrease in the amount of interacting proteins may thus accelerate the abnormal phenotype of tlg1 ts mutants. Under identical conditions, overexpression of Sec22p, Bet1p, Vam3p, or Tlg2p in tlg1 ts cells does not have any discernible phenotype, indicating that the growth inhibition is specific to Sed5p overexpression. Multiple copies of Tlg1p-expressing vectors in sec22-1, bet1-1, Δvam3, and sed5-1 cells at 38°C were not able to rescue their ts defects.
Immunofluorescence studies suggest that Tlg1p colocalizes quite well with early Golgi-located proteins Sed5p and Mnt1p, because the majority of the punctate staining of Tlg1p does coincide with both the cis-Golgi Sed5p and the medial-Golgi Mnt1p. We have also shown by sucrose gradient fractionation that the majority of Tlg1p cofractionates with Sed5p, Mnt1p, and the Golgi fraction of Vti1p. Together, these results suggest that the majority of Tlg1p is located to the Golgi apparatus. Some degree of colocalization was also detected between the late Golgi marker Kex2p and Tlg1p (Holthuis et al., 1998a). Tlg2p has been suggested by Holthuis et al. (1998a) to be a TGN protein and is possibly involved in anterograde transport, whereas others have suggested that it functions in retrograde transport from the endosome to the late Golgi as well as in the endocytic pathway (Abeliovich et al., 1998). Tlg2p forms a SEC18-dependent SNARE complex with Snc2p, a v-SNARE that functions in the Golgi to plasma membrane protein pathway (Protopopov et al., 1993). Seron et al. (1998) also suggested a role for Tlg2p in endocytosis and early endosome biogenesis.
Composition of the Tlg1p–SNARE Complex
The accumulation of docked vesicles in sec18-1 cells grown at the restrictive temperature leads to an accumulation of Sec17p and v/t-SNARE complex (Sogaard et al., 1994; Abeliovich et al., 1998; Holthuis et al., 1998a). Sec17p and Sec18p are considered part of the general machinery required for the fusion of various transport events (Sogaard et al., 1994; Whiteheart and Kubalek, 1995; Pelham, 1997). However, recent studies have suggested that Sec18p and Sec17p may act at the docking stage of the v/t-SNAREs (Mayer and Wickner, 1997; Nichols et al., 1997; Colombo et al., 1998; Weber et al., 1998). When extracts derived from sec18-1 cells incubated at 37°C were used for immunoprecipitation with antibodies against Sed5p, several other proteins were precipitated, including Sft1p, Gos1p, Bet1p, Sec22p, Bos1p, Ykt6p, and Sec17p (Sogaard et al., 1994; McNew et al., 1997). The interactions of the v-SNARE Vti1p with t-SNAREs Pep12p and Sed5p as well as the association of Ufe1p with Sec20p and Tip20p were established under similar experimental conditions (Fischer von Mollard et al., 1997; Lewis et al., 1997). Using this approach, at least five proteins that appear to be specifically associated with Tlg1p were immunoprecipitated with antibodies against Tlg1p. Amino acid microsequencing identified four of these proteins as Vti1p, Sec17p, Vps45p, and Tlg2p (Piper et al., 1994; Cowles et al., 1994; Fischer von Mollard et al., 1997; Lupashin et al., 1997; Abeliovich et al., 1998; Holthuis et al., 1998a) in addition to Tlg1p itself.
Vps45p is a member of the Sec1p family, which has been implicated in controlling fusion of post-Golgi transport vesicles with prevacuolar endosomes through its interaction with other proteins such as Pep12p, Vac1p, Pep7p, and Vps21 (Burd et al., 1997; Webb et al., 1997). It is also involved in vacuolar protein sorting (Cowles et al., 1994; Piper et al., 1994). The presence of Vps45p and Tlg2p in the Tlg1p-SNARE complex may be explained by the data of Holthuis et al. (1998a), showing the participation of Tlg1p in the endosomal pathway. Both Tlg1p and Tlg2p act in the Golgi and the endosome and interact with Vti1p, Snc1p, and/or Snc2p, which are possibly regulated by Vps45p (Abeliovich et al., 1998; Holthuis et al., 1998a). This would allow Vti1p and other cognate v-SNAREs to be recycled between the Tlg1p/Tlg2p intermediates present from different compartments in a Vps45p-dependent manner. Another example is Sly1p, a Sec1p family member participating in ER to Golgi transport in the Sed5p complex. Sly1p functions as a brake, whereas a Rab protein, Ypt1p, acts as a throttle of membrane fusion, directly opposing each other by allowing or preventing v-SNARE pairing with t-SNAREs, thus giving a level of specificity to the fusion process (Lupashin and Waters, 1997; Rothman and Sollner, 1997). Along with this hypothesis, Fischer von Mollard et al. (1997) have suggested that Vti1p may be regulated by a Rab/Ypt interaction and a Sec1p protein family member, such that a single v-SNARE can control the fusion of two different classes of vesicles. We believe that the Tlg1p–SNARE complex may be regulated in a similar manner by Vps45p.
ACKNOWLEDGMENTS
We thank Prof. Y.H. Tan for support and members of the Hong laboratory for their critical reading of the manuscript. We are grateful to Tom Stevens, Hugh Pelham, Scott Emr, James Rothman, Steven Aust, and Randy Schekman for their generosity in antibodies, yeast strains, and other reagents.
REFERENCES
- Abeliovich H, Grote E, Novick P, Ferro-Novick S. Tlg2p, a yeast syntaxin homolog that resides on the Golgi and endocytic structures. J Biol Chem. 1998;273:11719–11727. doi: 10.1074/jbc.273.19.11719. [DOI] [PubMed] [Google Scholar]
- Banfield DK, Lewis MJ, Pelham HRB. A SNARE-like protein required for traffic through the Golgi complex. Nature. 1995;375:806–809. doi: 10.1038/375806a0. [DOI] [PubMed] [Google Scholar]
- Becherer KA, Rieder SE, Emr SD, Jones EW. Novel syntaxin homologue, Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol Biol Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berber G, Dumont J, Gilliquet V, Bolle P-A, Higler F. The YDp plasmids, a uniform set of vectors bearing a versatile gene disruption cassettes for Saccharomyces cerevisiae. Yeast. 1991;7:475–477. doi: 10.1002/yea.320070506. [DOI] [PubMed] [Google Scholar]
- Bock JB, Lin RC, Scheller RH. A new syntaxin family member implicated in targeting of intracellular transport vesicles. J Biol Chem. 1996;271:17961–17965. doi: 10.1074/jbc.271.30.17961. [DOI] [PubMed] [Google Scholar]
- Bock JB, Scheller RH. Protein transport. A fusion of new ideas. Nature. 1997;387:133–135. doi: 10.1038/387133a0. [DOI] [PubMed] [Google Scholar]
- Brennwald P, Kearns B, Champion K, Keranen S, Bankaitis V, Novick P. Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Brew K, Shaper JH, Olsen KW, Trayer IP, Hill RL. Cross-linking of the components of lactose synthetase with dimethylpimelimidate. J Biol Chem. 1975;25:1434–1444. [PubMed] [Google Scholar]
- Burd CG, Peterson M, Cowles CR, Emr SD. A novel Sec18p/NSF-dependent complex required for Golgi-to-endosome transport in yeast. Mol Biol Cell. 1997;8:1089–1104. doi: 10.1091/mbc.8.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER, An S, Barton N, Jahn R. SNAP-25, a t-SNARE which binds to both syntaxin and synaptobrevin via domains that form coiled-coils. J Biol Chem. 1994;269:27427–27432. [PubMed] [Google Scholar]
- Clary DO, Rothman JE. Purification of three related peripheral membrane proteins needed for vesicular transport. J Biol Chem. 1990;265:10109–10117. [PubMed] [Google Scholar]
- Colombo MI, Gelberman SC, Whiteheart SW, Stahl PD. N-Ethylmaleimide-sensitive factor-dependent α-SNAP release, an early event in the docking/fusion process, is not regulated by Rab GTPases. J Biol Chem. 1998;273:1334–1338. doi: 10.1074/jbc.273.3.1334. [DOI] [PubMed] [Google Scholar]
- Cowles CR, Emr SD, Horazdovsky BF. Mutations in the VPS45P gene, a SEC1 homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J Cell Sci. 1994;107:3449–3459. doi: 10.1242/jcs.107.12.3449. [DOI] [PubMed] [Google Scholar]
- Cowles CR, Odorizzi G, Payne GS, Emr SD. The AP-3 adaptor complex is essential for cargo-selective transport to the yeast vacuole. Cell. 1997;91:109–118. doi: 10.1016/s0092-8674(01)80013-1. [DOI] [PubMed] [Google Scholar]
- Darsow T, Rieder SE, Emr SD. A multispecificity syntaxin homoloque, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer von Mollard G, Nothwehr SF, Stevens TH. The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. J Cell Biol. 1997;137:1511–1524. doi: 10.1083/jcb.137.7.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzusoff A, Schekman R. Functional compartments of the yeast Golgi apparatus are defined by the sec7 mutation. EMBO J. 1989;8:2695–2702. doi: 10.1002/j.1460-2075.1989.tb08410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromant M, Blanquet S, Plateau P. Direct random mutagenesis of gene-sized DNA fragment using PCR. Anal Biochem. 1995;224:347–353. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- Gaynor EC, te Heesen S, Graham TR, Aebi M, Emr SD. Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J Cell Biol. 1994;127:653–665. doi: 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino AI. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-bp restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Graham TR, Emr SD. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J Cell Biol. 1991;114:207–218. doi: 10.1083/jcb.114.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Krasnov VA. Sorting of yeast alpha 1,3 mannosyltransferase is mediated by a lumenal domain interaction and a transmembrane domain signal that can confer clathrin-dependent Golgi localization to a secreted protein. Mol Biol Cell. 1995;6:809–824. doi: 10.1091/mbc.6.7.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Seger M, Payne GS, MacKay VL, Emr SD. Clathrin-dependent localization of alpha 1,3 mannosyltransferase to the Golgi complex of Saccharomyces cerevisiae. J Cell Biol. 1994;127:667–678. doi: 10.1083/jcb.127.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Hay JC, Chao DS, Kuo CS, Scheller RH. Protein interactions regulating vesicle transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Cell. 1997;89:1–20. doi: 10.1016/s0092-8674(00)80191-9. [DOI] [PubMed] [Google Scholar]
- Hemmings BA, Zubenko GS, Hasilik A, Jones EW. Mutant defective in processing of an enzyme located in the lysosome-like vacuole of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1981;78:435–439. doi: 10.1073/pnas.78.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis JCM, Nichols BJ, Dhruvakumar S, Pelham HRB. Two syntaxin homologues in the TGN/endosomal system of yeast. EMBO J. 1998a;17:113–126. doi: 10.1093/emboj/17.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis JCM, Nichols BJ, Pelham HRB. The syntaxin Tlg1p mediates trafficking of chitin synthase III to polarized growth sites in yeast. Mol Cell Biol. 1998b;9:3383–3397. doi: 10.1091/mbc.9.12.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icho T, Wickner RB. The double-stranded RNA genome of yeast virus L-A encodes its own putative RNA polymerase by fusing two open reading frames. J Biol Chem. 1989;264:6716–6723. [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cation. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann J, Munro S. Multi-protein complexes in the cis-Golgi of Saccharomyces cerevisiae with α-1,6-mannosyltransferase activity. EMBO J. 1998;17:423–434. doi: 10.1093/emboj/17.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, Lin RC, Hsu SC, Scheller RH. Distinct domains of syntaxin are required for synaptic vesicle fusion complex formations and dissociation. Neuron. 1995;14:991–998. doi: 10.1016/0896-6273(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structure rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J, Kuliawat R, Griffith JM, Genje HJ, Arvan P. Mannose 6-phosphate receptors are sorted from immature secretory granules via adaptor protein AP-1, clathrin, and syntaxin 6-positive vesicles. J Cell Biol. 1998;141:359–371. doi: 10.1083/jcb.141.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M, Pelham HRB. SNARE mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell. 1996;85:205–215. doi: 10.1016/s0092-8674(00)81097-1. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Rayner JC, Pelham HRB. A novel SNARE complex implicated in vesicle fusion with the endoplasmic reticulum. EMBO J. 1997;16:3017–3024. doi: 10.1093/emboj/16.11.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier M, Sdicu A-M, Ketela T, Bussey H. Localization and targeting of the Saccharomyces cerevisiae KRE2/MNT1 α-1,2-mannosyltransferase to a medial Golgi compartment. J Cell Biol. 1995;131:913–927. doi: 10.1083/jcb.131.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupashin VV, Pokrovskaya ID, McNew J, Waters MG. Characterization of a novel yeast SNARE protein implicated in Golgi retrograde traffic. Mol Biol Cell. 1997;8:2659–2676. doi: 10.1091/mbc.8.12.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupashin VV, Waters MG. t-SNARE activates through transient interaction with a rab-like guanosine triphosphatase. Science. 1997;276:1255–1258. doi: 10.1126/science.276.5316.1255. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Mayer A, Wickner W. Docking of yeast vacuoles is catalyzed by the ras-like GTPase Ypt7p after symmmetric priming by Sec18p (NSF) J Cell Biol. 1997;136:307–317. doi: 10.1083/jcb.136.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- McNew JA, Sogaard M, Lampen NM, Machida S, Ye RR, Lacomis L, Tempst P, Rothman JE, Sollner TH. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J Biol Chem. 1997;272:17776–17783. doi: 10.1074/jbc.272.28.17776. [DOI] [PubMed] [Google Scholar]
- Nagahama M, Orci L, Ravazzola M, Amherdt M, Lacomis L, Tempst P, Rothman JE, Sollner TH. A v-SNARE implicated in intraGolgi transport. J Cell Biol. 1996;133:507–516. doi: 10.1083/jcb.133.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols BJ, Ungermann C, Pelham HRB, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–201. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Numata K, Ueki T, Naito N, Yamada N, Kamasawa N, Oki T, Osumi M. Morphological changes of Candida albicans induced by BMY-28864, a highly water soluble Pramidicin derivative. J Electron Microsc. 1993;42:147–155. [PubMed] [Google Scholar]
- Ossig R, Dascher C, Trepte HH, Schmitt HD, Gallwitz D. The yeast SLY gene products, suppressors of defects in the essential GTP-binding Ypt1 protein, may act in endoplasmic reticulum-to-Golgi transport. Mol Cell Biol. 1991;11:2980–2993. doi: 10.1128/mcb.11.6.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HRB. SNAREs and the organization of the secretory pathway. Eur J Cell Biol. 1997;74:311–314. [PubMed] [Google Scholar]
- Pevsner J, Scheller RH. Mechanism of vesicle docking and fusion: insights from the nervous system. Curr Opin Cell Biol. 1994;6:555–560. doi: 10.1016/0955-0674(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Piper RC, Whitters EA, Stevens TH. Yeast Vps45p is a Sec1p-like protein required for the consumption of vacuole-targeted, post Golgi transport vesicles. Eur J Cell Biol. 1994;65:305–318. [PubMed] [Google Scholar]
- Protopopov V, Govindan B, Novick P, Gerst JE. Homologs of the synaptobrevin/VAMP family of synaptic vesicle proteins function on the late secretory pathway in S. cerevisiae. Cell. 1993;74:855–861. doi: 10.1016/0092-8674(93)90465-3. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Sollner TA. Throttles and dampers: controlling the engine of membrane fusion. Science. 1997;276:1212–1213. doi: 10.1126/science.276.5316.1212. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Rowe T, Dascher C, Bannykh S, Plutner H, Balch WE. Role of vesicle-associated syntaxin 5 in the assembly of preGolgi intermediates. Science. 1998;279:696–700. doi: 10.1126/science.279.5351.696. [DOI] [PubMed] [Google Scholar]
- Sapperstein SK, Lupashin VV, Schmitt HD, Waters MG. Assembly of the ER to Golgi SNARE complex requires Uso1p. J Cell Biol. 1996;132:755–767. doi: 10.1083/jcb.132.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman R, Mellman I. Does COPI go both ways? Cell. 1997;90:197–200. doi: 10.1016/s0092-8674(00)80326-8. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schimmoller F, Itin C, Pfeffer S. Get your coat. Curr Biol. 1997;7:235–237. doi: 10.1016/s0960-9822(06)00109-6. [DOI] [PubMed] [Google Scholar]
- Seron K, et al. A yeast t-SNARE involved in endocytosis. Mol Biol Cell. 1998;9:2873–2889. doi: 10.1091/mbc.9.10.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard M, Tani K, Ye RR, Geromanos S, Tempst P, Kirchhausen T, Rothman JE, Sollner T. A Rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell. 1994;78:937–948. doi: 10.1016/0092-8674(94)90270-4. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Stepp JD, Huang K, Lemmon SK. The yeast adaptor protein complex, AP-3, is essential for the efficient delivery of alkaline phosphatase by the alternate pathway to the vacuole. J Cell Biol. 1997;139:1761–1774. doi: 10.1083/jcb.139.7.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T, Esmon B, Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Subramaniam VN, Peter F, Philp R, Wong SH, Hong WJ. GS28, a 28-kilodalton Golgi SNARE that participates in ER-Golgi transport. Science. 1996;272:1161–1163. doi: 10.1126/science.272.5265.1161. [DOI] [PubMed] [Google Scholar]
- Wada Y, Anraku Y. Genes for directing vacuolar morphogenesis in Saccharomyces cerevisiae. Vam7, a gene for regulating morphogenic assembly of the vacuoles. J Biol Chem. 1992;267:18671–18675. [PubMed] [Google Scholar]
- Webb GC, Hoedt M, Poole LJ, Jones E. Genetic interactions between a pep7 mutation and the PEP12 and VPS45P genes: evidence for a novel SNARE component in transport between the Saccharomyces cerevisiae Golgi complex and endosome. Genetics. 1997;147:467–478. doi: 10.1093/genetics/147.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Low SH, Chapin SJ, Mostov KRE, Butcher P, Hofmann K. A conserved domain is present in different families of vesicular fusion proteins—a new superfamily. Proc Natl Acad Sci USA. 1997;94:3046–3051. doi: 10.1073/pnas.94.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- Whiteheart SW, Kubalek EW. SNAPs and NSF, general members of the fusion apparatus. Trends Cell Biol. 1995;5:64–68. doi: 10.1016/s0962-8924(00)88948-5. [DOI] [PubMed] [Google Scholar]
- Whitters EA, McGee TP, Bankaitis V. Purification and characterization of a late-Golgi compartment from Saccharomyces cerevisiae. J Biol Chem. 1994;269:28106–28117. [PubMed] [Google Scholar]
- Xu Z, Mayer A, Muller E, Wickner W. A heterodimer of Thioredoxin and IB2 cooperates with Sec18p (NSF) to promote yeast vacuole inheritance. J Cell Biol. 1997;136:299–306. doi: 10.1083/jcb.136.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Matern HT, Gallwitz D. Specific binding to a novel and essential Golgi membrane protein (Yip1p) functionally links that transport GTPases Ypt1p and Ypt31p. EMBO J. 1998;17:4954–4963. doi: 10.1093/emboj/17.17.4954. [DOI] [PMC free article] [PubMed] [Google Scholar]