

Abstract
The structure of the 1,N2-εdG adduct, arising from the reaction of vinyl chloride with dG, was determined in the oligonucleotide duplex 5′-d(CGCATXGAATCC)-3′•5′-d(GGATTCCATGCG)-3′ (X=1,N2-εdG) at pH 8.6 using high resolution NMR spectroscopy. The exocyclic lesion prevented Watson-Crick base-pairing capability at the adduct site and resulted in an ~17 °C of the C decrease in Tm oligodeoxynucleotide duplex. At neutral pH, conformational exchange resulted in spectral line broadening near the adducted site, and it was not possible to determine the structure. However, at pH 8.6, it was possible to obtain well-resolved 1H NMR spectra. This enabled a total of 385 NOE based distance restraints to be obtained, consisting of 245 intra- and 140 inter-nucleotide distances. The 31P NMR spectra exhibited two downfield-shifted resonances, suggesting a localized perturbation of the DNA backbone. The two downfield 31P resonances were assigned to G7 and C19. The solution structure was refined by molecular dynamics calculations restrained by NMR derived distance and dihedral angle restraints, using a simulated annealing protocol. The generalized Born approximation was used to simulate solvent. The emergent structures indicated that the 1,N2-εdG-induced structural perturbation was localized at the X6•C19 base pair, and its 5′-neighbor T5•A20. Both 1,N2-εdG and the complementary dC adopted the anti conformation about the glycosyl bonds. The 1,N2-εdG adduct was inserted into the duplex but was shifted towards the minor groove as compared to dG in a normal Watson-Crick C•G base pair. The complementary cytosine was displaced toward the major groove. The 5′-neighbor T5•A20 base pair was destabilized with respect to Watson-Crick base pairing. The refined structure predicted a bend in the helical axis associated with the adduct site.
Introduction
Etheno DNA adducts (1) arise from the reactions of electrophilic epoxides derived from vinyl halides and other vinyl monomers with dC, dA, and dG nucleotides in DNA (2–9). They also arise as a consequence of endogenous exposure to lipid peroxidation products (9–11). The 1,N2-etheno-dG (1,N2-εdG) adduct, first reported by Sattsangi et al. (12), and later examined by Kusmierek and Singer (13), and Guengerich and coworkers (14), is one of two possible εdG lesions, the other being the N2,3-εdG adduct. The mechanism for formation of 1,N2-εdG was worked out by Guengerich and coworkers (14, 15). It has been detected in DNA treated with vinyl chloride metabolites (16). Among the four etheno DNA adducts, 1,N2-εdG is anticipated to be present at the lowest concentrations in cellular DNA, and it has thus far eluded detection in vivo.
The 1,N2-εdG adduct is mutagenic, both in Escherichia coli (17) and in mammalian (18) cells. When bypassed by E. coli polymerases I exo- and II exo-, T7 polymerase exo-, HIV-1 reverse transcriptase, and rat polymerase β, it produces misincorporation errors (19). The bypass of the 1,N2-εdG adduct by the Y-family Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) was dependent upon DNA sequence. When the template was 3′-(1,N2-εdG)TACT-5′, dATP was preferentially incorporated, whereas with the template 3′-(1,N2-εdG)CACT-5′, both dGTP and dATP were incorporated (20). The replicative human polymerase pol δ was blocked by the 1,N2-εdG adduct (21). Human polymerase ηconducts error-prone replication past 1,N2-εdG, preferentially incorporating dGTP opposite the adduct, irrespective of the identity of the base 5′ of 1,N2-εdG in the template (21). The human polymerases ℓ and κ showed similar rates of incorporation of dTTP and dCTP (21).
Presently, we have completed a detailed structural examination of the 1,N2-εdG adduct in duplex DNA, although it was reported that 1,N2-εdG is readily accommodated, causing localized structural perturbations when placed opposite dC in a DNA duplex (22). In the present work, the 1,N2-εdG adduct was inserted site-specifically into the oligodeoxynucleotide duplex 5′-d(CGCATXGAATCC)-3′•5′-d(GGATTCCATGCG)-3′ (X=1,N2-εdG), and its structure was determined using high-resolution 1H NMR spectroscopy. Since the 1,N2-εdG-containing duplex showed a mixture of conformations at neutral pH, the NMR measurements were carried out at pH 8.6, where the duplex showed single conformation. Under these conditions in duplex DNA, the 1,N2-εdG-induced structural perturbation was localized at the X6•C19 base pair, and its 5′-neighbor T5•A20. Both 1,N2-εdG and the complementary dC adopted the anti conformation about the glycosyl bonds. The 1,N2-εdG adduct was inserted into the duplex but was shifted towards the minor groove as compared to dG in a normal Watson-Crick C•G base pair. The complementary cytosine was displaced toward the major groove. The 5′-neighbor T5•A20 base pair was destabilized with respect to Watson-Crick base pairing. The refined structure predicted a bend in the helical axis associated with the adduct site.
Materials and Methods
Oligodeoxynucleotide Synthesis and Characterization
The oligodeoxynucleotides 5′-d(CGCATGGAATCC)-3′ and 5′-d(GGATTCCATGCG)-3′ were synthesized and purified by anion-exchange chromatography by the Midland Certified Reagent Company (Midland, TX). The phosphoramidite of 1,N2-εdG (23) was incorporated into 5′-d(CGCATXGAATCC)-3′ (X=1,N2-εdG) oligodeoxynucleotide on an Expedite 8909 DNA synthesizer (PerSeptive Biosystems) with tert-butylphenoxyacetyl-protected cyanoethyl phosphoramidites on a 1-μmol scale using the manufacturer’s standard synthetic protocols. The modified oligodeoxynucleotide was cleaved from the solid support and the exocyclic amino groups deprotected in a single step using 0.1 M aq NaOH at room temperature overnight. The deprotection reaction was neutralized and the oligodeoxynucleotide purified by HPLC using gradient 2 (vide infra). The oligdeoxyonucleotide was salted and desalted with a Sephadex G-25 column using a BioRad FPLC system and analyzed by CGE (99.5%), MALDI-TOF MS, and enzymatic digestion. 5′-d(CGC ATX GAA TCC)-3′, MALDI-TOFMS: calcd for [M - H]- m/z 3652.6, found m/z 3653.0.
HPLC
The oligodeoxynucleotide purification and enzymatic digestion analysis were performed on a Beckman HPLC system (32 Karat software version 5.0, pump module 125) with a diode array UV detector (module 168) monitoring at 260 nm using Waters YMC ODS-AQ columns (250 mm × 4.6 mm i.d., 1.5 mL/min for enzymatic digestion gradient 1 and 250 mm × 10 mm i.d., 5 mL/min for purification of oligonucleotides gradient 2) 0.1 M aqueous ammonium formate (A) and acetonitrile (B). HPLC Gradient 1: Initially 1% acetonitrile; then a 15 min linear gradient to 10% acetonitrile; a 5 min linear gradient to 20% acetonitrile; 5 min isocratic at 20% acetonitrile; 3 min linear gradient to 80% acetonitrile; 2 min isocratic at 80% B; then a 3 min linear gradient to initial conditions. Gradient 2: initially 1% acetonitrile; then a 5 min linear gradient to 8% acetonitrile; 15 min linear gradient to 11% acetonitrile; 2 min linear gradient to 80% acetonitrile; 1 min isocratic at 80% acetonitrile; then a 2 min linear gradient to initial conditions.
Capillary Gel Electrophoresis
Electrophoretic analyses were carried out using a Beckman P/ACE MDQ instrument system (32 Karat software, version 7.0) monitored at 260 nm on a 31.2 cm × 100 μm eCAP capillary with samples applied at 10 kV and run at 9 kV. The capillary was packed with the manufacturer’s 100-R gel (for ss-DNA) using a Tris-borate buffer system containing 7 M urea.
Enzymatic Digestion
Enzymatic hydrolysis was carried out in one step. Oligodeoxynucleotide (0.5 A260 units) was dissolved in 30 μL buffer (pH 7.0, 10 mM Tris-HCl, 10 mM MgCl2) and incubated with DNase I (8 units, Promega), snake venom phosphodiesterase I (0.02 units, Sigma), and alkaline phosphatase (1.7 units, E. coli; Sigma) at 37 °C for 24 h. The mixture was analyzed by reverse phase HPLC gradient 1. Adducted nucleosides were identified by comparison with authentic samples based on retention times, co-injection, and UV spectrum.
Mass Spectrometry
MALDI-TOF mass spectra (negative ion) of modified oligodeoxynucleotides were obtained on a Voyager Elite DE instrument (Perseptive Biosystems) at the Vanderbilt Mass Spectrometry Facility using a 3-hydroxypicolinic acid matrix containing ammonium hydrogen citrate (7 mg/mL) to suppress multiple sodium and potassium adducts.
Thermal Denaturation Studies
Experiments were carried out on a Cary 4E spectrophotometer (Varian Associates, Palo Alto, CA). The samples contained 0.24 A260 unit of duplex oligodeoxynucleotide dissolved in 1 mL of buffer containing 10 mM Na2HPO4/NaH2PO4, 1.0 M NaCl, and 5 μM Na2EDTA (pH 7.0). The UV absorbance at 260 nm was monitored at 1 min intervals with a 2.5 °C/min temperature gradient. The temperature was cycled between 20 and 85 °C. The first derivative of the melting curve was used to establish the Tm values.
NMR Sample Preparation
Oligodeoxynucleotide concentrations were determined from UV spectrophotometer using calculated extinction coefficients at 260 nm (24). The modified oligodeoxynucleotide and its complimentary strand were annealed in a 1:1 molar ratio, in aqueous solution, and the resulting DNA duplex was eluted from DNA Grade Biogel hydroxyapatite (Bio-Rad Laboratories, Richmond, CA) buffer at pH 7.0. It was then desalted using a gradient from 10 to 100 mM NaH2PO4 using Sephadex G-25. The sample was lyophilized and re-dissolved in 500 μL of aqueous buffer containing 10 mM NaH2PO4 at pH 7.
Nuclear Magnetic Resonance
One- and two-dimensional NMR spectra were recorded on Bruker (AMX) spectrometers operating at 600 and 800 MHz, at 7 ºC. Samples were dissolved to a duplex concentration of 1 mM in 500 μL of 10 mM NaH2PO4, 100 mM NaCl, and 5 μM Na2EDTA (pH 7.0). Samples for the observation of non-exchangeable protons were exchanged with D2O and suspended in 500 μL of 99.99% D2O. Samples for the observation of exchangeable protons were dissolved to a concentration of 1 mM in 500 μL of 1 mM NaH2PO4, 100 mM NaCl, and 5 μM Na2EDTA containing 9:1 H2O:D2O. The pH of the NMR samples was adjusted to 8.6 by titration with NaOD. Chemical shifts of the proton resonances were referenced to water. 1H NOESY experiments in D2O were conducted at 7 ºC. For each t1 increment, 32 scans were averaged with pre-saturation of the HDO resonance. To derive distance restraints, spectra were recorded consecutively using States-TPPI phase cycling with mixing times of 70, 150, 200 and 250 ms. These were recorded with 2048 complex points in the acquisition dimension and 1024 real data points in the indirect dimension covering 9615.385 Hz. The relaxation delay was 1.5 s. The data in the d2 dimension were zero-filled to give a matrix of 2K × 2K real points. NOESY spectra for observation of exchangeable protons were recorded at 7 °C, in 9:1 H2O:D2O, using a field gradient Watergate pulse sequence (25) for water suppression. The spectra, consisting of 128 transients, were obtained with a cryogenic probe using States-TPPI phase cycling with mixing time 250 ms. A squared sine-bell with 72º shift apodization was applied in d1 dimension while cosine-squared bell apodization was applied in d2 dimension. A total of 1536 real data points in the d1dimension and 512 points in d2 dimension were acquired. Double quantum-filtered 1H correlation (DQF-COSY) spectra (26, 27) were collected at 25 °C with 2048 complex points in the acquisition dimension and 512 points in the d2 dimension covering 6009.615 Hz and zero-filled to 1024 points to give a matrix of 1024 × 2048 real points. For each d1 increment, 64 or 84 transients were averaged with pre-saturation of the HDO resonance. A squared sine-bell apodization function was applied in both dimensions. Chemical shifts of proton resonances were referenced to water. NMR data were processed on Silicon Graphics Octane workstations and assigned using FELIX2000 (Accelrys, Inc., San Diego, CA).
Distance Restraints
The volumes of NOE cross-peaks (drawn as square boxes) for the NOESY spectrum recorded at a mixing time of 250 ms were obtained using the program FELIX2000. Similarly, the volumes were measured from the NOESY spectra recorded at other mixing time of 200 and 150 ms. The NOE-derived distances were obtained from the NOE volumes using the program MARDIGRAS v5.2 (28). The RANDMARDI algorithm (29) carried out 50 iterations for each set of data, randomizing peak volumes within limits specified by the input noise level. The molecular motion was assumed to be isotropic. The volume error was one-half the volume of the weakest cross peak. Calculations were performed using DNA starting structures generated using the program INSIGHT II (Accelyris), and NOE intensities derived from experiments at three mixing time, and with three isotropic correlation times (2, 3 and 4 ns), yielding 18 sets of distances. Analysis of these data yielded the experimental distance restraints and standard deviations for the distance restraints used in subsequent restrained molecular dynamics calculations. For partially overlapped crosspeaks, the upper bounds on the distances were increased.
Restrained Molecular Dynamics
The rMD calculations were carried out with the program AMBER 8.0 (30) using a simulated annealing protocol (vide infra). Classical B-DNA and A-DNA (31) were used as the reference structures to create starting structures for the refinement. The 1,N2-εdG adduct was constructed in both B-DNA and A-DNA forms by bonding the etheno group to N1 and N2 at G6 using the program INSIGHT II. The coordinates, connectivity and parameters for the current models were obtained from the program xLEaP. The restrained electrostatic potential (RESP) charges for the 1,N2-εdG adduct were calculated using the program GAUSSIAN98 (32). The input data for the GAUSSIAN98 calculations were obtained from the program ANTECHAMBER (33). The total charge on the 1,N2-εdG nucleotide was maintained at -1. Initially constructed A and B-DNA starting structures were energy-minimized by the conjugate gradients method for 250 iterations using the AMBER 8.0 force field to relieve poor van der Walls contacts using the program SANDER (34). Five hundred cycles of potential energy minimization using the steepest descent algorithm were used. The energy minimization was performed without experimental restraints to give the starting IniA and IniB structures used for the subsequent relaxation matrix analysis and molecular dynamics calculations. In subsequent rMD calculations, the restraint energy function included terms describing distance restraints as square-well potentials. The generalized Born solvent model was used for rMD simulated annealing calculations with 0.1 M salt concentration, and the SHAKE algorithm was used (35, 36). The van der Waals energy term used the Lennard-Jones potential energy function. The electrostatic term used the Coulomb function, based on a full set of partial charges (-1 per residue) and a distance-dependent dielectric constant of 4r. The nonbonded pair list was updated if any atom moved more than 0.5 Å, and the cutoff radius for nonbonded interactions was 18 Å. The effective energy function included terms describing distance restraints, in the form of square-well potentials. The partial charges assigned to the 1,N2-εdG adduct by RESP protocol are shown in Figure S2 of the Supporting Information. Calculations were initiated by coupling to a heating bath, with a target temperature of 600 K. The force constants were 20 kcal mol−1 Å−2 for empirical hydrogen bonding and 32 kcal mol−1 Å−2 for class 1, class 2, class 3, class 4, and class 5 NOE restraints. Those force constants were increased up 1.5 times during first 3 ps of heating, and reduced back to original values during the rest of calculations. The target temperature was reached and maintained for 5 ps. The molecules were cooled to 100 K over 12 ps for equilibrium dynamics. During the final 8 ps the temperature was reduced to 0 K. Coordinate sets were archived every 0.2 ps, and 10 structures from the last 5 ps were averaged in total. An average structure was subjected to 500 iterations of conjugate gradient energy minimization to obtain the final structure.
Complete Relaxation Matrix Calculations
The NOE intensities obtained from the structures emergent from rMD simulations were compared with the experimental intensities using CORMA v. 5.3 (37). Input volumes (intensities) were normalized for the intensities of protons with fixed internuclear distances (cytosine H5-H6 protons). An isotropic correlation time (τc) of 3 ns was used. The rotation of thymidine CH3 groups was modeled using a 3-jump site model. The sixth root residual (R1 x) factor was calculated for the averaged structure. Helicoidal parameters were examined using the program CURVES (38, 39).
Results
Characterization of the Modified Duplex
At neutral pH, the melting point (Tm) of the modified duplex was 39 °C in 10 mM NaH2PO4, 100 mM NaCl, 5 μM Na2EDTA. The corresponding Tm for the unmodified duplex under the same conditions was 53 °C. Thus, at neutral pH, the 1,N2-εdG induced a 14 °C drop in Tm. It was not possible to examine the1,N2-εdG adduct positioned opposite dC in the 5′-TXG-3′ sequence at neutral pH by NMR due to the existence of broad peaks in the modified region of the duplex. This was attributed to the presence of multiple conformations of the 1,N2-εdG adduct at neutral pH. However, it was found that increasing the pH of the sample to pH 8.6 yielded a well-resolved NMR spectrum. Therefore, the work described focuses on the conformation of the 1,N2-εdG adduct at pH 8.6.
The stability of the 1,N2-ε values of dG adduct was monitored at pH 8.6. The pKa values of the 1,N2-εdG nucleoside were reported to be at pH 9.2 and 2.2 and its fluorescence spectrum was reported at pH 12, and the adducted nucleoside was reported to be stable as the exocyclic adduct (13). The deglycosylation rate in 0.1 N HCl was slower than dG (13). These observations were consistent with the reported formation of the 1,N2-εdG adduct from dibromoethanol at pH 9.2 (14). However, when the 1,N2-εdG adduct was incubated for 7 days at pH 9.2 and monitored by NMR, the H7 proton slowly exchanged, suggesting that there was etheno ring opening and closing, though the opened form did not accumulate (14). This would be anticipated to occur slowly at basic pH. At the higher pH, the adducted oligodeoxynucleotide duplex was further destabilized with respect to DNA melting, as compared to neutral pH. Thus, the Tm decreased to 35 °C in buffer containing 10 mM NaH2PO4, 100 mM NaCl, 5 μM and Na2EDTA. At pH 8.6, the NMR data indicated that the 1,N 2-εdG adduct contained within the oligodeoxynucleotide duplex existed as the exocyclic adduct.
Resonance Assignments
a) Non-exchangeable DNA Protons
The sequential assignment for the 1,N2-εdG duplex was accomplished using standard protocols (40, 41). An expanded plot of 1H NOE sequential connectivities between the base protons (7.0– 8.6 ppm) and the sugar H1′ and dC H5 protons (5.0–6.4 ppm) is plotted in Figure 1. It exhibited well-resolved cross peaks. Seven strong NOE peaks accounted for all cytosine residues of the duplex. These assignments were confirmed by a COSY spectrum recorded under identical conditions. Each base proton exhibited NOE peaks to its own and 5′-flanking H1′ deoxyribose protons (Figure 1A and 1B). The NOESY connectivities were continuous from C1→C12 in the 1,N2-εdG containing strand and from G13→G24 in the complimentary strand. For T5, X6, and G7, the NOE cross peak intensities between the base protons and the sugar H1′ protons of the attached deoxyribose moieties were of the same relative magnitudes as those between other bases and the sugar H1′ protons of the attached deoxyribose moieties (Figure S1 in the Supporting Information). The T5 H1′→X6 H2 and the C19 H1′→A20 H8 sequential NOE cross peaks were weak (Figure 1A and 1B). Sequential NOE peaks were observed for the A4 H2→T5 H1′, A8 H2→A9 H1′, A9 H2→T10 H1′, A15 H2→T16 H1′, and A20 H2→T21 H1′ protons (peaks A, B, and C Figure 1A, and G and H, Figure 1B, respectively) and G2 H8→C3 H5, T10 H6→C11 H5, C11 H6→C12 H5, T17 H6→C18 H5, and G22 H8→C23 H5 protons (peaks D, E, and F, Figure 1A and I and J, Figure 1B, respectively). The NOE cross peak for C18 H6→C19 H5 overlapped with that of C18 H6→C18 H5. All the NOE cross peaks between A4 H2→A4 H1′, A8 H2→A8 H1′, A9 H2→A9 H1′, A15 H2→A15 H1′, and A20 H2→A20 H1′ were observed (Figure 1). The normally observed NOE sequential connectivities between H2′ and H2″ protons and the 3′-neighbor nucleotide H6 or H8 protons were observed from C1→C12 and G13→G24. The chemical shifts of all of the H2′ and H2″ protons were in the anticipated 1.6–2.8 ppm range. These H2′ and H2″ proton resonances were identified by comparing their NOE intensities at a mixing time of 150 ms. The stronger peaks were attributed to H1′→H2′ correlations and the weaker peaks to H1′→H2″ NOEs. The H2″ proton was consistently found to resonate downfield of the H2′ proton. The H3′ resonance assignments were obtained from NOEs to the nucleobase H6 or H8 (in the case of X6, H2; Scheme 1) protons. Sequential NOE connectivities were observed from aromatic protons to their 3′-neighboring aromatic protons for nucleotides C1→C12 in the modified strand and for nucleotides G13→G24 in the complimentary strand. In the complimentary strand, the C19 H6→A20 H8 NOE was weak. In the 70 ms mixing time NOESY spectrum, weak intra-nucleotide connectivities were detected between the base and H1′ protons. The assignments of the nonexchangeable protons are listed in Table S1 of the Supporting Information.
Figure 1.
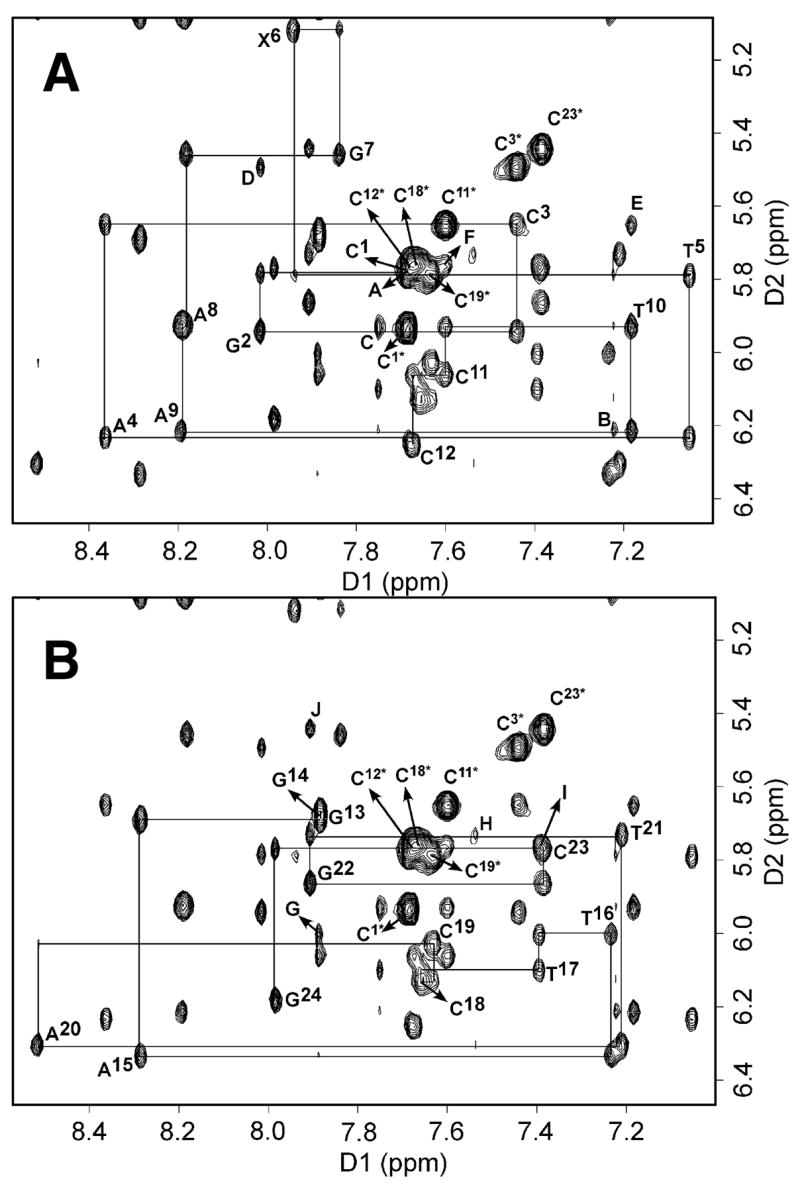
1H NOESY spectra showing sequential NOE connectivity from anomeric H1′ to 3′-neighbor aromatic protons for the modified and complimentary strands of the 1,N2-εdG oligonucleotide duplex. The spectra were recorded at 7 °C, at 250 ms mixing time (pH 8.6). A. The 1,N2-εdG modified strand. Labeled peaks are A, A4 H2→T5 H1′; B, A8 H2→A9 H1′; C, A9 H2→T10 H1′; D, G2 H8→C3 H5; E, T10 H6→C11 H5; and F, C11 H6→C12 H5. B. The complimentary strand. Labeled peaks are G, A15 H2→T16 H1′ H, A20 H2→T21 H1′, I, T17 H6→C18 H5 and J, G22 H8→C23 H5. Asterisks indicate the cytosine H5-H6 cross peaks.
Scheme 1.
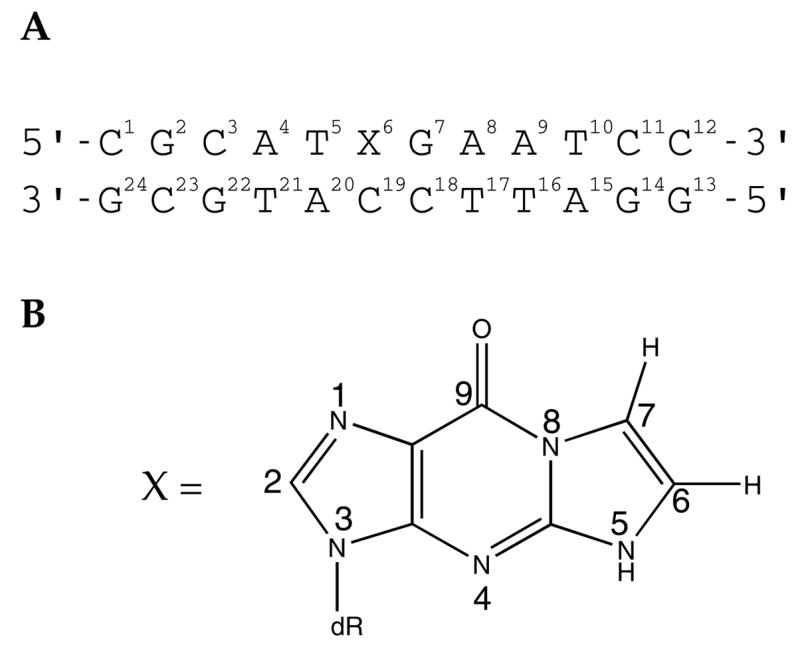
A. 1,N2-εdG-Modified Oligodeoxynucleotide Numbering Scheme. B. Structure and Numbering Scheme for 1,N2-εdG Adduct.
1, N2-etheno-2′-deoxyguanosine (1,N 2-εdG)
*The imidazole proton of the 1,N2-εdG nucleotide is designated as H2, corresponding to the H8 proton in guanine.
(b) Exchangeable DNA Protons
The one-dimensional spectrum of the 5.8–15 ppm imino and amino proton resonance region of the 1,N2-εdG modified duplex recorded in H2O buffer, pH 8.6 at 7 °C, is shown in Figure 2A. Eight well-resolved peaks between 11.8 and 14 ppm and one broad peak at 13.4 ppm accounted for all of the imino protons of the 1,N2-εdG duplex, except the two terminal imino protons that were not observable, presumably due to fast exchange with the solvent. A contour plot of an expanded region of the NOESY spectrum showing connectivities between imino and amino protons is shown in Figure 2B. The spectrum was recorded at 7 °C. The imino protons of the 1,N2-εdG-modified duplex were assigned from NOEs between adjacent base pairs and between their corresponding base-paired amino protons (42). The assignment of all dA H2 protons, from NOESY spectra recorded in D2O buffer, allowed identification of the A•T base pairs in the duplex (Figure 2B). Similarly, the assignment of all dC amino protons, observed from their intra-residue connectivity to the H5 protons, detected in a 250 ms mixing time NOESY spectrum, allowed identification of the G•C base pairs (Figure 2B). From Figure 2C, the expected pattern of NOE cross-peaks was observed for base pairs G2•C23, C3•G22, and A4•T21, which are located in the 5′ direction from the 1,N2-εdG adduct. Likewise, NOE connectivities were observed between base pairs G7•C18, A8•T17, A9•T16, T10•A15, and C11•G14, which are located in the 3′ direction from the 1,N2-εdG adduct. A weak NOE cross peak was observed between the A4•T21 and T5•A20 base pairs when the threshold of the counter plot of NOESY spectrum was decreased. NOE cross peaks were observed between dA H2 protons and flanking base pairs at the C3•G22→A4•T21, A4•T21→T5•A20, G7•C18→A8•T17, A8•T17→A9•T16, A9•T16→T10•A15, and T10•A15→ C11•G14 steps of the duplex (Figure 2B). The T5 N3H imino resonance was broadened, presumably due to solvent exchange. The amino proton of the modified 1,N2-εdG nucleotide (Scheme 1) was not observed, suggesting rapid exchange with solvent. The resonances for the C19 amino protons were not observed, suggesting fast exchange with solvent. The amino protons of the remaining cytosines in the duplex were observed in the anticipated 6.5–8.5 ppm region. The resonance assignments for the imino and amino protons are found in Table S2 of the Supporting Information.
Figure 2.
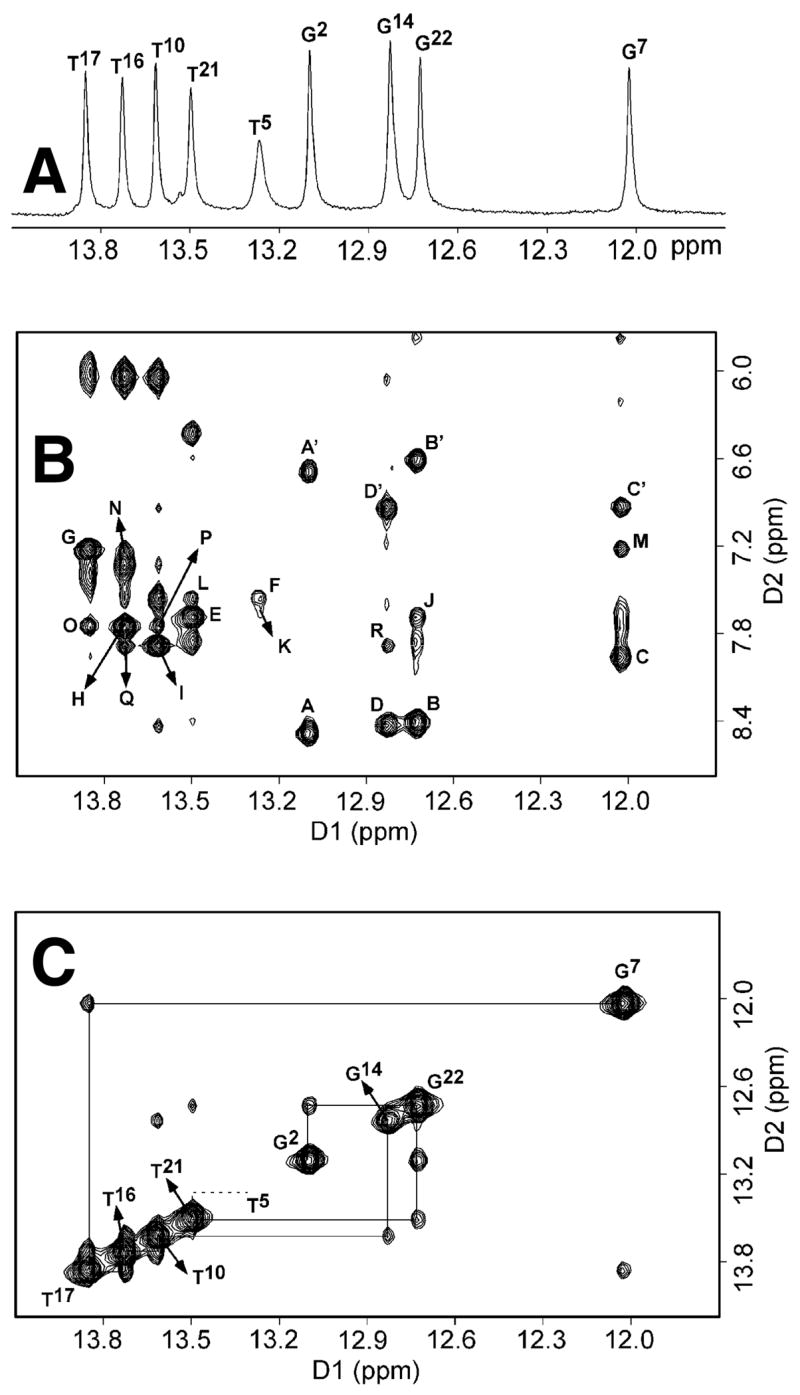
A. Downfield region of the 1H NMR spectrum. B. 1H NOESY spectrum showing the NOE connectivity between the imino protons and the amino and base protons. Labeled peaks are A,A′, G2 N1H→C23 N4H, h/n; B,B′, G22 N1H→C3 N4H, h/n; C,C′, G7 N1H→C18 N4H, h/n; D,D′, G14 N1H→C11 N4H, h/n; E, T21 N3H→A4 H2; F, T5 N3H→A20 H2; G, T17 N3H→A8 H2; H, T16 N3H→A9 H2; I, T10 N3H→A15 H2; J, G22 N1H→A4 H2; K, T5 N3H→A4 H2; L, T21 N3H→A20 H2; M, G7 N1H→A8 H2; N, T16 N3H→A8 H2; O, T17 N3H→A9 H2; P, T10 N3H→A9 H2; Q, T16 N3H→A15 H2; R, G14 N1H→A15 H2. The symbols h and n stand for hydrogen-bonded and non-hydrogen-bonded amino protons of cytosine. C. 1H NOESY spectrum showing resonances for the thymine and guanine imino protons and sequential NOE connectivity for the imino protons of the base pairs G2•C23→G14•C11 for the 1,N2-εdG oligodeoxynucleotide duplex. The labels designate the imino proton of the indicated nucleotide. The spectra were recorded in buffer solution containing 9:1 H2O:D2O at 7 °C (pH 8.6).
(c) Assignments of 1,N2-εdG Protons and NOEs to DNA Protons
Figure 3 shows the assignment of the excyclic H6 and H7 1,N2-εdG protons and also shows the NOEs between these protons and DNA. The etheno protons were identified by their strong interaction observed in all NOESY spectra, irrespective of mixing time (Figure 3). The etheno protons did not show a cross peak in COSY experiments, presumably because of their small J-coupling constant (43, 44). The unequivocal assignment of H6 and H7 was made based upon the observation that the H7 proton slowly exchanges in deuteriated solvent, as a consequence of transient opening of the etheno ring (14). This assignment could, in theory, also be made by observation of the NOE between the H6 proton and the amino proton of the 1,N2-εdG adduct. Unfortunately, it was not possible to assign the amino proton in the NMR spectra, presumably due to rapid exchange with solvent. The assignment of the H6 and H7 1,N2-εdG protons in the oligodeoxynucleotide duplex at δ 6.2 ppm and δ 6.5 ppm (with respect to the water resonance), respectively, for H6 and H7, was in agreement with their assignments at the base and nucleoside levels using 15N-labeled samples and NOE measurements (14). This suggested an absence of ring current effects in the modified duplex sufficiently strong so as to reverse the relative assignments of these two protons, which differed in chemical shift by 0.3 ppm.
Figure 3.
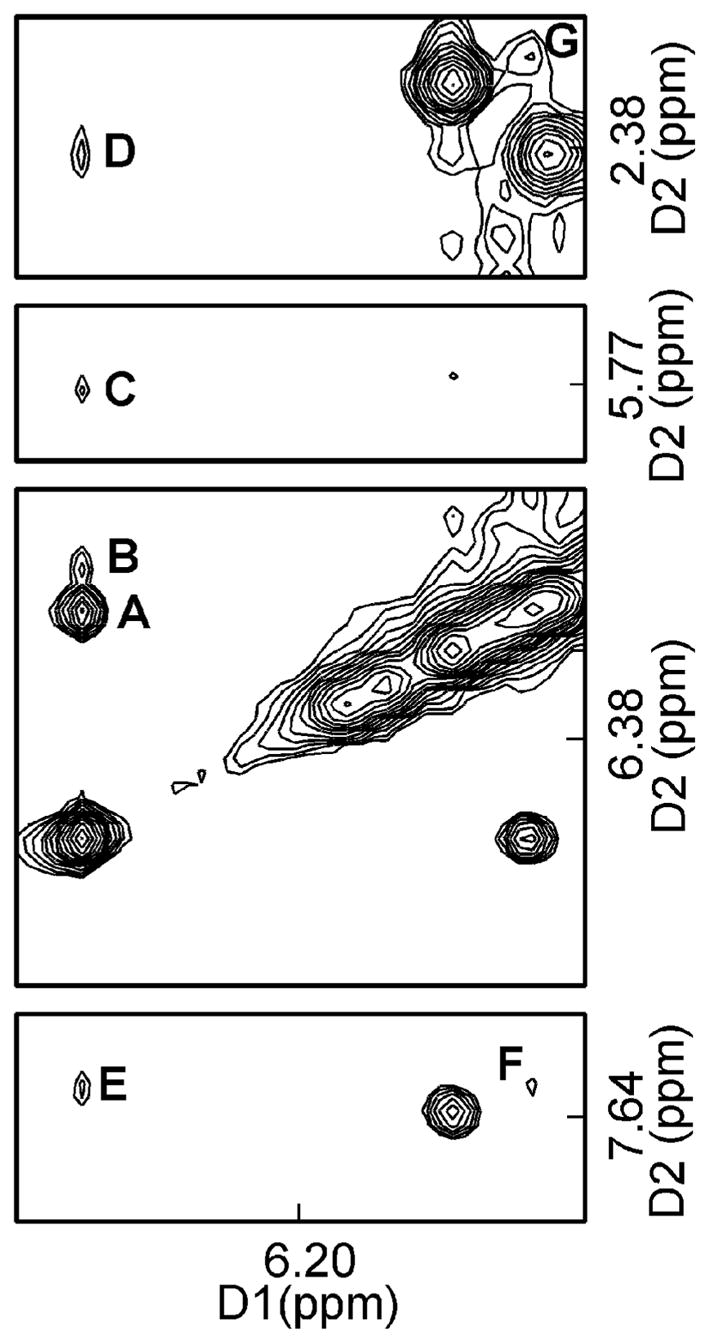
1H NOESY spectrum showing the assignment of the exocyclic 1,N2-εdG protons H6 and H7, and NOEs between the etheno and DNA protons. The assignments are A, X6 H7→X6 H6; B, X6 H7→C18 H1′; C, X6 H7→C19 H5; D, X6 H7→C18 H2″; E, X6 H7→C19 H6; F, X6 H6→C19 H6; and G, X6 H6→C19 H2″. The spectrum was recorded in 10 mM NaH2PO4, 100 mM NaCl, 5 μM Na2EDTA, at 7 °C (pH 8.6).
Phosphorus Spectra
The proton decoupled phosphorus one and two -dimensional spectra of the 1,N2-εdG duplex in D2O buffer, pH 8.6, at 25 °C, are shown in Figure 4. The 1D spectrum showed two phosphorous resonances shifted downfield when compared to the unmodified duplex. The two downfield 31P resonances were assigned to G7 and C19, from proton-decoupled 2D spectra in D2O buffer, pH 8.6 at 25 °C.
Figure 4.
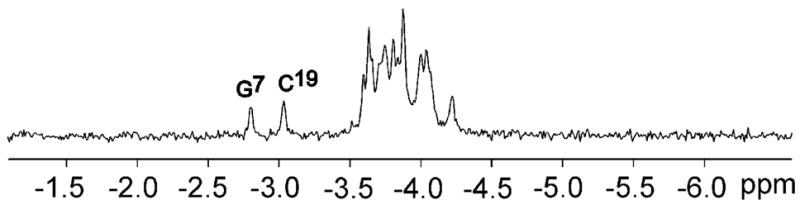
A. Proton-decoupled 31P NMR of the 1,N2-εdG-modified oligodeoxynucleotide duplex. Labeled peaks, G7 and C19, were identified from their two dimensional H3′-31P spectrum where it showed cross peak for X6 H3′→G7 31P, and C18 H3′→C19 31P. The spectrum was recorded at 25 °C in 10 mM NaH2PO4, 100 mM NaCl, and 5 μM Na2EDTA (pH 8.6).
Chemical Shift Comparisons
Figure 5 shows chemical shift differences between the 1,N2-εdG duplex and the corresponding unmodified duplex at pH 8.6. Major chemical shift changes were observed for the X6 H1′, X6 H2′, X6 H2″, and C19 H1′ resonances, as compared to their corresponding resonances in the unmodified duplex. The maximum change among these protons was observed for C19 H1′, which shifted downfield by 0.6 ppm.
Figure 5.

Chemical shift differences observed for 1,N2-εdG-modified duplex when compared to the corresponding unmodified duplex at pH 8.6. Δδ=[δunmodified oligonucleotide − δmodified oligonucleotide] (ppm).
Experimental and Empirical Restraints
A total of 678 restraints, which included distance, dihedral angle, sugar pucker, and Watson-Crick hydrogen bonding restraints, were used in the rMD calculations. A total of 385 NOE based distance restraints were obtained, consisting of 245 intra- and 140 inter-nucleotide distances. There was an average of approximately 15 distance restraints per nucleotide. The distribution of NOE restraints used in the rMD calculations is shown in Table S3 of the Supporting Information. Watson-Crick hydrogen bonding restraints were used for all the base pairs except for the X6•C19 base pair. The rationale for their inclusion was based on the NOESY experiments in H2O buffer in which the observation of imino and amino proton resonances was consistent with a right handed Watson-Crick paired duplex (Figure 3). Empirical sugar puckering restraints were not used for the X6•C19 base pair as the pattern of chemical shift perturbations (Figure 5) suggested a structural disturbance at the X6 and C19 nucleotides. Similarly, no empirical backbone torsion angles restraints were used for the X6•C19 base pair. Elsewhere, the backbone angles α, β, γ, δ, ε, and ζ were restrained to −60 ± 30°, 180 ± 30°, 60 ± 30°, 120 ± 30°, 180 ± 30°, and −90 ± 30°, respectively, to allow both A- and B-form DNA structures. A total of 23 distance restraints, including 11 intranucleotide, 6 internucleotide, and 6 internucleotide cross-strand distance restraints, respectively, were employed at nucleotide X6, which allowed the position of X6 in the duplex to be determined with confidence. Similarly, for the C19 nucleotide, 20 intranucleotide, internucleotide, and internucleotide cross-strand distance restraints were utilized, which allowed the C19 nucleotide to be positioned with confidence.
Structural Refinement
Randomly seeded rMD calculations were initiated from both A-form and B-form DNA starting structures, which had been energy minmized. The complementary base C19 was located in the center of the helix prior to energy minimization, but shifted toward the major groove as a consequence of the potential energy minimization calculations, which were carried out using the conjugate gradients algorithm. Subsequent restrained molecular dynamics calculations initiated from either starting structure (IniA and IniB) converged toward B-type DNA helices. A total of ten structures were calculated from each starting structure.
The precision of the rMD calculations was determined by pairwise rmsd calculations (Table 1). The rmsd between the set of structures emergent from the A-form vs. those emergent from the B-form calculations was 1.34 Å. Comparison of the individual sets of structures derived from A-form and B-form DNA between themselves resulted in rmsd values of 0.52 and 0.76 Å for A-form and B-form DNA, respectively. Comparison of starting structures with the average structures, which emerged from the rMD calculations starting from either, revealed that the rMD protocol resulted in a greater conformational change for the A-form starting structure than for B-form starting structure. Hence, the final structures were closer to B-form DNA geometry than A-form geometry. However, the B-form starting structure differed from the structures emergent from the rMD calculations, evidenced by an rmsd of 3.12 Å. This was attributed to the distortion of the duplex induced by the 1,N2-εdG adduct, and the observation of a bend near the adducted site.
Table 1.
Analysis of the rMD-Generated Structuresa of the 1,N2-εdG Adduct in the 5′-TXG-3′ Sequence at pH 8.6.
| NMR Restraints | |
| total number of distance restraints | 385 |
| Interresidue distance restraints | 140 |
| Intraresidue distance restraints | 245 |
| empirical restraints | |
| hydrogen bonding restraints | 45 |
| sugar pucker restraints | 100 |
| backbone torsion angle restraints | 148 |
| Initial structures | Atomic rmsd (Å) |
| IniA vs IniB | 5.77 |
| Rms shifts | |
| IniA vs <rMDA>b | 4.18 |
| IniB vs <rMDB>b | 3.12 |
| Rms distributions | |
| <rMDA> vs <rMDA> | 0.52 |
| <rMDB> vs <rMDB> | 0.76 |
| <rMDA> vs <rMDB> | 1.34 |
| <rMDA> vs rMDc | 0.26 |
| <rMDB> vs rMDc | 0.38 |
Root Mean Square Deviations (rmsd), Excluding the End Base Pairs, Between Various Initial Structures and Averaged Structures Emergent from rMD.
<rMDA> and <rMDB> represents the set of 5 structures that emergent from rMD calculations starting with IniA and IniB, respectively.
rMD represents the average minimized structure from all 10 rMD calculations
Figure 6 shows an ensemble of six structures of 1,N2-εdG duplex obtained from randomly seeded rMD calculations. The maximum pairwise rmsd of 0.91 Å was obtained, suggesting convergence. The final rMD structure was obtained by averaging the coordinates from 10 rMD structures using the program SUPPOSE, followed by 250 iterations of potential energy minimization using the conjugate gradient algorithm. The accuracies of these structures were verified using the program CORMA (37). This calculation was based on the comparison of the experimental NOE intensities with the NOE intensities calculated from CORMA. A plot of distribution of the sixth-root R factor, R1 x, measured from the CORMA calculation is shown in Figure 7. The overall intra- and inter-residue sixth root residual R1xvalues for 1,N2-εdG duplex structures were 9.63 × 10−2 and 1.14 × 10−2, respectively.
Figure 6.
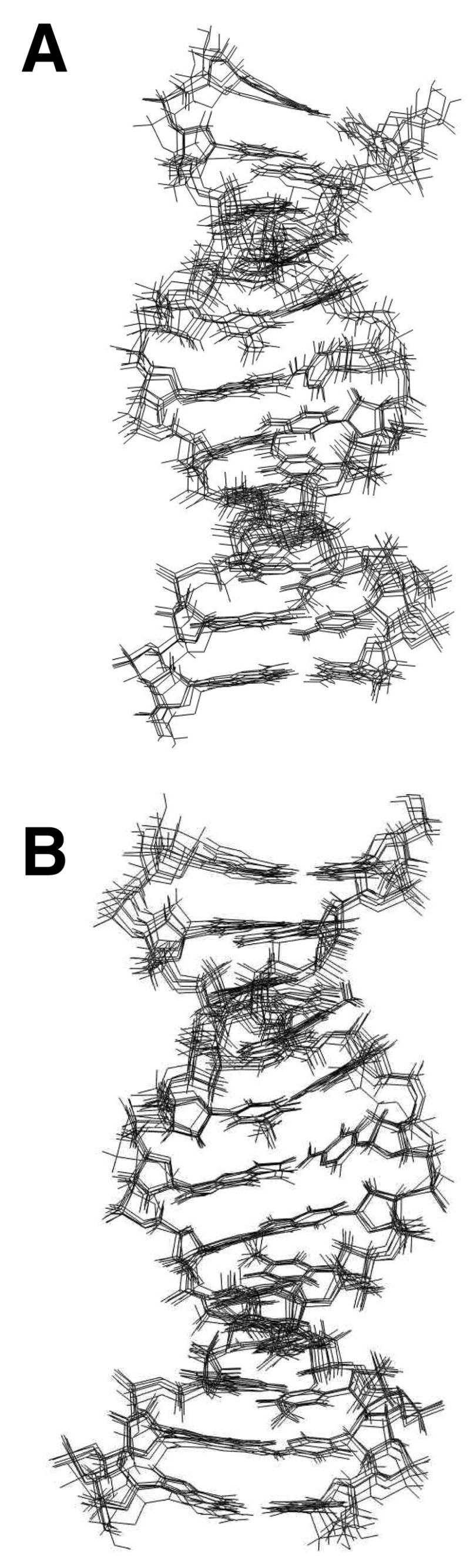
Superpositions of structures emergent from rMD calculations for the 1,N2-εdG-modified duplex at pH 8.6. A. Using a B-form DNA starting structure. B. Using an A-form DNA starting structure.
Figure 7.

Intra- (black) and inter-nucleotide (gray) sixth root R1x factors as a function of nucleotide position in the 1,N2-εdG duplex. A. The modified strand. B. The complementary strand.
A view normal to the helix axis and looking into the major groove of the central base pair segment (5′-T5X6G7-3′•-5′-C18C19A20-3′) is shown in Figure 8. The 1,N2-εdG adduct adopted the anti conformation about the glycosyl bond. The 1,N2-εdG ring was partially intercalated between T5 and G7. The 1,N2-εdG H6 etheno proton faced away from the center of the helix, whereas, the 1,N2-εdG H7 etheno proton faced towards center of the helix. The 1,N2-εdG adduct shifted in position toward the minor groove, whereas the complementary C19 shifted toward the major groove. Consequently, the X6 amino proton was oriented toward the minor groove, whereas the C19 amino protons were oriented toward the major groove. The helical rise between base pairs T5•A20 and X6•C19 was increased. The calculated 1,N2-εdG duplex structure exhibited a bend at the adduct site.
Figure 8.
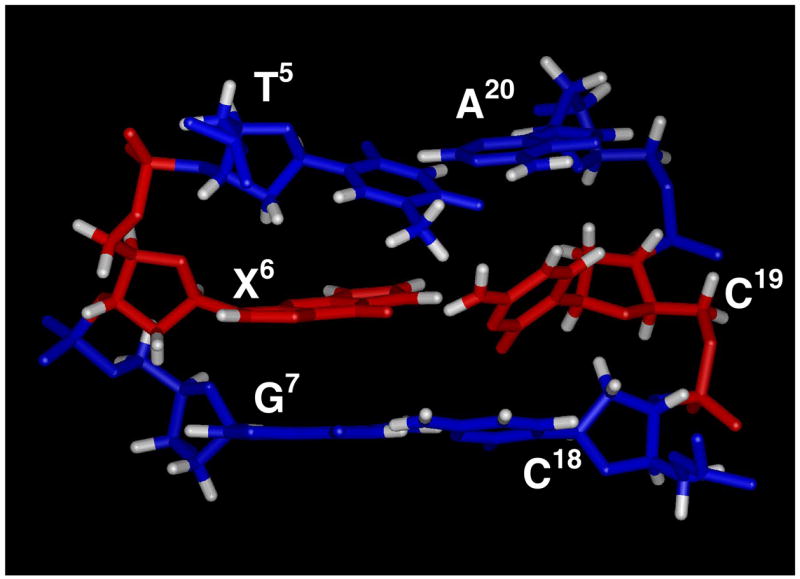
Stereoview of the central segment 5′-TXG-3′ of the 1,N2-εdG-modified 37 duplex as seen from the major groove.
Discussion
The structure of the 1,N2-εdG adduct is of interest due to the fact that etheno DNA adducts (1) arise both from exogenous exposures to vinyl halides and other vinyl monomers (2–9), and also as a consequence of endogenous exposure to lipid peroxidation products (9–11). The exocyclic etheno ring in the 1,N2-εdG adduct disrupts Watson-Crick base pairing, and, as anticipated, the 1,N2-εdG adduct is observed to be mutagenic, both in Escherichia coli (17) and in mammalian (18) cells. Presently, we have determined the solution structure of 1,N2-εdG adduct placed in the 5′-TXG-3′ sequence.
Structure of the 1,N2-εdG Duplex at pH 8.6
The refined structures calculated from rMD calculations indicate that nucleotides X6 and C19 both adopt the anti conformation about the glycosyl bond at the adduct site. This is corroborated by the similar intensities of the intranucleotide NOE peaks between all of the base and H1′ sugar protons (Figure 2S, in the Supporting Information). The X6 nucleotide is inserted into the helix but shifts toward the minor groove, while the complementary nucleotide C19 shifts towards the major groove. The pattern of NOEs between the εdG H6 and H7 protons and the DNA are consistent with the displacement or extrusion of C19 toward the major groove. The positioning of the 1,N2-εdG adduct is consistent with the pattern of NOEs arising from the H6 and H7 etheno protons, which showed few NOE cross peaks with DNA protons. The etheno protons are located toward the center of the helix and show internucleotide cross-strand NOEs to the C18 and C19 nucleotides (Figure 8). The observation that the H7 proton showed more cross peaks than did the H6 proton was consistent with the conclusion that the H7 proton was closer to the center of the helix than was H6. This supports the conclusion that that the 1,N2-εdG ring is inserted into the helix. As discussed, the assignment of the εdG H6 and H7 resonances was based upon extrapolation from unequivocal NMR assignments at the base and nucleoside levels (14). The relative chemical shifts of H6 and H7, which differ by 0.3 ppm, could be perturbed by the presence of strong ring-current shielding effects present in the oligodeoxynucleotide. However, the adducted base X6 does not show strong stacking with either of the flanking base pairs, X6•C19 and T5•A20, which can be observed in Figures 8 and 9. Additionally, the refined structures suggest that the X6•C19 and T5•A20 base pairs are ~5.0 Å apart, greater than the expectation distance of 3.4 Å in B- form DNA. Consequently, ring-current effects arising from G6 and A20 at the insertion site are anticipated to be small, and this is corroborated by the observation that the relative chemical shifts of the εdG H6 and H7 protons, which differ in value by only 0.3 ppm, remain the same as is observed at the nucleoside level (14).
Figure 9.
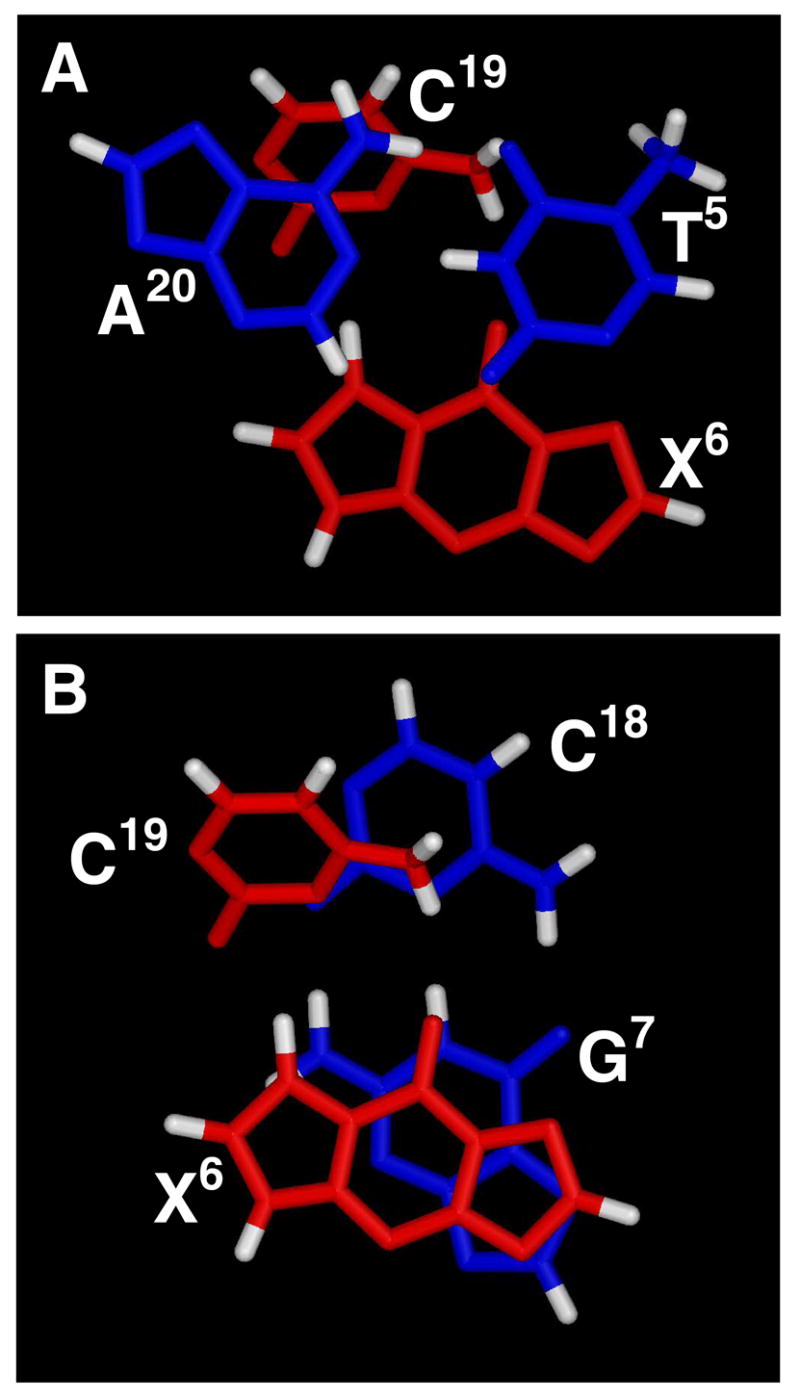
Stereoview from the top of the helix showing stacking interactions on the central segment of the duplex. A. The interaction between the T5•A20 and X6•C19 base pairs. B. The interaction between the X6•C19 and G7•C18 base pairs.
Watson-Crick base alignment is maintained for the base pairs at either side of the lesion, as indicated by the NOEs between dG N1H imino protons and hydrogen-bonded and non-hydrogen-bonded dC amino protons, and the dT imino (N3H) and dA H2 protons (Figure 2). The T5 N3H chemical shift, its NOE contacts, and the refined structure all indicate that it remains (on average on the NMR time scale) Watson-Crick hydrogen bonded. Nevertheless, the data indicate that the T5•A20 base pair is somewhat disrupted by the presence of the neighboring X6•C19 base pair, and consequently, on the NMR time scale, the T5 imino proton undergoes more rapid exchange with solvent. This is supported by the somewhat upfield shift of and broadening of the T5 imino resonance in Figure 2, and the observation of a broad cross-peak between T5 N3H and A20 H2 in the complementary strand. The greater-than-normal calculated distances between T5 H1′ and X6 H2 (the X6 imidazole proton), between C18 H1′ and C19 H6, and between C19 H1′ and A20 H8 are consistent with the weaker T5 H1′→X6 H2, C18 H1′→C19 H6, and C19 H1′→A20 H8 NOEs, while normal NOE intensity is observed for the X6 H1′→G7 H8 and C18 H1′→C19 H6 correlation (Figure 3).
The uninterrupted sequential NOESY connectivities between the aromatic base protons and the anomeric H1′ protons for nucleotides C1→C12 of the modified strand, and for nucleotides G13→G24 of the complementary strand support the conclusion that the oligodeoxynucleotide duplex containing the 1,N2-εdG adduct inserted into the 5′-TXG-3′ sequence adopts a right-handed helical DNA structure at pH 8.6. This is confirmed from the uninterrupted sequential NOESY connectivities between aromatic base protons and the sugar H2′ and H2″ protons, and between the aromatic base protons for both the modified and the complimentary strand. Thus, the structural perturbation induced by the insertion of 1,N2-εdG lesion is localized at base pair X6•C19. This conclusion is supported by the chemical shift differences (Figure 5), which show significant changes only for the X6 H1′, H2′, H2″, and C19 H1′ protons.
Comparisons to 1,N2-εdG-Modified Primer-Template Complexes in the Presence of the S. solfataricus DNA Polymerase Dpo4
The exocyclic ring masks the Watson-Crick base pair coding face of dG. Moreover, it presents a sterically bulky lesion. Thus, it is not surprising that the replicative human polymerase pol δ was blocked by the 1,N2-εdG adduct (21). When primer extension was done by the S. solfataricus Y-family DNA polymerase Dpo4 in the presence of a mixture of all four dNTPs, HPLC-mass spectrometry analysis of the products led to the conclusion that this lesion bypass enzyme uses several mechanisms, including A incorporation opposite 1,N2-εdG and also a variation of dNTP-stabilized misalignment, to generate both base-pair and frameshift mutations (20). On the other hand, the Dpo4 polymerase successfully inserts C opposite the 1,N2-εdG adduct approximately 18% of the time (20). While the similarities and differences between adduct conformations in duplex DNA and in complexes with specific DNA polymerases are not well understood, significantly, in each of these complexes with the Dpo4 enzyme, the 1,N2-εdG adduct also adopted the anti conformation about the glycosyl bond (20). Although at neutral pH when placed opposite dC in duplex DNA, multiple conformations are observed for the 1,N2-εdG adduct, it seems plausible that the Dpo4 enzyme selects for the anti conformation about the glycosyl bond during trans-lesion synthesis. The human Y-family polymerase ηconducted error-prone replication past 1,N2-εdG, preferentially incorporating dGTP opposite the adduct, irrespective of the identity of the base 5′ of 1,N2-εdG in the template (21). Obtaining structures of the 1,N2-εdG adduct in complex with human bypass polymerases will be of considerable future interest in delineating specific mechanisms by which this adduct induces mutations in human DNA.
Implications for Recognition of the 1,N2-εdG Adduct by DNA Repair Complexes
The presence of multiple conformations of the 1,N2-εdG adduct when placed opposite dC in duplex DNA at neutral pH and the prediction of an apparent bend in the duplex by the present rMD calculations on the pH 8.6 conformation of the adduct represent observations likely to be relevant with regard to the recognition of this lesion by damage-specific DNA glycosylases. The E. coli mismatch-specific uracil-DNA glycosylase and the human alkylpurine-DNA-N-glycosylase both release 1,N2-εdG adducts from DNA (45). Likewise, in mammalian cells, the alkylpurine-DNA-N-glycosylase repairs the 1,N6-εdA (46), whereas mismatch-specific thymine DNA glycosylase repairs 3,N4-εdC lesions (47). The “flipping” of damaged nucleotides out of the DNA, and into active site binding pockets, provides a common mechanism by which different DNA glycosylases interact with damaged DNA [for a review, see (48)]. It is thought that destabilization of the DNA duplex at lesion sites may play a role in damage recognition. It seems plausible that the 14 ºC decrease in Tm of the 1,N2-εdG-modified duplex, in combination with the conformational exchange in duplex DNA at neutral pH, may facilitate damage recognition, e.g., by human alkylpurine-DNA-N-glycosylase (45).
Structural Comparison to the 1,N2-Propano-2′-deoxyguanosine (PdG) Adduct
The PdG adduct provides a structural model for the six-member exocyclic 1,N2-propano lesions of dG (29), and thus provides a comparison to the five-member exocyclic 1,N2-εdG adduct. NMR studies reveal that PdG disrupts duplex DNA (39,40,43,44). At neutral pH, and similar to the present observations for the 1,N2-εdG adduct, it has not been possible to obtain a refined structure for PdG positioned opposite cytosine in duplex DNA. However, the spectrum of PdG opposite dC in duplex DNA sharpens considerably as pH is lowered (44). Likewise, when PdG is mismatched with dA in duplex DNA an ordered structure is stabilized at pH 5.8 (40,41). On the other hand, when PdG is mismatched with dG, an ordered structure is formed which is independent of pH (40). The pH dependent equilibrium observed when PdG is placed opposite from dC or dA in duplex DNA arises from the ability of the PdG base to rotate about the glycosyl bond into the syn conformation. In the syn conformation, PdG forms protonated hydrogen bonding interactions with dC and dA. When placed opposite dC, this allows formation of a protonated Hoogsteen pairing interaction (44). Preliminary data for the 1,N2-εdG adduct at pH 5.2 suggest similar behavior. Thus, the conformational interconversion at neutral pH observed in duplex DNA for the 1,N2-εdG adduct may arise from the presence of multiple rotamers about the glycosyl bond.
Comparisons to the Structures of εdC and εdA Adducts in Duplex DNA
Previous studies of etheno DNA adducts focused upon 3,N4-εdC and 1,N6-εdA. Structures of 3,N4-εdC paired with dG and in various mismatch combinations have been reported from crystallographic (49) and NMR data (43, 44, 50–52). As for the 1,N2-εdG adduct, Watson-Crick base pairing is blocked by the 3,N4-εdC adduct when paired with dG. The crystallographic data indicated the presence of a hydrogen bond involving O2 of 3,N4-εdC and N1 of dG (49). The NMR data for the 3,N4-εdC adduct yielded similar conclusions (52). The structure of the 3,N4-εdC•G pair was similar to a T•G wobble pair (49). When the εdC•G base pair was examined by NMR (43), both nucleotides were in the anti conformation about the glycosyl bond. On the other hand, in the εdC•T base pair, εdC existed in the syn orientation about the glycosyl bond, whereas the mismatched T remained in the anti conformation (43).
A structure of 1,N6-εdA opposite T in the complementary strand has been reported (53). Both nucleotides adopted glycosyl torsion angles in the anti range, and showed base stacking with flanking Watson-Crick pairs. The NMR data were consistent with a non-planar alignment of 1,N6-εdA and T (53). Additional studies of involving the 1,N6-εdA adduct mispaired with dG have been reported from crystallographic (54) and NMR (55) data. Both the crystallographic and NMR data indicated that the mispaired dG adopted the anti conformation, whereas 1,N6-εdA adopted the syn conformation about the glycosyl bond. The present structural data for the 1,N2-εdG adduct now extends our knowledge regarding the conformations of etheno adducts in duplex DNA.
Conclusions
At pH 8.6 the 1,N2-εdG adduct positioned opposite dC is inserted between the 5′-neighbor T and 3′-neighbor G in the 5′-TXG-3′ DNA sequence. Structural perturbations are localized at the X6•C19 base pair, and its 5′-neighbor T5•A20. Both 1,N2-εdG and the complementary dC adopt the anti conformation about the glycosyl bonds. The 1,N2-εdG adduct inserts into the duplex but shifts towards the minor groove as compared to dG in a normal Watson-Crick C•G base pair. The complementary cytosine is displaced toward the major groove. The 5′-neighbor T5•A20 base pair is destabilized, and the refined structure predicts a bend in the helical axis associated with the adduct site.
Supplementary Material
The Supporting Information consists of Table S1, chemical shifts of non-exchangeable protons for the 1,N2-εdG duplex; Table S2, chemical shifts of exchangeable protons for the 1,N2-εdG duplex; Table S3, distribution of experimental NOE restraints among nucleotide units of the 1,N2-εdG duplex; Figure S1, NOE cross peak intensities between the base protons and the sugar H1′ protons of the attached deoxyribose moieties; and Figure S2, partial charges assigned to the 1,N2-εdG adduct using the RESP protocol. This material is available free charge via the internet at http://pubs.acs.org.
Acknowledgments
Mr. Markus Voehler assisted with the collection of NMR data. This work was supported by NIH grants P01 ES005355 (C.J.R., and M.P.S.) and R01 ES010375 (F.P.G.). A.K.G. was supported by an NIH pre-doctoral traineeship (T32 ES007028). Funding for the NMR spectrometers was supplied by Vanderbilt University, the Vanderbilt Center in Molecular Toxicology, P30 ES000267, and by NIH grant RR005805. The Vanderbilt Ingram Cancer Center is supported by NIH grant P30 CA068485.
Footnotes
We congratulate Professor Lawrence J. Marnett upon the occasion of his sixtieth birthday.
Abbreviations: 1,N2-εdG, 3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,4-dihydro-9H-imidazo[1,2-a]purin-9-one or 1,N2-etheno-2′-deoxyguanosine; NOE, nuclear Overhauser enhancement; NOESY, two-dimensional NOE spectroscopy; COSY, correlation spectroscopy; DQF-COSY, double-quantum-filtered correlation spectroscopy; ppm, parts per million; TPPI, time proportional phase increment; 1D, one-dimensional; 2D, two-dimensional. A right superscript refers to numerical position in the sequence starting from the 5′-terminus of chain A and proceeding to the 3′-terminus of chain A and then from the 5′-terminus of chain B to the 3′-terminus of chain B. C2, C5, C6, C8, C1′, C2′, C2″, etc. represent specific carbon nuclei. H2, H5, H6, H8, H1′, H2′, H2″, etc. represent protons attached to these carbons.
References
- 1.Barbin A. Etheno-adduct-forming chemicals: From mutagenicity testing to tumor mutation spectra. Mutat Res. 2000;462:55–69. doi: 10.1016/s1383-5742(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 2.Barbin A, Bresil H, Croisy A, Jacquignon P, Malaveille C, Montesano R, Bartsch H. Liver-microsome-mediated formation of alkylating agents from vinyl bromide and vinyl chloride. Biochem Biophys Res Commun. 1975;67:596–603. doi: 10.1016/0006-291x(75)90854-2. [DOI] [PubMed] [Google Scholar]
- 3.Green T, Hathway DE. Interactions of vinyl chloride with rat-liver DNA in vivo. Chem Biol Interact. 1978;22:211–224. doi: 10.1016/0009-2797(78)90126-6. [DOI] [PubMed] [Google Scholar]
- 4.Eberle G, Barbin A, Laib RJ, Ciroussel F, Thomale J, Bartsch H, Rajewsky MF. 1, N6-etheno-2′-deoxyadenosine and 3, N4-etheno-2′-deoxycytidine detected by monoclonal antibodies in lung and liver DNA of rats exposed to vinyl chloride. Carcinogenesis. 1989;10:209–212. doi: 10.1093/carcin/10.1.209. [DOI] [PubMed] [Google Scholar]
- 5.Fedtke N, Boucheron JA, Walker VE, Swenberg JA. Vinyl chloride-induced DNA adducts. II: Formation and persistence of 7-(2′-oxoethyl)guanine and N2,3-ethenoguanine in rat tissue DNA. Carcinogenesis. 1990;11:1287–1292. doi: 10.1093/carcin/11.8.1287. [DOI] [PubMed] [Google Scholar]
- 6.Guengerich FP, Mason PS, Stott WT, Fox TR, Watanabe PG. Roles of 2-haloethylene oxides and 2-haloacetaldehydes derived from vinyl bromide and vinyl chloride in irreversible binding to protein and DNA. Cancer Res. 1981;41:4391–4398. [PubMed] [Google Scholar]
- 7.Guengerich FP, Kim DH. Enzymatic oxidation of ethyl carbamate to vinyl carbamate and its role as an intermediate in the formation of 1, N6-ethenoadenosine. Chem Res Toxicol. 1991;4:413–421. doi: 10.1021/tx00022a003. [DOI] [PubMed] [Google Scholar]
- 8.Guengerich FP. Roles of the vinyl chloride oxidation products 1-chlorooxirane and 2-chloroacetaldehyde in the in vitro formation of etheno adducts of nucleic acid bases [corrected] Chem Res Toxicol. 1992;5:2–5. doi: 10.1021/tx00025a001. [DOI] [PubMed] [Google Scholar]
- 9.Bartsch H, Barbin A, Marion MJ, Nair J, Guichard Y. Formation, detection, and role in carcinogenesis of ethenobases in DNA. Drug Metab Rev. 1994;26:349–371. doi: 10.3109/03602539409029802. [DOI] [PubMed] [Google Scholar]
- 10.Nair J, Barbin A, Guichard Y, Bartsch H. 1, N6-ethenodeoxyadenosine and 3, N4-ethenodeoxycytine in liver DNA from humans and untreated rodents detected by immunoaffinity/32P-postlabeling. Carcinogenesis. 1995;16:613–617. doi: 10.1093/carcin/16.3.613. [DOI] [PubMed] [Google Scholar]
- 11.Guichard Y, el Ghissassi F, Nair J, Bartsch H, Barbin A. Formation and accumulation of DNA ethenobases in adult Sprague-Dawley rats exposed to vinyl chloride. Carcinogenesis. 1996;17:1553–1559. doi: 10.1093/carcin/17.8.1553. [DOI] [PubMed] [Google Scholar]
- 12.Sattsangi PD, Leonard NJ, Frihart CR. 1, N2-Ethenoguanine and N2,3-ethenoguanine synthesis and comparison of electronic spectral properties of these linear and angular triheterocycles related to Y bases. J Org Chem. 1977;42:3292–3296. doi: 10.1021/jo00440a020. [DOI] [PubMed] [Google Scholar]
- 13.Kusmierek JT, Singer B. 1, N2-ethenodeoxyguanosine: properties and formation in chloroacetaldehyde-treated polynucleotides and DNA. Chem Res Toxicol. 1992;5:634–638. doi: 10.1021/tx00029a007. [DOI] [PubMed] [Google Scholar]
- 14.Guengerich FP, Persmark M, Humphreys WG. Formation of 1, N2- and N2,3-ethenoguanine from 2-halooxiranes: Isotopic labeling studies and isolation of a hemiaminal derivative of N2-(2-oxoethyl)guanine. Chem Res Toxicol. 1993;6:635–648. doi: 10.1021/tx00035a008. [DOI] [PubMed] [Google Scholar]
- 15.Guengerich FP, Persmark M. Mechanism of formation of ethenoguanine adducts from 2-haloacetaldehydes: 13C-Labeling patterns with 2-bromoacetaldehyde. Chem Res Toxicol. 1994;7:205–208. doi: 10.1021/tx00038a014. [DOI] [PubMed] [Google Scholar]
- 16.Morinello EJ, Ham AJ, Ranasinghe A, Sangaiah R, Swenberg JA. Simultaneous quantitation of N2,3-ethenoguanine and 1, N2-ethenoguanine with an immunoaffinity/gas chromatography/high-resolution mass spectrometry assay. Chem Res Toxicol. 2001;14:327–334. doi: 10.1021/tx0002076. [DOI] [PubMed] [Google Scholar]
- 17.Langouet S, Mican AN, Muller M, Fink SP, Marnett LJ, Muhle SA, Guengerich FP. Misincorporation of nucleotides opposite five-membered exocyclic ring guanine derivatives by Escherichia coli polymerases in vitro and in vivo: 1, N2-ethenoguanine, 5,6,7,9-tetrahydro-9-oxoimidazo[1,2-a]purine, and 5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-a]purine [published erratum appears in Biochemistry 1998 Jun 16;37(24):8816] Biochemistry. 1998;37:5184–1593. doi: 10.1021/bi972327r. [DOI] [PubMed] [Google Scholar]
- 18.Akasaka S, Guengerich FP. Mutagenicity of site-specifically located 1, N2-ethenoguanine in Chinese hamster ovary cell chromosomal DNA. Chem Res Toxicol. 1999;12:501–507. doi: 10.1021/tx980259j. [DOI] [PubMed] [Google Scholar]
- 19.Langouet S, Muller M, Guengerich FP. Misincorporation of dNTPs opposite 1, N2-ethenoguanine and 5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-a]purine in oligonucleotides by Escherichia coli polymerases I exo-and II exo-, T7 polymerase exo-, human immunodeficiency virus-1 reverse transcriptase, and rat polymerase beta. Biochemistry. 1997;36:6069–6079. doi: 10.1021/bi962526v. [DOI] [PubMed] [Google Scholar]
- 20.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4. Analysis and crystal structures of multiple base-pair substitution and frameshift products with the adduct 1, N2-ethenboguanine. J Biol Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 21.Choi JY, Zang H, Angel KC, Kozekov ID, Goodenough AK, Rizzo CJ, Guengerich FP. Translesion synthesis across 1, N2-ethenoguanine by human DNA polymerases. Chem Res Toxicol. 2006;19:879–886. doi: 10.1021/tx060051v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de los Santos C, Zaliznyak T, Bonala RR, Johnson F, Lukin M. Structural characterization of DNA duplexes having exocyclic 2′-deoxyguanosine adducts. The 232nd American Chemical Society National Meeting, Division of Chemical Toxicology, Trevor M Penning, Chair; San Francisco, CA. September 10–14, 2006.2006. [Google Scholar]
- 23.Goodenough AK, Kozekov ID, Zang H, Choi JY, Guengerich FP, Harris TM, Rizzo CJ. Site specific synthesis and polymerase bypass of oligonucleotides containing a 6-hydroxy-3,5,6,7-tetrahydro-9H-imidazo[1,2-a]purin-9-one base, an intermediate in the formation of 1, N2-etheno-2′-deoxyguanosine. Chem Res Toxicol. 2005;18:1701–1714. doi: 10.1021/tx050141k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavaluzzi MJ, Borer PN. Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 2004;32:e13. doi: 10.1093/nar/gnh015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 26.Piantini U, Sorensen OW, Ernst RR. Multiple quantum filters for elucidating NMR coupling networks. J Am Chem Soc. 1982;104:6800–6801. [Google Scholar]
- 27.Rance M, Sorensen OW, Hodenhausen G, Wagner G, Ernst RR, Wuthrich K. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem Biophys Res Comm. 1983;177:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 28.Borgias BA, James TL. MARDIGRAS--a procedure for matrix analysis of relaxation for discerning geometry of an aqueous structure. J Magn Reson. 1990;87:475–487. [Google Scholar]
- 29.Liu H, Spielmann HP, Ulyanov NB, Wemmer DE, James TL. Interproton distance bounds from 2D NOE intensities: Effect of experimental noise and peak integration errors. J Biomol NMR. 1995;6:390–402. doi: 10.1007/BF00197638. [DOI] [PubMed] [Google Scholar]
- 30.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The AMBER biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnott S, Hukins DWL. Optimised parameters for A-DNA and B-DNA. Biochem Biophys Res Comm. 1972;47:1504–1509. doi: 10.1016/0006-291X(72)90243-4. [DOI] [PubMed] [Google Scholar]
- 32.Frisch MJ, Trucks GW, et al. GAUSSIAN98, Gaussian, Inc; Pittsburgh, PA: 1998. [Google Scholar]
- 33.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Banci L, Bertini I, Savellini GG, Romagnoli A, Turano P, Cremonini MA, Luchinat C, Gray HB. Pseudocontact shifts as constraints for energy minimization and molecular dynamics calculations on solution structures of paramagnetic metalloproteins. Proteins. 1997;29:68–76. doi: 10.1002/(sici)1097-0134(199709)29:1<68::aid-prot5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 35.Bashford D, Case DA. Generalized Born models of macromolecular solvation effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 36.Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2000;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 37.Keepers JW, James TL. A theoretical-study of distance determinations from NMR - two-dimensional nuclear Overhauser effect spectra. J Magn Reson. 1984;57:404–426. [Google Scholar]
- 38.Lavery R, Sklenar H. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J Biomol Struct Dyn. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- 39.Stofer E, Lavery R. Measuring the geometry of DNA grooves. Biopolymers. 1994;34:337–346. doi: 10.1002/bip.360340305. [DOI] [PubMed] [Google Scholar]
- 40.Reid BR. Sequence-specific assignments and their use in NMR studies of DNA structure. Q Rev Biophys. 1987;20:2–28. doi: 10.1017/s0033583500004212. [DOI] [PubMed] [Google Scholar]
- 41.Patel DJ, Shapiro L, Hare D. DNA and RNA: NMR studies of conformations and dynamics in solution. Q Rev Biophys. 1987;20:35–112. doi: 10.1017/s0033583500004224. [DOI] [PubMed] [Google Scholar]
- 42.Boelens R, Scheek RM, Dijkstra K, Kaptein R. Sequential assignment of imino- and amino-proton resonances in 1H NMR spectra of oligonucleotides by two-dimensional NMR spectroscopy. Application to a lac operator fragment. J Magn Reson. 1985;62:378–386. [Google Scholar]
- 43.Cullinan D, Korobka A, Grollman AP, Patel DJ, Eisenberg M, de los Santos C. NMR solution structure of an oligodeoxynucleotide duplex containing the exocyclic lesion 3, N4-etheno-2′-deoxycytidine opposite thymidine: Comparison with the duplex containing deoxyadenosine opposite the adduct. Biochemistry. 1996;35:13319–13327. doi: 10.1021/bi9605705. [DOI] [PubMed] [Google Scholar]
- 44.Korobka A, Cullinan D, Cosman M, Grollman AP, Patel DJ, Eisenberg M, de los Santos C. Solution structure of an oligodeoxynucleotide duplex containing the exocyclic lesion 3, N4-etheno-2′-deoxycytidine opposite 2′-deoxyadenosine, determined by NMR spectroscopy and restrained molecular dynamics. Biochemistry. 1996;35:13310–13318. doi: 10.1021/bi9605696. [DOI] [PubMed] [Google Scholar]
- 45.Saparbaev M, Langouet S, Privezentzev CV, Guengerich FP, Cai H, Elder RH, Laval J. 1, N2-ethenoguanine, a mutagenic DNA adduct, is a primary substrate of Escherichia coli mismatch-specific uracil-DNA glycosylase and human alkylpurine-DNA-N-glycosylase. J Biol Chem. 2002;277:26987–26993. doi: 10.1074/jbc.M111100200. [DOI] [PubMed] [Google Scholar]
- 46.Singer B, Antoccia A, Basu AK, Dosanjh MK, Fraenkel-Conrat H, Gallagher PE, Kusmierek JT, Qiu ZH, Rydberg B. Both purified human 1, N6-ethenoadenine-binding protein and purified human 3-methyladenine-DNA glycosylase act on 1, N6-ethenoadenine and 3-methyladenine. Proc Natl Acad Sci USA. 1992;89:9386–9390. doi: 10.1073/pnas.89.20.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hang B, Medina M, Fraenkel-Conrat H, Singer B. A 55-kDa protein isolated from human cells shows DNA glycosylase activity toward 3, N4-ethenocytosine and the G/T mismatch. Proc Natl Acad Sci USA. 1998;95:13561–13566. doi: 10.1073/pnas.95.23.13561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: New twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 49.Freisinger E, Fernandes A, Grollman AP, Kisker C. Crystallographic characterization of an exocyclic DNA adduct: 3, N4-etheno-2′-deoxycytidine in the dodecamer 5′-CGCGAATTεCGCG-3′. J Mol Biol. 2003;329:685–697. doi: 10.1016/s0022-2836(03)00445-5. [DOI] [PubMed] [Google Scholar]
- 50.Cullinan D, Johnson F, de los Santos C. Solution structure of an 11-mer duplex containing the 3, N4-ethenocytosine adduct opposite 2′-deoxycytidine: Implications for the recognition of exocyclic lesions by DNA glycosylases. J Mol Biol. 2000;296:851–861. doi: 10.1006/jmbi.1999.3490. [DOI] [PubMed] [Google Scholar]
- 51.Cullinan D, Eisenberg M, de los Santos C. Solution structures of DNA duplexes containing the exocyclic lesion 3, N4-etheno-2′-deoxycytidine. IARC Sci Publ. 1999;150:179–189. [PubMed] [Google Scholar]
- 52.Cullinan D, Johnson F, Grollman AP, Eisenberg M, de los Santos C. Solution structure of a dna duplex containing the exocyclic lesion 3, N4-etheno-2′-deoxycytidine opposite 2′-deoxyguanosine. Biochemistry. 1997;36:11933–11943. doi: 10.1021/bi9705725. [DOI] [PubMed] [Google Scholar]
- 53.Kouchakdjian M, Eisenberg M, Yarema K, Basu A, Essigmann J, Patel DJ. NMR studies of the exocyclic 1, N6-ethenodeoxyadenosine adduct (εdA) opposite thymidine in a DNA duplex. Nonplanar alignment of εdA(anti) and dT(anti) at the lesion site. Biochemistry. 1991;30:1820–1828. doi: 10.1021/bi00221a014. [DOI] [PubMed] [Google Scholar]
- 54.Leonard GA, McAuley-Hecht KE, Gibson NJ, Brown T, Watson WP, Hunter WN. Guanine-1, N6-ethenoadenine base pairs in the crystal structure of d(CGCGAATT(EdA)GCG) Biochemistry. 1994;33:4755–4761. doi: 10.1021/bi00182a002. [DOI] [PubMed] [Google Scholar]
- 55.de los Santos C, Kouchakdjian M, Yarema K, Basu A, Essigmann J, Patel DJ. NMR studies of the exocyclic 1, N6-ethenodeoxyadenosine adduct (εdA) opposite deoxyguanosine in a DNA duplex. εdA(syn):dG(anti) pairing at the lesion site. Biochemistry. 1991;30:1828–1835. doi: 10.1021/bi00221a015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supporting Information consists of Table S1, chemical shifts of non-exchangeable protons for the 1,N2-εdG duplex; Table S2, chemical shifts of exchangeable protons for the 1,N2-εdG duplex; Table S3, distribution of experimental NOE restraints among nucleotide units of the 1,N2-εdG duplex; Figure S1, NOE cross peak intensities between the base protons and the sugar H1′ protons of the attached deoxyribose moieties; and Figure S2, partial charges assigned to the 1,N2-εdG adduct using the RESP protocol. This material is available free charge via the internet at http://pubs.acs.org.