

Abstract
Small heat shock proteins (sHSPs) and the related α-crystallins are ubiquitous chaperones linked to neurodegenerative diseases, myopathies, and cataract. To better define their mechanism of chaperone action, we used hydrogen/deuterium exchange and mass spectrometry (HXMS) to monitor conformational changes during complex formation between the structurally defined sHSPs, pea PsHsp18.1, and wheat TaHsp16.9, and the heat-denatured model substrates malate dehydrogenase (MDH) and firefly luciferase. Remarkably, we found that even when complexed with substrate, the highly dynamic local structure of the sHSPs, especially in the N-terminal arm (>70% exchange in 5 s), remains unchanged. These results, coupled with sHSP-substrate complex stability, indicate that sHSPs do not adopt new secondary structure when binding substrate and suggest sHSPs are tethered to substrate at multiple sites that are locally dynamic, a feature that likely facilitates recognition and refolding of sHSP-bound substrate by the Hsp70/DnaK chaperone system. Both substrates were found to be stabilized in a partially unfolded state that is observed only in the presence of sHSP. Furthermore, peptide-level HXMS showed MDH was substantially protected in two core regions (residues 95–156 and 228–252), which overlap with the MDH structure protected in the GroEL-bound MDH refolding intermediate. Significantly, despite differences in the size and structure of TaHsp16.9-MDH and PsHsp18.1-MDH complexes, peptide-level HXMS patterns for MDH in both complexes are virtually identical, indicating that stabilized MDH thermal unfolding intermediates are not determined by the identity of the sHSP.
The small heat shock proteins (sHSPs)2 and related vertebrate α-crystallins are a ubiquitous class of molecular chaperones associated with diverse cellular activities (1, 2). In addition to attaining high levels of expression during high temperature stress, sHSPs are induced by other stresses (i.e. oxidative stress, heavy metals, ischemic injury) and are constitutive components of specific tissues in many different organisms. sHSPs have been found to modulate a wide range of biological processes, including cytoskeletal dynamics, cell differentiation, aging, and apoptosis (3, 4). Furthermore, expression and/or mutation of specific sHSPs is linked to neurodegenerative diseases, myopathies, and cataract (5–7), and sHSPs have been suggested to have therapeutic potential for amyotrophic lateral sclerosis (8) and multiple sclerosis (9). The mechanism of sHSP chaperone action and interaction with substrates, therefore, has wide-ranging implications for understanding cellular stress and disease processes.
The sHSPs are defined by a signature α-crystallin domain of ∼100 amino acids, flanked by a short C-terminal extension and an N-terminal arm of variable length and divergent sequence (1). sHSP monomers range from ∼12 to 42 kDa, but in their native state, the majority of sHSPs form oligomers of 12 to >32 subunits. X-ray crystallographic structures of the 24-subunit Methanococcus jannaschii sHSP, MjHsp16.5 (10) and dodecameric Triticum aestivum (wheat) TaHsp16.9 (11) reveal that the α-crystallin domain comprises a β-sandwich with topology identical to the Hsp90 co-chaperone p23, and participates in strand exchange to form a dimeric building block. A conserved IX(I/V) motif in the C-terminal extension makes essential oligomeric contacts by “patching” one edge of the α-crystallin β-sandwich. All of the N-terminal arms in the MjHsp16.5 structure were disordered (10), as were six in TaHsp16.9, but the remaining six had helical structure and were entwined to stabilize the oligomer (11). Remarkably, although sHSPs are observed as stable oligomeric structures by many analytical techniques, kinetic studies reveal the oligomers undergo rapid subunit exchange (12–14), which is potentially critical to their function.
The model for sHSP chaperone action has been developed from studies of diverse sHSPs and their interactions with multiple model substrates in vitro, and is supported by in vivo studies (1,2). sHSPs bind denaturing substrate proteins in an ATP-independent fashion and have a very high substrate capacity, binding up to an equal weight of some proteins. Proteins bound in the resulting high molecular weight sHSP-substrate complexes can be refolded by the ATP-dependent chaperone action of the Hsp70/DnaK system, assisted by Hsp100/ClpB and GroEL in cells/compartments where these latter chaperones co-occur (15–17). Interaction with substrate is believed to involve hydrophobic binding sites on the sHSP, which are exposed by an increased rate of subunit exchange, by heat or phosphorylation-induced shift of the sHSP oligomer equilibrium to a dimeric form, or through more subtle structural rearrangements.
Defining the chaperone mechanism of sHSPs requires a more complete understanding of how sHSPs recognize and bind substrates. Detailed study, however, of the sHSP-substrate interaction is challenging, because of the difficulties associated with investigating heterogeneous protein mixtures exemplified by sHSP-substrate complexes (1, 2). Both the N-terminal arm and β-4 strand of the α-crystallin domain have been implicated as sHSP substrate binding sites, but overlap of proposed binding sites with structural elements required for sHSP oligomerization (and therefore structural integrity) complicates data interpretation (18, 19). Virtually nothing is known about sHSP structural rearrangements that must accompany formation of sHSP-substrate complexes. sHSP subunit exchange can continue in the presence of bound substrate (12, 20), and the chaperone remains protease-accessible (21, 22), although a recent study indicates that protease sites on the sHSP N-terminal arm are protected in substrate complexes (23). There is more, though still limited, information about organization of the denatured substrate. The structure of substrates associated with sHSPs has been monitored by fluorescence dye binding, intrinsic fluorescence, CD, 1H NMR, and spin labeling, primarily using proteins that aggregate upon reduction, such as the insulin β-chain and α-lactalbumin, but heat aggregation of rhodanese, carbonic anhydrase, and other proteins have also been investigated (1, 24, 25). Results generally agree that substrates bind when in an aggregation-prone, partially unfolded molten globule form, although both early and late unfolding intermediates have been identified as binding structures.
To gain more detailed insight into sHSP-substrate interactions, we have employed solution phase hydrogen/deuterium exchange (HX) coupled with mass spectrometry (MS) to investigate the conformational changes of sHSPs and model substrates in sHSP-substrate complexes. The ability to perform HXMS with complex mixtures in physiological buffers and with minimal material make this a valuable approach to structural studies of systems not amenable to other high resolution techniques, and it is now being used to probe amyloid and other complex structures (26–28). HXMS monitors exchange of protein backbone amide hydrogens with deuterium in the D2O solvent. The rate of exchange depends on amide hydrogen access to D2O and involvement in internal hydrogen bonds (29). Comparison of exchange rates for proteins in different states can reveal differences in protein conformation, and importantly, by measuring HX at the peptide level by MS, structural differences can be located to specific protein regions (30–32).
To enable data interpretation in the context of structure, for our studies we used TaHsp16.9 and its closely related homolog from Pisum sativum (pea), PsHsp18.1, complexed with the well-characterized chaperone substrates malate dehydrogenase (MDH) and firefly luciferase. Results here provide novel insights into how sHSPs affect the structure of denaturing substrates and how sHSPs bind substrate in a manner that may facilitate release to other chaperones.
MATERIALS AND METHODS
Pig heart mitochondrial malate dehydrogenase (Roche Applied Science, PDB 1MLD) and firefly luciferase (Promega, PDB 1LCI) were purchased from the manufacturer and used without further purification.
Protein Purification—Triticum aestivum (wheat) TaHsp16.9 (PDB 1GME) and Pisum sativum (pea) PsHsp18.1 (GenBank™ accession no. P19243) were expressed in Escherichia coli and purified as described previously (33). Protein concentrations were determined using the calculated extinction coefficient of E280 = 16500 m-1 cm-1 for both proteins. The expected molecular masses of both proteins were confirmed by mass spectrometry.
Size Exclusion Chromatography (SEC)—sHSPs and substrate proteins were incubated at concentrations, temperatures, and times specified in the figure legends or text. Protein mixtures were cooled on ice after heating and centrifuged for 15 min at 13,000 rpm. The supernatant (100 μl) was loaded onto a Bio-Sil SEC 400-5 column (Bio-Rad) at a flow rate of 1 ml/min. The mobile phase used in all the experiments was 25 mm sodium phosphate and 150 mm KCl, pH 7.5. Standards for chromatography were: thyroglobulin 670 kDa, γ-globulin 158 kDa, ovalumin 44 kDa, carbonic anhydrase 29 kDa, and myoglobin 17 kDa (Bio-Rad).
Peptide Mapping of MDH and sHSPs by HPLC-Tandem Mass Spectrometry—To map the peptides produced by digestion of sHSPs and MDH with pepsin, a total of 100 pmol of protein stock was diluted into 25 mm sodium phosphate and 150 mm KCl in H2O (pH 7.5) to a final concentration of 20 μm, followed by addition of 25% formic acid to adjust the pH to pH 2.5 (by pH paper). The sample was applied to a column of immobilized pepsin (2 mm × 50 mm, packed in-house (34), using water and 0.05% trifluoroacetic acid as the mobile phase at a flow rate of 50 μl/min. The protein digest was collected by a micropeptide trap (Michrome BioResources, Auburn, CA) and washed for 2 min at a flow rate of 300 μl/min. Peptides in the trap were then eluted onto a microbore C-18 HPLC column (1 mm × 50 mm, Micro-Tech Scientific, Vista, CA) coupled to a Waters micro Q-TOF (Milford, MA) with a typical ESI voltage of 3 kV for accurate parent mass measurements (35, 36). Peptides were eluted from the column over 12 min. using a gradient of 15–45% acetonitrile at a flow rate of 50 μl/min. The micropeptide trap and HPLC column were immersed in ice water during the entire process. The same experiment was repeated using a Finnigan LCQ Classic quadrupole ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) in data-dependent mode to acquire product (tandem) MS spectra. The typical ESI voltage used in LCQ was 4.5 kV. Both parent mass and tandem mass spectra were used for peptide identification. The Waters micro Q-TOF was subsequently used for all H/D exchange measurements.
Hydrogen/Deuterium Exchange (HX)—HX exchange experiments were initiated by diluting the protein sample (∼20-fold) into the labeling solution (D2O, 25 mm sodium phosphate and 150 mm KCl, pD 7.5). Incubation times are specified in the text. All pH and pD values reported were taken directly from the pH meter and were not corrected for isotope effects (37). At each time point, an aliquot of 200 μl of protein was taken out of the exchange tube and quenched by mixing the solution with 25% D2 formic acid in D2O to pD 2.5 and frozen in liquid nitrogen. The amount of formic acid required to achieve the desired pD was first estimated by titrating 20 ml of protein buffer with 25% formic acid to pH 2.5 using a pH meter. The required aliquots of 25% D2 formic acid in D2O were then added to individual quenching tubes for direct mixing with protein exchange samples, followed by a pH reading using pH paper. After freezing in liquid N2, quenched samples were transferred to a -80 °C freezer and stored until analysis. All samples were analyzed within 24 h of the HX experiments. Frozen samples were transferred to a dry ice container and removed directly prior to LC-MS analysis. Individual 200-μl samples were defrosted within <1 min at 37 °C before loading directly onto the pepsin column. It is important to note that the pH of the quenched samples was kept at ∼pH 2.5. This pH is maintained during the HPLC step by using 0.05% trifluoroacetic acid as the modifier. In addition, the temperature of the whole digestion/separation setup was maintained at ∼4 °C, by immersion of peptide/protein trap, LC column, and the connecting tubings in an ice bath to further decrease exchange rate. It is well documented that the combination of low pH and low temperature decreases the exchange reaction rate by five orders of magnitude compared with that at neutral pH and 25 °C (29, 37, 38). Furthermore, correction for back-exchange during the LC-MS step can be done using a fully deuterated controlled sample, as described below.
HX Analysis by HPLC-ESI—Samples for peptide analysis were treated as described for unlabeled protein in the peptide mapping section. Intact protein samples were analyzed similarly to the protein digest, with three exceptions. The pepsin column was not used for the MS experiment with intact protein, the micropeptide trap was replaced by a microprotein trap (Michrom BioResources), and 60% acetonitrile was used for protein elution. Mass spectrometry analyses of all samples within each comparison set were done on the same day with the same instrumental conditions. Deconvolution of intact protein spectra was performed with the program MaxEnt1 (Waters, MA). The mass of each peptide was taken as the centroid mass of the isotopic envelope with the program MagTran (39).
To account for the exchange of deuterium during the HPLC step (back exchange), and the use of only 20-fold excess D2O during the labeling step, which limits the forward exchange reaction, an experimental correction was applied. A 100% deuterated protein control (100D reference) was prepared by fully denaturing the sHSPs or substrates in 6 m GuDCl and diluting 20-fold into D2O for >24 h prior to mass determination. The corrected extent of deuterium incorporation was then calculated according to Equation 1 (38, 40),
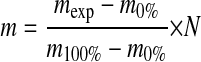 |
(Eq. 1) |
where mexp is the experimental centroid mass of the peptide at a certain time point, m0% is the centroid mass of the undeuterated control, m100% is the centroid mass of the 100% deuterated control, and N is the number of amide hydrogens for each protein/peptide characterized. All exchange results presented here have been corrected, with intact proteins correction factors ranging from 85 to 90%, and peptide correction factors ranging from 75 to 90%.
RESULTS
Complex Formation between sHSPs and Substrate—To investigate structural properties of sHSP-substrate complexes, we prepared complexes with thermally denatured MDH. MDH has been used extensively as a model substrate for the chaperone GroEL (41–44) and for sHSPs, including TaHsp16.9 and PsHsp18.1 (16, 18). sHSP-MDH complexes were formed by incubating either 24 μm PsHsp18.1 or TaHsp16.9 with MDH at an sHSP/MDH molar ratio of 2.4:1 or 3:1, respectively, at 45 °C for 0–120 min (Fig. 1, a and b). These sHSP/MDH ratios were chosen because they afford complete protection of MDH from heat insolubilization, and on a molar basis PsHSP18.1 is more effective than TaHsp16.9 (18). Accumulation of high molecular weight complexes comprising MDH with either sHSP increased over time at 45 °C as observed by size exclusion chromatography performed at room temperature. When MDH was heated in the absence of sHSPs, the protein aggregated, as shown by the gradual decrease in the MDH native peak (Fig. 1c). In contrast, when the sHSPs are heated alone, the majority of the sHSP elutes identically to unheated protein at the position of the dodecamer, with a minor fraction in the case of TaHsp16.9 eluting at position predicting to be a dimer (supplemental Fig. S1).
FIGURE 1.

Complex formation between sHSP and substrate. a, SEC of PsHsp18.1-MDH complex formation. 24 μm PsHsp18.1 and 10 μm MDH were incubated from 0 to 120 min at 45 °C (legend in panel c), and then separated on Bio-Select SEC-5 column (Bio-Rad). Protein was monitored by absorbance at 220 nm. Elution times for void volume, as well as two protein markers are as labeled. b, SEC of TaHsp16.9-MDH complex formation with 24 μm TaHsp16.9 and 8 μm MDH as in a. c, SEC of 10 μm MDH incubated as in a.
Although both sHSPs fully protect MDH, there are significant differences in the sHSP-substrate complexes formed. PsHsp18.1-MDH complexes eluted later, corresponding to an apparent size of 600–700 kDa, while TaHsp16.9-MDH complexes eluted in the void volume (>1000 kDa), consistent with previous observations (18). Increasing the ratio of TaHsp16.9 to MDH did not reduce apparent complex size (not shown). In addition, while virtually all of the MDH and sHSP in the PsHsp18.1 samples are incorporated into the complex peak after 2 h at 45°C, after the same treatment, significant TaHsp16.9 elutes at the position of free sHSP, with no change on further heating (not shown). Thus, TaHsp16.9 either initially associates less effectively with MDH or is less stably complexed with MDH and dissociates prior to or during chromatography. Both sHSP-substrate complexes are heterogeneous in size and/or shape, based on the broadness of the complex peak. Altogether the stoichiometry of protection and the nature of the complexes formed suggest that TaHsp16.9 and PsHsp18.1 have somewhat different modes of interaction with MDH.
HX of sHSP in the sHSP-Substrate Complex—To monitor structural changes during heat-induced sHSP-substrate complex formation, we used the protocol for HX as diagramed in Fig. 2a. Each protein mixture was heated at 45 °C for 0–120 min. Samples were removed every 30 min, cooled to 25 °C, and then subjected to 5-s pulse labeling in D2O. The HX information of both partners in the sHSP-substrate complexes can be monitored simultaneously by MS, because of their significant difference in monomeric mass. The HX data for PsHsp18.1 and TaHsp16.9 in the presence of MDH are shown in Fig. 2b. In the native state, prior to heating, both sHSPs exchanged 58% of backbone hydrogens. Surprisingly, throughout the heating time course, the extent of HX remained at 58% for both PsHsp18.1 and TaHsp16.9. If the interactions between the sHSPs and MDH leave a “fingerprint” by HX, one would expect to observe a two-population pattern for sHSP as more and more sHSP interacts with heat-denaturing MDH, but this was not observed.
FIGURE 2.

Global HXMS of sHSP and substrate. a, experimental scheme for examining HX during sHSP-MDH complex formation. Concentrations of sHSP and MDH are as in Fig. 1. b, mass spectra of sHSP global exchange pattern as a function of time monitored by HXMS. Left, PsHsp18.1 in the presence of MDH. Three populations of PsHsp18.1 were present in the sample used in the experiment: unmodified protein, protein with N-terminal methylation and a very minor fraction of N-terminally acetylated protein. All forms bound substrate equally well (not shown). Right, TaHsp16.9 in the presence of MDH. Two populations of TaHsp16.9 were present in the protein used in the experiment: unmodified protein and a very minor fraction of N-terminally acetylated protein. c, mass spectra of MDH global HX pattern as a function of time. Left, MDH thermal unfolding in the presence of PsHsp18.1. Right, MDH thermal unfolding in the presence of TaHsp16.9.
To test if this result was specific to complexes between sHSP and MDH, we preformed HX on complexes formed by heating PsHsp18.1 together with firefly luciferase (Luc) for 7 min at 42 °C. At an sHSP to substrate ratio of 4 to 1, PsHsp18.1 fully protects Luc from insolubilization, and the vast majority of both sHSP and Luc are complexed (supplemental Fig. S2a). TaHsp16.9 does not protect or form stable complexes with Luc at this temperature (18), again indicating differential interaction with substrate compared with PsHsp18.1, and was therefore not used in this experiment. As we saw with MDH, there was no change in total PsHsp18.1 HX in complex with Luc; HX remained at 58% of amide protons (supplemental Fig. S2b). We conclude that total sHSP amide hydrogen exchange is unchanged by interaction between sHSP and substrate.
Although the total percentage of amide hydrogens exchanged remained the same throughout the 120 min of complex formation with MDH, to test the possibility that there were changes in distribution of exchanged hydrogens between free and complexed sHSP, analysis of mass changes at the peptide level was performed. We have previously documented HX at the peptide level for native TaHsp16.9 in the absence of substrate (45). Interestingly, those regions of the sHSP that are directly involved in oligomeric contacts, the N-terminal arm and C-terminal extension, were the most rapidly exchanged (>70% within 5 s), reflecting the dynamic nature of the sHSP oligomer, and the essential intrinsic disorder of the N-terminal arm. We reexamined HX for native TaHsp16.9 in parallel with native PsHsp18.1 and for both sHSPs after complexing with MDH by 120 min of heating (Fig. 3). Peptide coverage of TaHsp16.9 and PsHsp18.1 was 100% for both proteins. The pattern of HX across native PsHsp18.1 was similar to that observed for TaHsp16.9, with the most rapid exchange in those regions known to form critical oligomeric contacts. Analysis of HX on the peptide level for the sHSP-MDH complexes, even when monitored over a time scale of 5 s to 8 h (sHSP-MDH complexes remain unchanged over 8 h as observed by SEC, not shown) did not reveal any differences compared with the native sHSPs alone (supplemental Fig. S3). There was also no evidence that any sHSP peptide existed in more than one mass form; we estimate it would have been possible to detect minor mass species representing on the order of 5% of the major detected species. Thus, interaction with substrate does not involve formation of any new stable hydrogen-bonded structures within the sHSP or between sHSP and substrate, nor do substrate interactions change solvent access to any part of the sHSP.
FIGURE 3.

Peptide-level HXMS for PsHSP18.1 and TaHsp16.9, with and without MDH. sHSP secondary structural elements, as determined from the x-ray crystal structure of TaHsp16.9 (PDB 1GME), are shown above the sequence. Each peptide is represented by a colored bar, with the color indicating percentage of exchange, as shown in the legend. Eleven peptides and twelve peptides are presented for TaHsp16.9 and PsHsp18.1, respectively. Peptides are numbered from N terminus to C terminus just below the color bar.
HX of MDH in the HSP-MDH Complex—To determine how heat denaturation and complex formation impacted the structure of MDH as probed by HXMS, we first examined the change in total mass of MDH over the course of the experiment diagrammed in Fig. 2a. In contrast to the sHSPs, MDH showed a two-state incorporation of deuterium in both the PsHsp18.1-MDH and TaHsp16.9-MDH complexes (Fig. 2c). In the native form, prior to heating the mixture, a 5-s deuterium pulse increased MDH mass by 82 ± 3 Da (28% exchange). Dramatically, during heat treatment, a new population of MDH that exchanged 202 ± 4 hydrogens (70% exchange), representing an unfolded form of MDH, increased as a function of duration of heating. In parallel, the native form disappeared from the sHSP-MDH mixture, although a significant level remained in the PsHsp18.1-MDH mixture. Because the total MDH is unchanged throughout the experiment, the increase of the 70% exchanged population results from a decrease of the ratio of native to unfolded forms as the heat treatment progresses. For TaHsp16.9-MDH complexes, the native form disappeared relatively faster. However, this difference was determined to be due to the difference in MDH concentration, rather than differences in MDH interaction with the two sHSPs. When 8 μm MDH was used to form complexes with 24 μm PsHsp18.1 or 24 μm TaHsp16.9, the rate of disappearance of the MDH native form was similar for both sHSPs (supplemental Fig. S4).
Under the pulse-labeling conditions used (pD 7.5 for 5 s), a completely unstructured peptide would have all of its amide hydrogens exchanged (29). The fact that even the high mass population of MDH only reached 70% exchange indicates the presence of some MDH secondary structure or that the interaction of sHSP and MDH slows down backbone amide exchange. Importantly, in the absence of sHSP and under the same heating conditions, monitoring MDH by HXMS showed that only the MDH native form was present, which gradually disappeared with heating (not shown), correlating well with the observation that the protein aggregates and falls out of solution (18). Therefore, the MDH form observed by HXMS in sHSP-MDH complexes is stable only in the presence of the sHSP.
We were also able to observe stabilization of an unfolded form of Luc in complex with PsHsp18. In the absence of sHSP at room temperature Luc exchanged 38% of backbone amide hydrogen, while in heat-induced complex with PsHsp18.1 Luc exchanged 60% of amide hydrogens (supplemental Fig. S2c). As for MDH, this latter, unfolded form is only stable in the presence of the sHSP.
Identification of Core-protected Regions in Heated MDH Complexed with sHSP—Because ∼30% of MDH amide hydrogens are resistant to HX in the sHSP-substrate complexes, it was of interest to determine if this was due to protection of a specific region of MDH and if there were differences between TaHsp16.9- and PsHsp18.1-complexed MDH. To address these issues, exchanged sHSP-MDH complexes were subjected to peptide-level analysis. Online digestion of unlabeled MDH with pepsin was performed, and eighteen peptides were used in the subsequent data analysis, covering 78% of the sequence (Fig. 4). Peptides ranged from 6 to 22 amino acids, with an average length of 12 residues. We were unable to perform the same analysis with Luc, because of the higher ratio of sHSP to Luc required for protection, making the Luc peptides difficult to identify in the background of sHSP peptides.
FIGURE 4.

A core structure of MDH is protected in complex with sHSP. Percent HX for MDH peptides after 5-s pulse labeling at pD 7.5 for native MDH, PsHsp18.1-MDH complex and TaHsp16.9-MDH complex. MDH secondary structural elements (PDB 1MLD) are shown above the sequence. Each peptide is represented by a colored bar, with the color indicating percentage of exchange, as shown in the legend. A total of eighteen peptides are presented in the figure. Peptides are numbered from N terminus to C terminus just below the color bar.
From the peptide level data, it is apparent that amide hydrogen protection of MDH complexed with sHSPs is significantly different than that observed for native MDH, and that protection is unevenly distributed along the protein backbone (Fig. 4). There are two core regions of MDH, residues 95–156 and 228–252, which are mostly protected against exchange after heat denaturation in the presence of sHSP. Representative mass spectra of three peptides outside and three within the core-protected regions are presented in Fig. 5. Peptide spectra in the left column show examples of regions where protection from exchange is almost completely lost once MDH is heated in the presence of sHSP. Peptide spectra on the right are examples of regions where substantial protection remains even after MDH was thermally denatured in the presence of sHSP. Considering the exchange conditions (pD 7.5 at 25 °C for 5 s), the level of exchange for these core regions (Fig. 5, right) is substantially lower than the intrinsic exchange rates for peptides if no structure were present (29).
FIGURE 5.

MDH HX data for representative peptides throughout the MDH backbone. Comparison of masses after 5-s pulse labeling (pD 7.5) at room temperature of native MDH, 10 μm MDH heated with 24 μm PsHsp18.1 for 2 h, and 8 μm MDH heated with 24 μm TaHsp16.9 for 2 h. The 100D reference, representing denatured protein labeled in D2O for 24 h is also shown.
To further confirm protection in these regions, a time course analysis was carried out in which nonheated or heated MDH in the presence of PsHsp18.1 was subjected to HX for times ranging from 5 s to 8 h. A comparison of exchange profiles for the seven peptides in the two protected regions is summarized in Fig. 6. It should be noted that all of the peptides remain partially protected even after 8 h. This is particularly obvious for peptides 149–156 and 228–236, both of which exchanged less than 50% after 8 h.
FIGURE 6.

Percent HX of MDH peptides as a function of exchange time. Regions of MDH that are most protected against exchange are shown in the figure. Black closed square, native MDH exchange as a function as time. Open triangle, MDH heated in the presence of PsHsp18.1 for 2 h, HX at room temperature.
A second important observation is that the overall protection pattern of the partially unfolded MDH does not correlate with that of the native protein (Figs. 4 and 7). The N and C termini of MDH display a low level of exchange in the native state. In the sHSP-bound state, both these regions lose their protection, reaching over 80% exchange for a 5-s pulse labeling at pD 7.5 (except peptide 32–41, which exchanges only 70% upon 5-s labeling).
FIGURE 7.

MDH HX profile comparison between native state and sHSP-protected, denatured state (PDB 1MLD) formed by heating PsHsp18.1 and MDH together at 45 °C for 2 h. HX data are from 5-s pulse labeling at room temperature. a, HX profile for the native MDH. b, HX profile for partially unfolded MDH bound to PsHsp18.1.
A third striking result derived from Figs. 4 and 5, along with the global exchange data, is that the MDH protection patterns were similar whether it was complexed with PsHsp18.1 or TaHsp16.9, despite the clear difference the sHSP-MDH complexes revealed in Fig. 1. On the peptide level, not only the overall mass shifts, but also the appearances of the mass envelope for individual peptides appear to be similar.
DISCUSSION
sHSPs Are Dynamic Holdases—The ability of HXMS to provide peptide-level data reporting conformational properties of both sHSP and substrate in sHSP-substrate complexes provides new insights into the mechanism of sHSP chaperone action. A surprising result was our inability to detect peptide-level changes in HX in either TaHsp16.9 or PsHsp18.1 on association with denaturing MDH. We have previously demonstrated for TaHsp16.9, and show here for PsHsp18.1, that backbone amide hydrogens of the N-terminal arm and C-terminal extension are highly exchangeable (>70% in 5 s), despite the involvement of both of these regions in oligomer assembly (1, 11). Considerable data also implicate the N-terminal arm as important for substrate binding, although substrate interaction with multiple sites on the sHSP is likely (18, 19, 46). We expected that association with the substrate would at a minimum alter the dynamic behavior of the N-terminal arm, which would occur whether substrate recognition involved primarily hydrophobic amino acid side chains and/or direct backbone amide interactions. The absence of any change in the peptide-level HX, in both the highly flexible regions of the sHSP, as well as the more rigid α-crystallin domain, is most consistent with the interpretation that sHSP-MDH interactions are highly dynamic, with an off-rate much faster than the time scale detectable by HX. Thus, unlike amyloid protein aggregates, which arise from formation of stable β-sheet structures (29), sHSP-substrate aggregate formation does not involve adoption of new, stable secondary structure. However, although interactions with substrate appear to be dynamic, this appears to be on a local scale, as sHSP-MDH complexes are quite stable and sHSP-associated Luc or MDH cannot be transferred to free sHSP or to GroEL trap (16, 20), indicating that substrates are tethered at multiple sites which prevents release of substrate to solvent. This behavior explains how sHSPs can effectively hold substrates, but at the same time can allow substrates to be accessible to the Hsp70/DnaK folding machinery.
Substrate HX Protection Represents Residual Substrate Structure—In contrast to the sHSPs, both MDH and Luc showed a major change in amide hydrogen protection during denaturation and association with the sHSP. Both substrates also retained highly protected amide hydrogens. We cannot directly determine how much of the remaining amide hydrogen protection of sHSP-bound substrate arises from protection of a binding surface between sHSP and substrate. The fact, however, that protection was not observed on the partner sHSP suggests that solvent occlusion by a binding interface is not a major contributing factor to the residual protection seen for substrate. In addition, HX has been employed to study the conformation of MDH when bound to GroEL (43, 47). In these studies, MDH protection against HX was attributed to the structure of MDH in the bound form. In particular, Chen et al. (47) applied HXMS to monitor the process of MDH refolding mediated by GroEL. In that study, 45 hydrogens were protected from exchange when MDH was bound to GroEL. Upon addition of ATP/GroES, MDH was released from the inside wall of GroEL, and a deprotection of 13 broadly distributed amide hydrogens was observed. Hence the original protection was attributed mainly to the structure of MDH in the bound state. We conclude, based on similar HXMS studies reported for MDH, as well as the lack of additional protection on sHSP upon binding to MDH observed in our study, that at least the bulk of the HX protection observed in our study represents residual substrate structure.
Region and Degree of Substrate Protection—Our finding that the sHSP-bound MDH and Luc are partially protected against exchange is in basic agreement with previous studies that have concluded sHSPs bind substrate in some form of aggregation-prone molten globule state (1, 25, 48). Unlike previous studies, however, we were able to directly compare peptide-level structural changes in a substrate using HXMS. Our HXMS results show that the bound MDH structure is not native-like; neither does it maintain the same relative stability profile as in the native state. The highly stable N and C termini are destabilized, leaving only the core structure. Moreover, in the absence of sHSP, no stable heat-denatured species can be detected by size exclusion chromatography or HX. We were also unable to detect any stable non-native intermediate by CD during heat denaturation of MDH (supplemental Fig. S5). Thus, any MDH unfolding intermediate is extremely aggregation-prone, and its life time is too short to be detectable by the techniques employed here, or alternatively the sHSP stabilizes a novel intermediate on the heat denaturation pathway.
It is interesting that the regions of sHSP-bound MDH show the most protection against exchange overlap with those found in GroEL-bound MDH (47). Fig. 7 shows a comparison of exchange profiles between native MDH and PsHsp18.1-bound MDH. Regions that remain most protected include residues 95–156 and residues 228–252, which cover secondary structure αd-αE, βE, α1F, βF, α2F, α3G, and βK, according to the MDH x-ray structure. βD, αd-αE, βE, α1F, βF, α2F were the most protected against exchange when MDH was bound to GroEL (47). In studies with GroEL, the substrate protein is typically fully denatured and then diluted into a solution of GroEL to initiate binding and refolding, while in our studies, MDH was thermally denatured in the presence of sHSP. So in the bound state, the substrate structure may well be different. Moreover, the protection mechanism of GroEL is most definitely different from that of sHSPs. In the GroEL/GroES system, GroEL binds only one molecule of substrate at a time, and substrate is captured inside the GroEL internal cavity (43). This has two consequences. First, GroEL-bound substrate does not have any opportunity to interact with other substrate molecules. This is in sharp contrast to the sHSP-MDH complex, which likely consists of multiple copies of both sHSP and MDH. Second, the space within the GroEL cavity limits the conformational flexibility of the substrate protein. In the case of sHSP-MDH complex, it remains to be determined if MDH motion is limited to the same extent as with GroEL.
MDH Structure in sHSP-MDH Complexes Is Independent of the sHSP—Judging from SEC and the sHSP to MDH ratio required to maintain substrate solubility, TaHsp16.9-MDH and PsHsp18.1-MDH complexes are significantly different, suggesting a different overall organization of sHSP and substrate. Nevertheless, peptide-level HXMS of MDH in these two complexes was remarkably similar, which indicates similar stability of MDH secondary structural elements, and that the identity of the sHSP does not determine the substrate unfolding pathway. The difference in sHSP-MDH complex size with PsHsp18.1 and TaHsp16.9 may be explained by the fact that thermal unfolding is a kinetic process. Although very similar in sequence, PsHsp18.1 and TaHsp16.9 have different protecting efficiencies for different substrates (21, 49). Once MDH reaches a partially unfolded state, exposing hydrophobic regions, sHSP molecules as well as other copies of partially unfolded MDH will compete for interacting with these regions. Because PsHsp18.1 is more efficient in protecting MDH, aggregation of MDH is more likely to be stopped earlier, resulting in a smaller complex size compared with complexes formed with TaHsp16.9. The conclusion that the unfolding of MDH in the presence of sHSP is not determined by the identity of sHSP is in agreement with a study of the interaction of α-crystallin with α-lactalbumin reported by Carver et al. (24). They observed refolding of apo α-lactalbumin upon dilution from denaturant by real-time NMR, and the presence of α-crystallin did not show any effect toward the refolding process. Hence, sHSPs are shown to be chaperones that do not direct the path of protein folding/unfolding.
Many critical questions remain concerning sHSP-substrate recognition. While HXMS can reveal important conformational properties of the sHSP-substrate interaction, it cannot directly detect binding interfaces. To address this issue, we are currently investigating sHSP-substrate contacts using site-specific cross-linking. It is also important to define the precise features of sHSP-substrate interactions that lead to the difference in effectiveness of substrate protection seen for such similar proteins as PsHsp18.1 and TaHsp16.9. Understanding these basic substrate binding properties is key to defining how sHSPs can play diverse roles in normal growth, stress, and disease.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM42762 (to E. V.) and Grant GM051387 (to V. H. W.). This work was also supported by the 2005–2006 Pfizer Graduate Research Fellowship in Analytical Chemistry (to G. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: sHSP, small heat shock protein; HXMS, hydrogen/deuterium exchange and mass spectrometry; MDH, malate dehydrogenase; SEC, size exclusion chromatography; PDB, Protein Data Bank; Luc, luciferase.
References
- 1.Van Montfort, R., Slingsby, C., and Vierling, E. (2002) Adv. Protein Chem. 59 105-156 [DOI] [PubMed] [Google Scholar]
- 2.Haslbeck, M., Franzmann, T., Weinfurtner, D., and Buchner, J. (2005) Nat. Struct. Mol. Biol. 12 842-846 [DOI] [PubMed] [Google Scholar]
- 3.Dierick, I., Irobi, J., De Jonghe, P., and Timmerman, V. (2005) Ann. Med. (Basingstoke, UK) 37 413-422 [DOI] [PubMed] [Google Scholar]
- 4.Arrigo, A.-P. (2007) Adv. Exp. Med. Biol. 594 14-26 [DOI] [PubMed] [Google Scholar]
- 5.Brady, J. P., Garland, D., Dublas-Tabor, Y., Robison, W. G., Jr., Groome, A., and Wawrousek, E. F. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 884-889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun, Y., and MacRae, T. H. (2005) Cell Mol. Life Sci. 62 2460-2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjoerkdahl, C., Sjoegren, M. J., Winblad, B., and Pei, J.-J. (2008) J. Neurosci. Res. 86 1342-1352 [DOI] [PubMed] [Google Scholar]
- 8.Sharp, P. S., Akbar, M. T., Bouri, S., Senda, A., Joshi, K., Chen, H.-J., Latchman, D. S., Wells, D. J., and de Belleroche, J. (2008) Neurobiol. Dis. 30 42-55 [DOI] [PubMed] [Google Scholar]
- 9.Ousman, S. S., Tomooka, B. H., van Noort, J. M., Wawrousek, E. F., O'Conner, K., Hafler, D. A., Sobel, R. A., Robinson, W. H., and Steinman, L. (2007) Nature 448 474-479 [DOI] [PubMed] [Google Scholar]
- 10.Kim, K. K., Kim, R., and Kim, S.-H. (1998) Nature 394 595-599 [DOI] [PubMed] [Google Scholar]
- 11.Van Montfort, R. L. M., Basha, E., Friedrich, K. L., Slingsby, C., and Vierling, E. (2001) Nat. Struct. Biol. 8 1025-1030 [DOI] [PubMed] [Google Scholar]
- 12.Bova, M. P., Ding, L.-L., Horwitz, J., and Fung, B. K. K. (1997) J. Biol. Chem. 272 29511-29517 [DOI] [PubMed] [Google Scholar]
- 13.Bova, M. P., McHaourab, H. S., Han, Y., and Fung, B. K. K. (2000) J. Biol. Chem. 275 1035-1042 [DOI] [PubMed] [Google Scholar]
- 14.Sobott, F., Benesch, J. L. P., Vierling, E., and Robinson, C. V. (2002) J. Biol. Chem. 277 38921-38929 [DOI] [PubMed] [Google Scholar]
- 15.Lee, G. J., and Vierling, E. (2000) Plant Physiol. 122 189-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mogk, A., Schlieker, C., Friedrich, K. L., Schoenfeld, H.-J., Vierling, E., and Bukau, B. (2003) J. Biol. Chem. 278 31033-31042 [DOI] [PubMed] [Google Scholar]
- 17.Veinger, L., Diamant, S., Buchner, J., and Goloubinoff, P. (1998) J. Biol. Chem. 273 11032-11037 [DOI] [PubMed] [Google Scholar]
- 18.Basha, E., Friedrich, K. L., and Vierling, E. (2006) J. Biol. Chem. 281 39943-39952 [DOI] [PubMed] [Google Scholar]
- 19.Giese, K. C., Basha, E., Catague, B. Y., and Vierling, E. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 18896-18901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedrich, K. L., Giese, K. C., Buan, N. R., and Vierling, E. (2004) J. Biol. Chem. 279 1080-1089 [DOI] [PubMed] [Google Scholar]
- 21.Lee, G. J., Roseman, A. M., Saibil, H. R., and Vierling, E. (1997) EMBO J. 16 659-671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haslbeck, M., Walke, S., Stromer, T., Ehrnsperger, M., White, H. E., Chen, S., Saibil, H. R., and Buchner, J. (1999) EMBO J. 18 6744-6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aquilina, J. A., and Watt, S. J. (2007) Biochem. Biophys. Res. Commun. 353 1115-1120 [DOI] [PubMed] [Google Scholar]
- 24.Carver, J. A., Lindner, R. A., Lyon, C., Canet, D., Hernandez, H., Dobson, C. M., and Redfield, C. (2002) J. Mol. Biol. 318 815-827 [DOI] [PubMed] [Google Scholar]
- 25.Lindner, R. A., Treweek, T. M., and Carver, J. A. (2001) Biochem. J. 354 79-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carulla, N., Caddy, G. L., Hall, D. R., Zurdo, J., Gairi, M., Feliz, M., Giralt, E., Robinson, C. V., and Dobson, C. M. (2005) Nature 436 554-558 [DOI] [PubMed] [Google Scholar]
- 27.Kheterpal, I., Cook, K. D., and Wetzel, R. (2006) Methods Enzymol. 413 140-166 [DOI] [PubMed] [Google Scholar]
- 28.Wu, Z., Wagner, M. A., Zheng, L., Parks, J. S., Shy, J. M., III, Smith, J. D., Gogonea, V., and Hazen, S. L. (2007) Nat. Struct. Mol. Biol. 14 861-868 [DOI] [PubMed] [Google Scholar]
- 29.Bai, Y., Milne, J. S., Mayne, L., and Englander, S. W. (1993) Proteins Struct. Funct. Genet. 17 75-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komives, E. A. (2005) Int. J. Mass Spectrom. 240 285-290 [Google Scholar]
- 31.Rist, W., Graf, C., Bukau, B., and Mayer, M. P. (2006) J. Biol. Chem. 281 16493-16501 [DOI] [PubMed] [Google Scholar]
- 32.Shi, J., Koeppe, J. R., Komives, E. A., and Taylor, P. (2006) J. Biol. Chem. 281 12170-12177 [DOI] [PubMed] [Google Scholar]
- 33.Lee, G. J., Pokala, N., and Vierling, E. (1995) J. Biol. Chem. 270 10432-10438 [DOI] [PubMed] [Google Scholar]
- 34.Wang, L., Pan, H., and Smith, D. L. (2002) Mol. Cell Proteom. 1 132-138 [DOI] [PubMed] [Google Scholar]
- 35.Cheng, G., Wysocki, V. H., and Cusanovich, M. A. (2006) J. Am. Soc. Mass Spectrom. 17 1518-1525 [DOI] [PubMed] [Google Scholar]
- 36.Cheng, G., Cusanovich, M. A., and Wysocki, V. H. (2006) Biochemistry 45 11744-11751 [DOI] [PubMed] [Google Scholar]
- 37.Englander, J. J., Rogero, J. R., and Englander, S. W. (1985) Anal. Biochem. 147 234-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, D. L., Deng, Y., and Zhang, Z. (1997) J. Mass Spectrom. 32 135-146 [DOI] [PubMed] [Google Scholar]
- 39.Zhang, Z., and Marshall, A. G. (1998) J. Am. Soc. Mass Spectrom. 9 225-233 [DOI] [PubMed] [Google Scholar]
- 40.Wales, T. E., and Engen, J. R. (2006) Mass Spectrom. Rev. 25 158-170 [DOI] [PubMed] [Google Scholar]
- 41.Farr, G. W., Furtak, K., Rowland, M. B., Ranson, N. A., Saibil, H. R., Kirchhausen, T., and Horwich, A. L. (2000) Cell 100 561-573 [DOI] [PubMed] [Google Scholar]
- 42.Horst, R., Bertelsen, E. B., Fiaux, J., Wider, G., Horwich, A. L., and Wuthrich, K. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 12748-12753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fenton, W. A., and Horwich, A. L. (2003) Quart. Rev. Biophys. 36 229-256 [DOI] [PubMed] [Google Scholar]
- 44.Elad, N., Farr, G. W., Clare, D. K., Orlova, E. V., Horwich, A. L., and Saibil, H. R. (2007) Mol. Cell 26 415-426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wintrode, P. L., Friedrich, K. L., Vierling, E., Smith, J. B., and Smith, D. L. (2003) Biochemistry 42 10667-10673 [DOI] [PubMed] [Google Scholar]
- 46.Haslbeck, M., Ignatiou, A., Saibil, H., Helmich, S., Frenzl, E., Stromer, T., and Buchner, J. (2004) J. Mol. Biol. 343 445-455 [DOI] [PubMed] [Google Scholar]
- 47.Chen, J., Walter, S., Horwich, A. L., and Smith, D. L. (2001) Nat. Struct. Biol. 8 721-728 [DOI] [PubMed] [Google Scholar]
- 48.Treweek, T. M., Lindner, R. A., Mariani, M., and Carver, J. A. (2000) Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1481 175-188 [DOI] [PubMed] [Google Scholar]
- 49.Basha, E., Lee, G. J., Demeler, B., and Vierling, E. (2004) Eur. J. Biochem. 271 1426-1436 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.