

Abstract
Recombinant human parainfluenza virus type 1 (rHPIV1) was modified to create rHPIV1-P(C−), a virus in which expression of the C proteins (C′, C, Y1, and Y2) was silenced without affecting the amino acid sequence of the P protein. Infectious rHPIV1-P(C−) was readily recovered from cDNA, indicating that the four C proteins were not essential for virus replication. Early during infection in vitro, rHPIV1-P(C−) replicated as efficiently as wild-type (wt) HPIV1, but its titer subsequently decreased coincident with the onset of an extensive cytopathic effect not observed with wt rHPIV1. rHPIV1-P(C−) infection, but not wt rHPIV1 infection, induced caspase 3 activation and nuclear fragmentation in LLC-MK2 cells, identifying the HPIV1 C proteins as inhibitors of apoptosis. In contrast to wt rHPIV1, rHPIV1-P(C−) and rHPIV1-CF170S, a mutant encoding an F170S substitution in C, induced interferon (IFN) and did not inhibit IFN signaling in vitro. However, only rHPIV1-P(C−) induced apoptosis. Thus, the anti-IFN and antiapoptosis activities of HPIV1 were separable: both activities are disabled in rHPIV1-P(C−), whereas only the anti-IFN activity is disabled in rHPIV1-CF170S. In African green monkeys (AGMs), rHPIV1-P(C−) was considerably more attenuated than rHPIV1-CF170S, suggesting that disabling the anti-IFN and antiapoptotic activities of HPIV1 had additive effects on attenuation in vivo. Although rHPIV1-P(C−) protected against challenge with wt HPIV1, its highly restricted replication in AGMs and in primary human airway epithelial cell cultures suggests that it might be overattenuated for use as a vaccine. Thus, the C proteins of HPIV1 are nonessential but have anti-IFN and antiapoptosis activities required for virulence in primates.
Human parainfluenza virus type 1 (HPIV1) is a member of the Paramyxoviridae family, which includes a number of other medically important human pathogens, such as HPIV2 and -3, respiratory syncytial virus (RSV), measles virus, mumps virus, and human metapneumovirus (33). The HPIVs are enveloped, nonsegmented, single-stranded, negative-sense RNA viruses that are classified in the genera Respirovirus (HPIV1 and HPIV3) and Rubulavirus (HPIV2). HPIV1, -2, and -3 are significant respiratory pathogens for infants and young children, with clinical manifestations ranging from mild disease, including rhinitis and pharyngitis, to more-severe disease, including croup, bronchiolitis, and pneumonia (12, 24, 25, 33, 44, 55). The contribution of HPIV infections to pediatric respiratory hospitalizations varies between studies and ranges from 7 to 21% overall for HPIV1, -2, and -3. The HPIVs collectively are the second leading cause of pediatric hospitalizations for viral respiratory disease, behind RSV and ahead of influenza (31, 33, 47). A licensed vaccine is currently not available for the prevention of HPIV disease, but experimental live attenuated candidate vaccines are under development for HPIV1, -2, and -3, with those for HPIV3 in clinical trials (3, 5, 22, 32, 34, 51).
The HPIV1 genome is 15,600 nucleotides in length and contains six genes in the order 3′-N-P/C-M-F-HN-L-5′ (50). Each gene encodes a single protein, with the exception of the P/C gene that encodes the phosphoprotein P in one open reading frame (ORF) and up to four accessory C proteins, C′, C, Y1, and Y2, in a second, overlapping ORF. The synthesis of the C proteins initiates at four separate translational start codons in the C ORF in the order C′, C, Y1, and Y2, and the four proteins are carboxy coterminal (33). However, it is unclear whether the Y2 protein is actually expressed during HPIV1 infection (54). C proteins are expressed by members of the Respirovirus, Morbillivirus, and Henipavirus genera, but not by viruses that belong to the Rubulavirus and Avulavirus genera. The paramyxovirus C proteins studied to date are nonessential accessory proteins that contribute significantly to virus replication and virulence in vivo (1, 30, 41, 42). The C proteins of Sendai virus (SeV), a member of the Respirovirus genus and the closest homolog of HPIV1, are the most extensively characterized.
The C proteins of SeV have been shown to have multiple functions that include inhibition of host innate immunity through antagonism of interferon (IFN) induction and/or signaling (17, 20, 38); regulation of viral mRNA synthesis by binding to the L polymerase protein (10, 13, 27, 42, 64); participation in virion assembly and budding via an interaction with AIP1/Alix, a cellular protein involved in apoptosis and endosomal membrane trafficking (23, 28, 56); and regulation of apoptosis (see below). SeV mutants containing deletions of all four C proteins are viable but are highly attenuated in vitro and in mice (19, 41, 42). To date, the HPIV1 C proteins have not been as extensively studied as those of SeV. However, the HPIV1 C proteins, like the SeV C proteins, play a role in evasion of host innate immunity through inhibition of type I IFN production and signaling (8, 65). Type I IFN was not detected during infection with wild-type (wt) HPIV1 in A549 cells, a human epithelial lung carcinoma cell line, but was induced during infection with a recombinant HPIV1 (rHPIV1) mutant bearing an F170S amino acid substitution in C, designated rHPIV1-CF170S (65). Wt HPIV1, but not the rHPIV1-CF170S mutant, inhibited the antiviral state induced by type I IFN, most likely due to inhibition of STAT1 nuclear translocation in human lung cells (8, 65).
Many viruses have evolved strategies to regulate host cell apoptotic responses to virus infection (reviewed in reference 45). Apoptosis, a process of programmed cell death mediated by the activation of a group of caspases, results in systematic cellular self-destruction in response to a variety of stimuli (reviewed in reference 14). There are two major apoptotic pathways, the extrinsic and intrinsic pathways, which converge at a step involving the activation of effector caspase 3. The activation of effector caspases, nuclear condensation and fragmentation, and cell death are the final steps in the apoptosis pathway. Viral proteins that are able to modulate the host apoptotic response include proapoptotic viral proteins, such as West Nile virus capsid protein (71) and bunyavirus NSs proteins (11), and antiapoptotic viral proteins, such as RSV NS proteins (6), Bunyamwera virus NSs (35), and Rift Valley fever virus NSm protein (69). However, these studies are complex and some viral proteins, such as influenza A virus NS1, have been reported to have both pro- and antiapoptotic functions (58, 73). SeV C proteins have been implicated in the regulation of apoptosis, but their role in this process remains incompletely defined (7, 26, 29, 39, 52). A role for HPIV1 proteins in apoptosis has not been investigated to date.
In the present study, an rHPIV1 mutant was generated in which the C protein ORF was modified by using reverse genetics to preclude the expression of any of the four C proteins while maintaining the expression of a wt P protein. This mutant, designated rHPIV1-P(C−), was evaluated for replication in vitro and in vivo in order to determine (i) whether C was essential for HPIV1 replication; (ii) what effect the deletion of C protein expression had on the host response to virus infection; and (iii) whether a C protein deletion mutant had properties that warranted further development as a live attenuated HPIV1 candidate vaccine. In contrast to wt HPIV1 (but similar to rHPIV1-CF170S), rHPIV1-P(C−) was found to induce a robust IFN response. In addition, in contrast to wt HPIV1 (and also in contrast to rHPIV1-CF170S), rHPIV1-P(C−) induced a potent apoptotic response. Both phenotypes appeared to contribute to attenuation in African green monkeys (AGMs) and in cultures of ciliated human airway epithelium (HAE).
MATERIALS AND METHODS
Cells and viruses.
LLC-MK2 cells (ATCC CCL 7.1) and HEp-2 cells (ATCC CCL 23) were maintained in Opti-Mem I (Gibco-Invitrogen, Inc., Grand Island, NY) supplemented with 5% fetal bovine serum (FBS) and gentamicin sulfate (50 μg/ml). A549 cells (ATCC CCL-185) were maintained in F-12 nutrient mixture (Ham) (Gibco-Invitrogen, Inc.) supplemented with 10% FBS, gentamicin sulfate (50 μg/ml), and l-glutamine (4 mM). Vero cells (ATCC CCL-81) were maintained in minimal essential medium (MEM; Gibco-Invitrogen, Inc.) supplemented with 10% FBS, gentamicin sulfate (50 μg/ml), and l-glutamine (4 mM). BHK-T7 cells, which constitutively express T7 RNA polymerase (9), were kindly provided by Ursula Buchholz, NIAID, and were maintained in Glasgow MEM (GMEM; Gibco-Invitrogen, Inc.) supplemented with 10% FBS, Geneticin (1 mg/ml), MEM amino acids, and l-glutamine (2 mM). Tracheobronchial HAE cells were isolated from airway specimens of patients without underlying lung disease provided by the National Disease Research Interchange (NDRI, Philadelphia, PA) or from excess tissue obtained during lung transplantation, provided by the University of North Carolina (UNC) Cystic Fibrosis Center Tissue Culture Core under protocols approved by the UNC Chapel Hill Institutional Review Board. Growth and differentiation of these cells on semipermeable Transwell inserts at the air-liquid interface generated ciliated HAE, as previously described (53). All infections were incubated at 32°C except where indicated otherwise.
Biologically derived wt HPIV1 Washington/20993/1964, the parent of rHPIV1, was isolated from a clinical sample in primary AGM kidney cells and passaged two more times in primary AGM kidney cells (48) and once in LLC-MK2 cells (2). This preparation has a wt phenotype in AGMs and will be referred to here as wt HPIV1, but it was previously referred to as HPIV1LLC1 (2). The wt rHPIV1 referred to in this study also contains a mutation in the HN gene, HNT553A, which has previously been shown not to have an effect on virus replication (1), and wt rHPIV1 is therefore considered the equivalent of wt virus. Wt rHPIV1 was generated as previously described (2, 50, 65). Wt rHPIV1 was used in all experiments, with the exception that the biological wt HPIV1 was used for the hamster challenge and in the AGM studies, as indicated below. The generation and characterization of the rHPIV1-CF170S mutant was also described previously (2); this virus contains a single-nucleotide substitution in the P/C gene that creates a phenylalanine-to-serine substitution at amino acid (aa) 170 (numbered relative to the C protein) that affects all four C proteins and is silent in the P protein. The medium used for the propagation and infection of wt HPIV1 and rHPIV1 mutants in LLC-MK2 cells did not contain FBS but contained 1.2% TrypLE Select, a recombinant trypsin (Gibco-Invitrogen, Inc.), in order to cleave and activate the HPIV1 fusion (F) protein, as described previously (50). Purified virus stocks were obtained by infection of LLC-MK2 cells, followed by centrifugation and banding of virus containing supernatant in a discontinuous 30%/60% (wt/vol) sucrose gradient, steps designed to minimize contamination with cellular factors, especially IFN. Recombinant vesicular stomatitis virus expressing the green fluorescent protein (VSV-GFP) was originally obtained from John Hiscott (61). Stocks of VSV were propagated in Vero cells and sucrose purified as indicated above.
Virus titers in samples were determined by 10-fold serial dilution of virus in 96-well LLC-MK2 monolayer cultures, using two or four wells per dilution. After 7 days of incubation, infected cultures were detected by hemadsorption with guinea pig erythrocytes, as described previously (59). Virus titers are expressed as log10 50% tissue culture infectious dose per ml (log10 TCID50/ml). VSV stock titers were determined by plaque assay on Vero cells under an 0.8% methyl cellulose overlay.
Antibodies.
Polyclonal antisera directed against the HPIV1 C or P proteins were generated by repeated immunization of rabbits with the following keyhole limpet hemocyanin-conjugated peptides: (i) QMREDIRDQYLRMKTERW (aa residues 153 to 170 of HPIV1 C′; directed against the carboxyl-terminal region of C′, C, Y1, and Y2) for Skia-31 and (ii) RDPEAEGEAPRKQES (aa 10 to 24 of P) for Skia-2. Antisera were generated at Spring Valley Laboratories, Inc. (Woodbine, MD). Two murine monoclonal antibodies directed against the HPIV1 HN protein, designated 8.2.2.A and 4.5, were kindly provided by Yasuhiko Ito (37).
Construction of mutant rHPIV1-P(C−) cDNA.
Nucleotide insertions, deletions, and substitutions were introduced into the P/C gene of wt rHPIV1 (Fig. 1A) (49) in order to silence the expression of the C′, C, Y1, and Y2 proteins without affecting the P protein (Fig. 1B and C). The 93 nucleotides between the P/C gene start signal and the P start codon, including the C′ start codon, were deleted (Fig. 1B and C, mutation 2) and replaced with a 6-nucleotide insertion to act as a “linker” (Fig. 1B and C, mutation 1). The sequence immediately upstream of the P start codon was modified by the addition of the “linker” CGA(ATG) to AAC(ATG) (nucleotide substitutions are underlined), making the P start site more efficient by Kozak's rules (40) and reducing translational initiation at the downstream start codons (Fig. 1B and C, mutation 1). The C start codon was modified (ATG to ACG) (Fig. 1B and C, mutation 3), and three codons were converted to stop codons, including one immediately downstream of the Y1 start codon (TCA to TGA) which will affect all of the C proteins except Y2 and two downstream of the Y2 start codon (TCG to TAG and TTG to TAG) which will affect all of the C proteins (Fig. 1B and C, mutations 4, 5, and 6). All of the introduced changes are silent in the P protein. These changes were achieved by using a modified PCR mutagenesis protocol described elsewhere (46) and an Advantage HF PCR kit (Clontech Laboratories, Palo Alto, CA). The entire PCR-amplified gene product was sequenced by using a Perkin-Elmer ABI 3100 sequencer with a BigDye sequencing kit (Perkin-Elmer Applied Biosystems, Warrington, United Kingdom) to confirm amplification of the desired sequence containing the introduced changes. Full-length antigenomic cDNA clones (FLCs) of HPIV1 containing the desired mutations were assembled in T7 polymerase-driven plasmids by using standard molecular cloning techniques (50), and the region containing the introduced mutation in each FLC was sequenced as described above to confirm the presence of the introduced mutation and absence of adventitious changes. Each virus was designed to conform to the rule of six, i.e., the nucleotide length of each genome was designed to be an even multiple of six (36), a requirement for efficient replication of HPIV1.
FIG. 1.

Designing the HPIV1-P(C−) viral cDNA. (A) The wt HPIV1 genome, shown 3′ to 5′, includes the P/C gene that encodes the phosphoprotein P from one ORF and the four carboxy-coterminal C proteins, C′, C, Y1 and Y2, from a second, overlapping ORF. The coding regions for these proteins are shown, with the initiation and termination codons numbered according to the P/C gene sequence. (B and C) Various mutations were introduced into the HPIV1 P/C gene to silence expression of the four C proteins without affecting the amino acid sequence of the P protein. Panel B shows the sequence of the upstream end of the P/C gene, with the transcription gene start signal and the translational start signal for each protein boxed. Nucleotide (nt) substitutions and an insertion in the rHPIV1-P(C−) sequence are indicated in boldface, and a deletion is indicated with a dotted line. Conversion of the C start codon and stop codons are indicated with the symbol ⊠. These mutations are identified with circled numbers that correspond with a description in panel C of the effect of each mutation. Briefly, 93 nt were deleted between the gene start signal and P start codon and replaced with a 6-nt spacer, CCCAAC (mutations 1 and 2), thus eliminating the first 11 codons of C′, including its start codon. The sequence immediately upstream of the P start codon was modified from CGA(ATG) to AAC(ATG), which also optimizes the Kozak sequence and reduces translational initiation at the downstream start codons (mutation 1). The methionine start codon of the C protein was converted to threonine (mutation 3), one stop codon was introduced downstream of the Y1 start codon (mutation 4), and two stop codons were introduced downstream of the Y2 start codon (mutations 5 and 6). The arrows labeled “M” and “T” indicate unaltered Y1 and Y2. This cDNA was used to recover infectious rHPIV1-P(C−).
Recovery of infectious rHPIV1-P(C−).
rHPIV1-P(C−) was recovered by using a reverse genetics system similar to previously described methods (50) in BHK-T7 cells constitutively expressing T7 polymerase (9) that were grown to 90 to 95% confluence in six-well plates. Cells were transfected with 5 μg of the FLC, 0.8 μg each of the N and P, and 0.1 μg of the L support plasmids in a volume of 100 μl of Opti-Mem per well. Transfection was carried out with Lipofectamine 2000 (Invitrogen, Inc., Carlsbad, CA) according to the manufacturer's directions. The transfection mixture was removed after a 24-h incubation period at 37°C. Cells were then washed and maintained in GMEM supplemented with amino acids and 1.2% TrypLE Select and transferred to 32°C. On day 2 following transfection, the supernatant was harvested. Virus was amplified by passage on LLC-MK2 cells and cloned biologically by two successive rounds of terminal dilution using LLC-MK2 monolayers on 96-well plates (Corning Costar, Inc., Acton, MA). To confirm that rHPIV1-P(C−) contained the appropriate mutations and lacked adventitious mutations, viral RNA was isolated from infected cell supernatants by using a QIAamp viral RNA mini kit (Qiagen, Inc., Valencia, CA), reverse transcribed by using a SuperScript first-strand synthesis system (Invitrogen, Inc., Carlsbad, CA), and amplified by using an Advantage HF cDNA PCR kit (Clontech Laboratories). The viral genome was sequenced in its entirety, confirming its sequence.
Western blotting.
LLC-MK2 monolayers grown in six-well plates (Costar) were mock infected or infected at an input multiplicity of infection (MOI) of 5 TCID50/cell with sucrose-purified wt rHPIV1 or rHPIV1-P(C−). Cell lysates were harvested 48 h postinfection (p.i.) with 200 μl of 1× loading dye solution sample buffer (Qiagen, Inc.) and purified on QIAshredder (Qiagen, Inc.) spin columns. Ten microliters (for Skia-31 probing) or 6 μl (for Skia-2 probing) of each sample was reduced, denatured, and loaded onto 10-well 10% Bis-Tris gels (Invitrogen, Inc.). Gels were run in morpholinepropanesulfonic acid (MOPS) buffer (Invitrogen, Inc.), and protein was transferred onto polyvinylidene difluoride membranes (Invitrogen, Inc.) and blocked overnight at 4°C in phosphate-buffered saline (PBS)-Tween (0.1%) containing 3% bovine serum albumin (BSA). The polyvinylidene difluoride membranes were incubated with 15 ml of a 1:1,000 dilution of primary antibody in PBS-Tween with 1% BSA at room temperature (RT) for 2 h and then were washed three times for 10 min with PBS-Tween. Membranes were incubated for 1 h at RT with a 1:20,000 dilution of peroxidase-labeled goat anti-rabbit immunoglobulin G (IgG) (KPL, Gaithersburg, MD) as the secondary antibody. After the membranes were washed three times for 10 min with PBS-Tween, SuperSignal West pico chemiluminescent substrate (Pierce, Rockford, IL) was added for 10 min at RT. Membranes were developed on Kodak MR films (Kodak, Rochester, NY).
Kinetics of replication of rHPIV1-P(C−).
Wt rHPIV1 and rHPIV1-P(C−) were compared in multicycle growth curves. Confluent monolayer cultures of LLC-MK2 cells in six-well plates were infected in triplicate at an MOI of 0.01 TCID50/cell. Virus adsorption was performed for 1 h in medium containing trypsin. The inoculum was then removed, and cells were washed three times, after which fresh medium containing trypsin was added and then harvested as the day zero sample and replaced with fresh medium containing trypsin. On days 1 to 7 p.i., the entire supernatant was removed for virus quantitation and was replaced with fresh medium containing trypsin. Supernatants containing virus were frozen at −80°C, and virus titers (log10 TCID50/ml) were determined with endpoints identified by hemadsorption. Cytopathic effect (CPE) was visually monitored. The amount of CPE observed under the microscope was given a score ranging from 1 to 5 based on the percentage of cells in the monolayer showing CPE. A CPE of less than 20% of cells was scored as 1, 21 to 40% as 2, 41 to 60% as 3, 61 to 80% as 4, and 81 to 100% as 5.
Immunostaining and confocal microscopy.
LLC-MK2 cells were seeded onto 24-well plates containing 12-mm glass coverslips, mock infected or infected with rHPIV1-P(C−) or wt rHPIV1 at an MOI of 10 TCID50/cell, and incubated for 72 h. Medium was removed, and coverslips were washed twice with PBS. Cells were then fixed with 3% formaldehyde solution in PBS for 40 min at RT, washed once with PBS, permeabilized with 0.1% Triton X-100 in PBS for 4 min at RT, and washed twice with PBS prior to blocking with PBS containing 0.25% BSA and 0.25% gelatin for 1 h at RT. HPIV1 HN staining was performed by using as primary antibody a 1:4,000 dilution of a mixture of HPIV1-HN 8.2.2.A and HPIV1-HN 4.5, two murine antibodies directed against the HPIV1 HN protein, kindly provided by Yasuhiko Ito, Mie University School of Medicine, Mie, Japan. After incubation at RT for 1 h, cells were washed twice with PBS and stained with a 1:1,000 dilution of Texas Red-conjugated donkey anti-mouse IgG (Jackson Immunochemicals, West Grove, PA) as secondary antibody for 1 h at RT. Activated caspase 3 was detected by using a 1:25 dilution of a fluorescein isothiocyanate (FITC)-conjugated rabbit anti-human activated caspase 3 antibody (BD Pharmingen, San Jose, CA). Cells were washed twice with PBS and immediately mounted onto slides with the 4′,6′-diamidino-2-phenylindole (DAPI)-containing antifade reagent ProLong gold (Invitrogen, Inc.). Slides were covered with foil, left to dry overnight at RT, and then stored at −20°C until microscopy was performed on a Leica SP5 confocal microscope.
FACS analysis.
LLC-MK2, Vero, or A549 cells in six-well plates were mock infected or infected with wt rHPIV1 or rHPIV1-P(C−) at an MOI of 5 TCID50/cell. Cells were harvested at 24, 48, and 72 h p.i. by scraping cells into 2 ml of fresh fluorescence-activated cell sorting (FACS) buffer (PBS, 1% FBS) and pelleting at 1,200 rpm for 10 min at 4°C. Cells were resuspended in 1 ml of 3% paraformaldehyde, fixed for 15 min on ice, and then rinsed twice in 3 ml FACS buffer. Cells were permeabilized and stained with the following antibodies diluted in FACS buffer containing 0.1% Triton X-100: (i) rabbit anti-human activated caspase 3 FITC (1:100; BD Pharmingen) and (ii) mouse anti-PIV1 HN (1:2,000 dilution of a 1:1 mix of HPIV1 HN 8.2.2.A and HPIV1 HN 4.5). Staining was performed for 45 min at RT in a dark environment, and then cells were rinsed twice with 2 ml FACS buffer and stained with allophycocyanin-conjugated goat anti-mouse IgG (1:1,000; Jackson ImmunoResearch Laboratories, West Grove, PA) diluted in FACS buffer containing 0.1% Triton X-100. Staining was carried out for 30 min at RT. Finally, cells were rinsed twice in FACS buffer and resuspended in 250 μl FACS buffer for analysis. Sample analysis was carried out on a FACSCalibur (BD Biosciences, San Jose CA) using CellQuestPro software. Further analysis was performed using FlowJo software (TreeStar, Inc., Ashland, OR).
Type I IFN bioassay.
The amount of type I IFN produced by HPIV1-infected A549 cell cultures was determined by an IFN bioassay, as previously described (65). Type I IFN concentrations were determined by measuring the ability of samples of the cell cultures to restrict replication of VSV-GFP on HEp-2 cell monolayers in comparison to a known concentration of a human beta interferon (IFN-β) standard (Avonex; Biogen, Inc., Cambridge, MA). Briefly, samples were treated at pH 2.0 to inactivate virus and acid-labile type II IFN prior to being serially diluted 10-fold in duplicate in 96-well plates of HEp-2 cells along with the IFN-β standard (5,000 pg/ml). After 24 h, the cells were infected with VSV-GFP at 6.5 × 104 PFU/well. After an additional 24 to 36 h, plates were read for GFP expression by using a Typhoon 8600 phosphorimager (Molecular Dynamics, Sunnyvale, CA). The dilution at which the level of GFP expression was approximately 50% of that in untreated cultures was determined as the end point. The end point of the Avonex standard was compared to the end point of the unknown samples, and IFN concentrations were determined and expressed as mean ± standard error (SE) (pg/ml).
Evaluation of replication of viruses in hamsters and efficacy against challenge.
Four- to 6-week-old Syrian golden hamsters in groups of five or six per virus were inoculated intranasally (i.n.) with 0.1 ml of Leibovitz's L-15 medium (Gibco-Invitrogen) containing 105.5 TCID50 of wt rHPIV1, rHPIV1-P(C−), or control (L-15 only) inoculum. On days 4 and 5 p.i, the nasal turbinates and lungs were collected as previously described (50). Virus present in the tissue homogenates was quantified by titration on LLC-MK2 monolayers. Infected cells were detected on day 7 p.i. by hemadsorption with guinea pig erythrocytes. The mean titer (log10 TCID50/g tissue) was calculated for each group of hamsters. The limit of detection was 1.5 log10 TCID50/g. On day 28 p.i., hamsters that had been previously immunized were challenged i.n. with 106 TCID50 of wt HPIV1 in 0.1 ml L-15. The nasal turbinates and lungs were collected for virus quantitation on day 4 p.i.
Evaluation of replication of viruses in AGMs and efficacy against challenge.
AGMs in groups of two to four animals at a time were inoculated i.n. and intratracheally (i.t.) with 106 TCID50 of either wt HPIV1 or mutant rHPIV1 in a 1-ml inoculum at each site. Nasopharyngeal (NP) swab samples were collected daily from days 0 to 10 p.i., and tracheal lavage (TL) fluid samples were collected on days 2, 4, 6, 8, and 10 p.i. The specimens were flash frozen and stored at −80°C until they were assayed in parallel. Virus present in the samples was titered in serial dilutions on LLC-MK2 cell monolayers in 96-well plates, and an undiluted 100-μl aliquot was also tested in 24-well plates. Following incubation for 7 days, virus was detected by hemadsorption, and the mean log10 TCID50/ml titer was calculated for each sample day. The limit of detection was 0.5 log10 TCID50/ml. The mean peak titer for each group was calculated by using the peak titer for each animal, irrespective of the day of sampling. On day 28 p.i., the AGMs were challenged i.n. and i.t. with 106 TCID50 of wt HPIV1 in 1 ml L-15 per site. NP swab and TL samples were collected for virus quantitation on days 2, 4, 6, and 8 postchallenge.
All animal studies were performed under protocols approved by the National Institute of Allergy and Infectious Disease (NIAID) Animal Care and Use Committee (ACUC).
Viral inoculation of HAE.
The apical surfaces of HAE were rinsed with PBS to remove apical surface secretions and fresh medium was supplied to the basolateral compartments prior to inoculation. The apical surfaces of HAE were inoculated with HPIV1s at a low input MOI (0.01 TCID50/cell) in a 100-μl inoculum, and the cultures were incubated at 37°C. The inoculum was removed 2 h p.i., and the apical surfaces rinsed for 5 min with PBS and then incubated at 37°C. At days 0 to 7 p.i., apical samples were collected by incubating the apical surface with 300 μl of medium for 30 min at 37°C, after which the medium was recovered. Samples were stored at −80°C prior to determination of virus titer.
Statistical analysis.
The Prism 5 (GraphPad Software, Inc., San Diego, CA) one-way analysis of variance test (Student-Newman-Keuls multiple comparison test) was used to assess statistically significant differences between data groups (P < 0.05).
RESULTS
Construction and recovery of an rHPIV1 mutant not expressing any of the four C proteins.
The P/C gene of wt HPIV1 encodes the phosphoprotein, P, in one ORF and four carboxy-coterminal C proteins, C′, C, Y1, and Y2, in a second, overlapping ORF (Fig. 1A). We engineered rHPIV1 to silence the expression of all four C proteins without affecting the P protein, creating the mutant virus rHPIV1-P(C−) (Fig. 1B and C). The changes introduced to silence the expression of the C proteins included the deletion of the 3′ portion of the P/C gene containing the C′ start, conversion of the C start to an ACG codon, and the introduction of three stop codons into the C ORF immediately downstream of the Y1 and Y2 start codons (Fig. 1B and C). Importantly, all of the introduced changes were silent in the P protein (Fig. 1B and C). The Y1 and Y2 start codons were not modified since any changes introduced at these sites would have altered P protein amino acid assignments. The AUG-to-ACG change at the start site of C would not necessarily silence its expression entirely, since ACG functions (inefficiently) as a start codon for the C′ protein of SeV, but other changes at this site could not be accommodated without affecting P coding, and in any event, any residual expression of C would be ablated by the three stop codons that were introduced downstream. The recombinant virus was recovered from this mutant cDNA in cell culture, and the virus replicated to 8.0 log10 TCID50/ml. Sequence analysis of the entire virus genome revealed that rHPIV1-P(C−) contained all the intended mutations and no unintended changes (data not shown).
The results of Western blot analysis of infected LLC-MK2 cell lysates using an antibody directed against the carboxy terminus of the C proteins demonstrated the expression of the C′ and C proteins in cells infected with wt HPIV1, but not rHPIV1-P(C−) (Fig. 2A). C′ was found to be the most abundant C protein, and Y1 and Y2 were not detected. An additional unidentified species, indicated with an asterisk in Fig. 2A, was detected in cells infected with rHPIV1-P(C−), but not in cells infected with wt rHPIV1 (Fig. 2A). This species was not detected by using an antibody directed against the amino terminus of the C′ and C proteins (data not shown). An ATG codon in the C ORF that is downstream of the last inserted stop codon in Y2 could potentially give rise to a truncated protein that would be carboxy coterminal with the C proteins and would be 157 aa in length, compared to 204 aa for the C protein and 175 aa for Y2 (Fig. 1A). This 47-aa difference in predicted size between C and the unknown protein in Fig. 2A would correspond to an approximately 5-kDa difference in the proteins' apparent molecular masses, consistent with the mobility difference observed in our Western blot (Fig. 2A). The P protein could be detected in both wt rHPIV1- and rHPIV1-P(C−)-infected cells (Fig. 2B). The ability to recover the rHPIV1-P(C−) mutant indicates that the four wt C proteins are not essential for replication in vitro, with the caveat that there was expression of a new species that may have been a truncated C protein.
FIG. 2.

Identification of HPIV1 C and P proteins in lysates from infected LLC-MK2 cells. Lysates were prepared 48 h p.i. from LLC-MK2 cells that were mock infected or infected with sucrose-purified wt rHPIV1 or rHPIV1-P(C−) at an input MOI of 5 TCID50/cell. Reduced, denatured cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and Western blots were prepared and analyzed by using rabbit antipeptide antisera against the HPIV1 C proteins (A) and the HPIV1 P protein (B). The asterisk in panel A indicates a new band of unknown identity, detected only in the rHPIV1-P(C−)-infected cell lysates.
The rHPIV1-P(C−) mutant replicates efficiently in vitro but causes increased CPE compared to wt rHPIV.
Multicycle replication of the rHPIV1-P(C−) mutant was assessed in LLC-MK2 cells infected at an MOI of 0.01 TCID50/cell (Fig. 3A). LLC-MK2 cells were chosen since wt HPIV1 replicates efficiently in this cell line. rHPIV1-P(C−) and wt rHPIV1 replicated to similar titers until day 3 p.i., when wt rHPIV1 continued to increase in titer, whereas rHPIV1-P(C−) decreased in titer. Concomitantly, rHPIV1-P(C−)-infected LLC-MK2 cells developed extensive CPE, while wt rHPIV1-infected cells did not (Fig. 3B). This also is evident in photomicrographs of LLC-MK2 cells taken 72 h following infection at MOIs of 0.01 or 5 TCID50/cell (Fig. 3B). In LLC-MK2 cells infected at an MOI of 5 TCID50/cell, increased CPE associated with rHPIV1-P(C−), but not wt rHPIV1, became evident at approximately 48 h p.i. (data not shown). Similarly, enhanced CPE associated with infection by the rHPIV1-P(C−) mutant was observed in A549 cells (data not shown), which were evaluated since they are a human respiratory tract-derived continuous cell line and in vivo HPIV1 replicates preferentially, if not exclusively, in respiratory epithelial cells. In summary, rHPIV1-P(C−) and wt rHPIV1 replicated with equal efficiency early in infection, but there was a subsequent decrease in rHPIV1-P(C−) titers that was temporally associated with the development of extensive CPE, a phenomenon not seen in wt rHPIV1-infected cells.
FIG. 3.
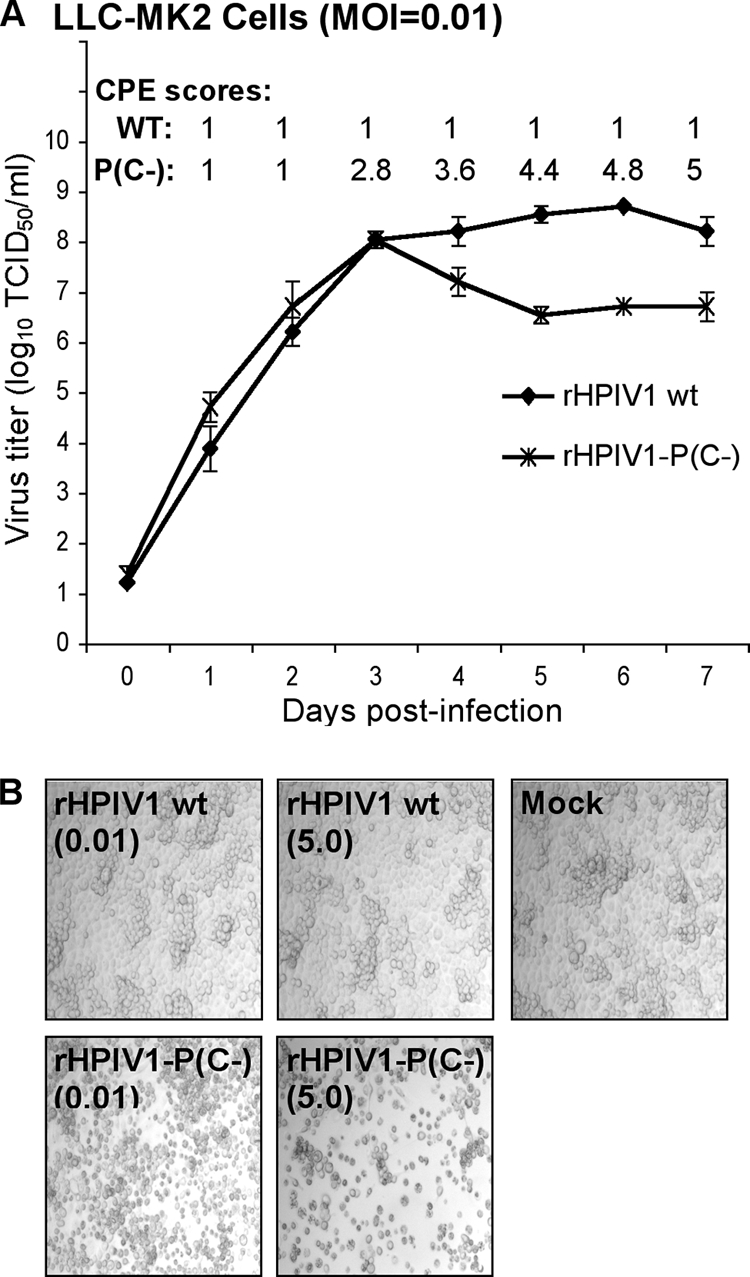
Comparison of the replication of wt rHPIV1 and rHPIV1-P(C−) in vitro. (A) Multicycle replication in LLC-MK2 cells infected at an MOI of 0.01 TCID50/cell. On days 0 to 7 p.i., the overlying medium was harvested for virus titration, shown as the means ± SE of the results for three replicate cultures. On days 1 to 7 p.i., the cell monolayers were monitored for CPE and assigned a score of 1 to 5 according to the extent of CPE (Materials and Methods), shown as the means of the results for the three replicate cultures. (B) LLC-MK2 cells were mock-infected or infected with wt rHPIV1 or rHPIV1-P(C−) at an MOI of 0.01 or 5 TCID50/cell, as indicated in parentheses below the virus names. Photomicrographs taken at 72 h p.i. show increased CPE in the rHPIV1-P(C−)-infected cultures (magnification, ×10).
Infection with rHPIV1-P(C−) infection induces apoptosis.
To further explore the basis of the enhanced CPE associated with the rHPIV1-P(C−) mutant, we assayed virus-infected LLC-MK2 cells for activation of caspase 3, the major effector caspase in the apoptotic pathway. Activated caspase 3 was visualized by immunofluorescence (Fig. 4A) and by FACS analysis (Fig. 4B) using an antibody that specifically recognizes the cleaved, activated form of the enzyme. Replicate LLC-MK2 monolayers were infected at an MOI of 10 TCID50/cell; incubated for 24, 48, and 72 h; fixed; permeabilized; and stained with antibodies for HPIV1 HN (Texas Red) and for activated caspase 3 (FITC). The HPIV1 HN antigen was detected in the vast majority of cells infected with either virus (Fig. 4A). By 72 h p.i., activated caspase 3 was detected in the majority of the rHPIV1-P(C−)-infected cells, but not wt rHPIV1-infected cells (Fig. 4A). In addition, cell rounding and nuclear condensation was seen in the majority of rHPIV1-P(C−)-infected cells, but not in wt rHPIV1-infected cells (Fig. 4A), consistent with the interpretation that the rHPIV1-P(C−)-induced CPE is the direct result of virus-induced apoptosis.
FIG. 4.

Infection with rHPIV1-P(C−) induces activation of caspase 3, indicative of apoptosis. Caspase 3 activation was evaluated by immunostaining and FACS analysis. (A) Evaluation of caspase 3 activation by immunofluorescence. LLC-MK2 cells were mock infected or infected with wt rHPIV1 or rHIV1-P(C−) at an MOI of 10 TCID50/cell. At 72 h p.i., cells were fixed, permeabilized, and stained for HPIV1 HN protein (red) and activated caspase 3 (green), and nuclei were stained with DAPI (blue). The asterisk indicates cell rounding and nuclear condensation seen in the majority of rHPIV1-P(C−)-infected cells, but not in wt rHPIV1-infected cells. Cells were visualized by confocal microscopy, and scale bars represent 10 μm. (B) Evaluation of caspase 3 activation by FACS analysis. LLC-MK2 cells were mock infected or infected with wt rHPIV1, rHPIV1-CF170S, or rHPIV1-P(C−) at an MOI of 5 TCID50/cell in triplicate. Cells were harvested at 24, 48, and 72 h p.i., fixed, permeabilized, and stained for HPIV1 HN and activated caspase 3 in FACS buffer prior to analysis. Sample analysis was carried out by using a FACSCalibur flow cytometer and FlowJo software. Dot plots of representative data for samples from the 48-h time point are shown. The frequency of events is indicated for each quadrant (% total events). APC, allophycocyanin. (C) Percentages of cells positive for activated caspase 3 at 24, 48, and 72 h p.i., as determined by FACS analysis. Error bars show SE.
We next determined the frequency of apoptosis in infected cells by using flow cytometry. The rHPIV1-CF170S mutant, which encodes an F170S substitution in C, and which has previously been associated with type I IFN induction and effective type I IFN signaling, but not with CPE in vitro, was included here for comparison. Replicate cultures of LLC-MK2 cells were infected with wt rHPIV1, rHPIV1-CF170S, or rHPIV1-P(C−) at an MOI of 5 TCID50/cell and, at 24, 48, and 72 h p.i., were fixed, permeabilized, and immunostained for HPIV1 HN protein and activated caspase 3 (Fig. 4B). More than 70% of cells in the rHPIV1-P(C−)-infected cultures were positive for activated caspase 3 by 72 h p.i., compared to approximately 5% and 7% in the wt rHPIV1- and rHPIV1-CF170S-infected cell cultures, respectively (Fig. 4C). The results of similar studies in Vero and A549 cells confirmed that rHPIV1-P(C−) was a potent activator of caspase 3 activation, while wt rHPIV1 was not, although the level of caspase 3 activation in these cells was lower than in LLC-MK2 cells (data not shown). Vero cells were also evaluated here since vaccine viruses to be used in clinical trials are prepared in this cell line, and therefore, it is important to determine the growth characteristics of potential vaccine viruses in this cell line. By 72 h p.i., approximately 12% and 18% of rHPIV1-P(C−)-infected Vero and A549 cell cultures, respectively, were positive for activated caspase 3, compared to approximately 4% of wt rHPIV1-infected cell cultures for both cell types.
rHPIV1-P(C−), but not wt rHPIV1, induces type I IFN production and signaling.
HPIV1 C proteins have been shown to inhibit production of and signaling by type I IFN (8, 65). We have previously demonstrated that type I IFN was not detected during infection of A549 cells with wt HPIV1 but was efficiently produced in response to rHPIV1-CF170S (65). To determine the relative effect of deleting all four C proteins on the ability of HPIV1 to inhibit the type I IFN response, we infected A549 cells at an MOI of 5 TCID50/cell with rHPIV1-P(C−), wt rHPIV1, or rHPIV1-CF170S and subsequently quantified type I IFN in medium supernatants by using a bioassay based on the inhibition of infection and GFP expression by VSV-GFP (Fig. 5A). As shown previously, wt rHPIV1 inhibited the IFN response effectively (Fig. 5A), with barely detectable levels of IFN-β appearing late in infection, at 72 h p.i. In contrast, both rHPIV1-P(C−) and rHPIV1-CF170S induced a robust type I IFN response, with IFN detectable in the supernatant as early as 24 h p.i. and until 72 h p.i., achieving a peak concentration of approximately 300 pg/ml at 48 h p.i. These data suggest that C proteins play a critical role in antagonism of type I IFN production but that IFN generation is unrelated to the apoptotic response seen with the C mutants, since the F170S mutant generates similar levels of IFN with no observable CPE beyond that of wt HPIV1.
FIG. 5.
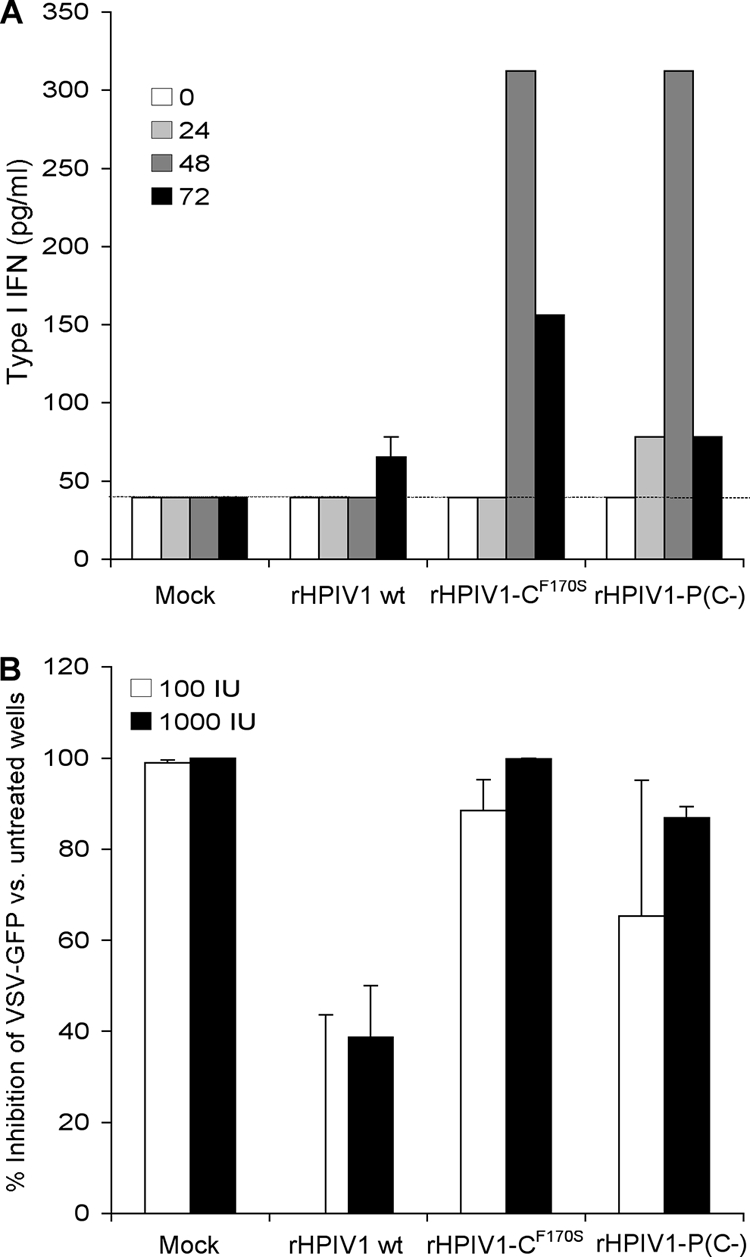
Wt rHPIV1, but not rHPIV1-P(C−), inhibits type I IFN induction and signaling. (A) Induction of type I IFN. A549 cell monolayers were either mock infected or infected with wt rHPIV1, rHPIV1-CF170S, or rHPIV1-P(C−) at an MOI of 5 TCID50/cell. Aliquots of the overlying medium were taken at 0, 24, 48, and 72 h p.i. and assayed on fresh cells for the ability to inhibit infection and GFP expression by VSV-GFP as measured with a phosphorimager. IFN concentrations were determined by comparison with a standard curve prepared in parallel with an Avonex IFN-β standard and are expressed in pg/ml ± SE based on the results for triplicate samples. The lower limit of detection was 39.1 pg/ml (dashed line). (B) Type I IFN signaling. Vero cells in six-well plates were infected with the indicated rHPIV1 at an MOI of 5 TCID50/cell and incubated for 24 h. Cells were then left untreated or were treated with 100 or 1,000 IU/ml IFN-β (one well per treatment per virus) for 24 h. The cells were then infected with VSV-GFP and incubated for 48 h. The VSV-GFP foci were visualized by using a phosphorimager and counted. The graph represents the percent inhibition of VSV-GFP replication in IFN-β-treated versus untreated cells based on the results of two independent experiments. Error bars show SE.
We have previously demonstrated that type I IFN signaling leading to the establishment of an antiviral state is inhibited following infection with wt HPIV1, but not with mutants that encode defective C proteins, e.g., rHPIV1-CF170S (65). To determine the relative effect of deleting all four C proteins on the ability of HPIV1 to inhibit type I IFN signaling, Vero cells were mock infected or infected with wt rHPIV1, rHPIV1-CF170S, or rHPIV1-P(C−) at an MOI of 5 TCID50/cell for 24 h, treated with 0, 100, or 1,000 IU of IFN-β for 24 h, and infected with 200 PFU/well of VSV-GFP. The number of VSV-GFP foci were counted 48 h later, and the percent inhibition due to IFN-β treatment was calculated relative to the inhibition in cells that did not receive IFN-β (Fig. 5B). In control cells that were not infected with rHPIV1, VSV-GFP replication was completely inhibited by IFN-β treatment. In contrast, infection with wt rHPIV1 ablated the ability of 100 IU/ml of IFN-β to inhibit VSV-GFP replication and blunted the inhibitory effect of 1,000 IU/ml of IFN-β, indicating that wt rHPIV1 can prevent IFN-β signaling and the induction of an antiviral state. Infection with rHPIV1-P(C−) did not inhibit the antiviral effect of IFN-β at either concentration, an effect similar to that for rHPIV1-CF170S (Fig. 5B). In summary, unlike wt rHPIV1, rHPIV1-P(C−) is unable to inhibit both the production of type I IFN and the induction of an antiviral state by IFN-β.
rHPIV1-P(C−) is highly attenuated in hamsters and confers protection against wt HPIV1 challenge.
Golden Syrian hamsters were inoculated i.n. with 105.5 TCID50 of rHPIV1-P(C−) or wt rHPIV1. Animals were sacrificed on days 4 and 5 p.i., and the level of virus replication in nasal turbinates and lungs was quantified by virus titration. In comparison to titers of wt rHPIV1, rHPIV1-P(C−) was restricted approximately 1,000-fold in the upper respiratory tract (URT) and 250-fold in the lower respiratory tract (LRT) on both sampling days (Table 1). Although the replication of rHPIV1-P(C−) was highly restricted in hamsters, the animals were protected against i.n. challenge with 106 TCID50 of wt HPIV1 28 days postvaccination (Table 2). The replication of challenge virus in rHPIV1-P(C−)-immunized hamsters was restricted 200-fold in the URT and 16-fold in the LRT compared to these levels of replication in nonimmunized hamsters. Previous infection with wt rHPIV1 restricted the replication of challenge virus even better than rHPIV1-P(C−), reducing challenge virus titers 1,000-fold in the URT and 160-fold in the LRT (Table 2).
TABLE 1.
Replication of rHPIV1-P(C−) in the URT and LRT of hamstersa
| Virus | Mean virus titer (log10 TCID50/g) ± SEb
|
|||
|---|---|---|---|---|
| Nasal turbinates on day
|
Lungs on day
|
|||
| 4 | 5 | 4 | 5 | |
| Wt rHPIV1c | 5.2 ± 0.2 (10) | 5.3 ± 0.4 (11) | 4.6 ± 0.2 (10) | 4.6 ± 0.4 (11) |
| rHPIV1-P(C−) | 2.0 ± 0.2d (5) | 2.2 ± 0.2d (5) | 2.2 ± 0.2d (5) | 2.2 ± 0.1d (5) |
Hamsters were inoculated i.n. with 105.5 TCID50 of the indicated virus.
The limit of detection was 1.5 log10 TCID50/g. The number of animals per group is indicated in parentheses.
The data for the wt rHPIV1 group represent the results of two independent experiments.
There was a statistically significant reduction in virus titer compared to that of the rHPIV1 wt group at the same time point (P < 0.001; Student-Newman-Keuls multiple-comparison test).
TABLE 2.
Protection against wt HPIV1 challenge in hamsters following immunization with rHPIV1-P(C−)a
| Immunizing virus or L-15 medium | Mean HPIV1 challenge virus titer (log10 TCID50/g) ± SEb
|
|
|---|---|---|
| Nasal turbinates | Lungs | |
| Wt rHPIV1 | ≤1.5 ± 0.0c | 1.7 ± 0.2c |
| rHPIV1-P(C−) | 2.2 ± 0.3c | 2.7 ± 0.2c |
| Control | 4.5 ± 0.2 | 3.9 ± 0.4 |
Hamsters were inoculated i.n. with 105.5 TCID50 of the indicated virus and challenged on day 28 p.i. with 106 TCID50 of wt HPIV1 i.n. There were five animals in each group.
The limit of detection was 1.5 log10 TCID50/g. Nasal turbinates and lungs from each group were harvested on day 4 postchallenge.
There was a statistically significant reduction in virus titer compared to that of the control group at the same time point (P < 0.01; Student-Newman-Keuls multiple-comparison test).
rHPIV1-P(C−) is highly attenuated in AGMs and confers protection against wt HPIV1 challenge.
The attenuation phenotype of rHPIV1-P(C−) was also evaluated in AGMs. Following i.n. and i.t. inoculation of AGMs with 106 TCID50 of rHPIV1-P(C−) or wt HPIV1 at each site, virus titers were determined in NP swab samples (representative of the URT) on days 0 to 10 p.i. and TL samples (representative of the LRT) on days 2, 4, 6, 8, and 10 p.i. Wt HPIV1 replication was robust in both the URT and the LRT of AGMs, with continued replication through day 10 p.i. (Fig. 6) and mean peak virus titers of 3.7 and 3.3 log10 TCID50/ml in the URT and LRT, respectively. In contrast, rHPIV1-P(C−) replication was undetectable in the URT and restricted but detectable in the LRT (Fig. 6). The level of replication of rHPIV1-P(C−) was compared with that of the previously described rHPIV1-CF170S (1), a mutant that shares the IFN induction phenotype of rHPIV1-P(C−) but, as shown above, differs from rHPIV1-P(C−) in that it does not induce apoptosis. rHPIV1-P(C−) was found to be more attenuated than rHPIV1-CF170S, and the difference in viral load between the two vaccination groups over time is indicated in Fig. 6. Despite its restricted replication, rHPIV1-P(C−) provided AGMs with protection against i.n. and i.t. challenge with 106 TCID50 wt HPIV1 per site at 28 days p.i. (Table 3). The replication of the wt HPIV1 challenge virus was restricted by approximately 100-fold in the URT and the LRT of rHPIV1-P(C−)-immunized monkeys compared to its levels of replication in nonimmunized monkeys (Table 3).
FIG. 6.

rHPIV1-P(C−) is attenuated for replication in both the URT and LRT of AGMs. Groups of AGMs were inoculated i.n. and i.t. with 106 TCID50 of wt HPIV1 (n = 16) or rHPIV1-P(C−) (n = 4) per site. Previously published data for rHPIV1-CF170S (n = 4) are also included for comparison (2). Mean daily virus titers ± SE in NP swabs (representative of the URT) (A) and TL fluid (representative of the LRT) (B) were determined for each sampling day (see Materials and Methods; the limit of detection is 0.5 log10 TCID50/ml). The area shaded in gray represents the additional reduction in replication observed for rHPIV1-P(C−) compared to that of rHPIV1-CF170S.
TABLE 3.
Protection against wt HPIV1 challenge in AGMs following immunization with rHPIV1-P(C−)a
| Immunizing virus | No. of animals | Mean peak titer (log10 TCID50/ml) ± SEb
|
|
|---|---|---|---|
| NP swabc | TL fluidd | ||
| Wt HPIV1 | 14 | 0.9 ± 0.2e | 0.8 ± 0.1e |
| rHPIV1-P(C−) | 4 | 2.8 ± 0.5e | 2.3 ± 0.4e |
| Mock immunized | 14 | 4.7 ± 0.3 | 4.4 ± 0.4 |
Monkeys were inoculated i.n. and i.t. with 106 TCID50 of the indicated virus in a 1-ml inoculum at each site. On day 28 p.i., monkeys were challenged i.n. and i.t. with 106 TCID50 wt HPIV1 in a 1-ml inoculum at each site.
The limit of detection was 0.5 log10 TCID50/ml.
NP samples were collected on days 0, 2, 4, 6, and 8 postchallenge. The titers on day zero were ≤0.5 log10 TCID50/ml.
TL samples were collected on days 2, 4, 6, and 8 postchallenge.
There was a statistically significant reduction in virus titer compared to that of the nonimmunized group at the same time point (P < 0.001; Student-Newman-Keuls multiple-comparison test).
Replication of rHPIV1-P(C−) is restricted in HAE cells in vitro.
Wt HPIV1 has previously been shown to infect ciliated HAE cells in an in vitro model of the HAE (4). Here, we characterized the ability of rHPIV1-P(C−) to infect HAE in a multiple-cycle growth curve. Following apical inoculation at a low input MOI (0.01 TCID50/cell), wt rHPIV1 replicated efficiently in HAE, reaching a peak titer of 7.4 log10 TCID50/ml in the apical wash fluid. However, the replication of rHPIV1-P(C−) was severely restricted in human ciliated cells, reaching a barely detectable peak of 1.8 log10 TCID50/ml (Fig. 7).
FIG. 7.

rHPIV1-P(C−) replicates very poorly in primary HAE cells compared to the replication of wt rHPIV1. HAE cultures were inoculated on the apical surface with the indicated virus at an MOI of 0.01 TCID50/cell, and virus titers in apical surface washes were determined at days 0 to 7 p.i. The results for triplicate cultures from two donors are shown as the means ± SE, and the limit of detection is 1.2 log10TCID50/ml.
DISCUSSION
The mutant rHPIV1-P(C−), which does not express any of the four wt C proteins but does express a wt P protein, was generated and characterized in vitro and in vivo. rHPIV1-P(C−) was found to replicate efficiently in vitro, implying that the HPIV1 C proteins are nonessential accessory proteins. However, rHPIV1-P(C−) expressed a novel protein not seen with rHPIV1 that may have been a truncated form of C, and thus, we cannot yet conclude that C-related proteins are completely dispensable. rHPIV1-P(C−) replicated with the same efficiency as wt HPIV1 early after infection of human- and monkey-derived cell lines, but its replication subsequently decreased coincident with the onset of extensive CPE that was not observed with wt rHPIV1. The C proteins of SeV have been extensively characterized as nonessential gene products with multiple functions (39, 41, 42). However, SeV and HPIV1 differ with regard to the genetic organization of their accessory proteins and the phenotypes specified by accessory protein mutations. First, SeV encodes a V protein in addition to the C proteins, whereas HPIV1 does not (33). Second, deletion of all four C proteins in SeV significantly restricted its replication in vitro (23, 39, 41), whereas loss of the wt forms of all four HPIV1 C proteins did not appear to reduce the replication efficiency of rHPIV1-P(C−) apart from the indirect effect of its enhanced CPE. Third, a 6-aa deletion in the N-terminal region of the SeV C protein had a profound effect on replication in its natural host, i.e., rodents (15), whereas a similar mutation in HPIV1 C did not affect replication in nonhuman primates, the closest available animal model to its natural human host (1). Fourth, the F170S mutation in SeV C induced apoptosis in primary mouse pulmonary epithelial cells (29), whereas the same mutation in HPIV1 failed to specify this phenotype in the present study. Since the genetic organization of the accessory proteins of SeV and HPIV1 differ and since the phenotypes of C protein mutants differ significantly in vitro and in vivo, the functions of the HPIV1 C proteins cannot be reliably inferred from findings obtained with SeV C protein mutants, and therefore, they must be determined directly. An amino acid alignment of the HPIV1 and SeV C proteins is provided in Fig. 8. In the case of HPIV1, this information has added importance since live attenuated vaccine candidates that are presently being prepared for clinical trials include mutations in the C protein.
FIG. 8.

CLUSTAL W alignment of the C protein amino acid sequences of SeV (Sendai C) and HPIV1 (GenBank accession numbers NP_056874 and NP_604436, respectively). The alignment was generated by using the MacVector software program (MacVector, Inc., Cary, NC), using default parameters. Conserved amino acids are indicated with asterisks.
Replication of rHPIV1-P(C−) in cell culture peaked early and then decreased steadily, coincident with the development of extensive CPE that was not observed with wt HPIV1. This CPE was associated with caspase 3 activation, cell rounding, nuclear condensation, and nuclear fragmentation, indicating that it was apoptotic in nature. For this report, we identified a novel protein expressed by rHPIV1-P(C−), and therefore, there is also the alternative possibility—albeit unlikely—that the observed apoptosis during infection with rHPIV1-P(C−) was a gain-of-function due to the novel protein. However, in other work in progress, we have generated additional HPIV1 mutants that also express this novel protein but do not induce apoptosis and do not specify an attenuation phenotype (data not shown). This indicates that the absence of the four C proteins, and not the presence of the additional protein, is associated with the apoptosis-inducing (and also the attenuation) phenotypes of rHPIV1-P(C−). Taken together, our data indicate that one function of the HPIV1 C proteins is to delay or prevent the apoptotic response of the infected cell. Apoptosis has also previously been associated with SeV C protein mutants. SeV C deletion mutants have been shown to induce apoptosis in vitro, whereas wt SeV did not (29, 39), suggesting that both SeV C proteins and HPIV1 C proteins act as inhibitors of apoptosis. In addition, the SeV mutant Ohita MVC11, containing the CF170S substitution, was observed to induce cell death in vitro, while wt SeV did not (29). Similar to our observations of rHPIV1-P(C−), Ohita MVC11 titers peaked early and decreased concomitant with the induction of apoptosis (29). However, data we have presented suggest that despite the relatively high degree of amino acid conservation (70% identity) between HPIV1 and SeV C proteins (Fig. 8), the induction of apoptosis and/or virus-mediated inhibition of apoptosis may be mediated by different mechanisms for HPIV1 and SeV C proteins. For example, although SeV Ohita MVC11 induces apoptosis, the HPIV1 mutant containing the homologous CF170S substitution, rHPIV1-CF170S, does not induce apoptosis. Furthermore, IRF-3 activation was recently shown to be required for apoptosis during SeV infection in human cell lines (52), but we have previously shown that rHPIV1-CF170S, which does not induce apoptosis, stimulates IRF-3 activation (65). Studies to define the mechanism of apoptosis inhibition by HPIV1 have been initiated.
The most extensively characterized function of paramyxovirus C proteins is their type I IFN antagonist activity. HPIV1 C proteins have been shown to disrupt the host type I IFN response by (i) inhibiting IRF-3 activation and thereby inhibiting the production of type I IFN and (ii) inhibiting STAT nuclear translocation (8) and thereby inhibiting the JAK-STAT signaling pathway (65). Our results support these previous findings by demonstrating that, in the absence of C proteins, type I IFN is produced in response to HPIV1 infection and is able to successfully establish an antiviral state in respiratory epithelial cells. In contrast, infection with wt HPIV1 inhibits type I IFN production, as well as the establishment of an antiviral state that results from the activation of the JAK-STAT pathway (18, 21, 43, 57, 63, 68). This pathway controls the transcription of a group of more than 300 genes termed the IFN-stimulated genes, which have antiviral, antiproliferative, immunomodulatory, and apoptosis-modulating functions (43). Therefore, it is not surprising that many viral proteins that have been characterized as IFN antagonists have also been identified as playing a role in the regulation of apoptosis. Examples of such proteins include the Bunyamwera virus NSs proteins (35, 67), the RSV NS1 and NS2 proteins (6, 60), and the influenza A virus NS1 protein (58, 62, 66, 73). Our data demonstrate that the HPIV1 C proteins also act both as type I IFN antagonists and as apoptosis antagonists. Interestingly, however, the anti-IFN and antiapoptosis activities of the HPIV1 C proteins were separable: while the rHPIV1-P(C−) and rHPIV1-CF170S mutants were indistinguishable with regard to the induction of IFN production and signaling in cell culture, only rHPIV1-P(C−) induced apoptosis.
Since the type I IFN response and apoptosis are both components of the host's innate antiviral response, viruses that have lost the ability to inhibit these responses are often attenuated in vivo. The attenuation phenotype of rHPIV1-P(C−) was evaluated in two in vivo models, i.e., in hamsters and AGMs, and an in vitro model of ciliated HAE. The replication of rHPIV1-P(C−) was restricted more than 1,000-fold in the URT and 250-fold in the LRT of hamsters following i.n. inoculation. Despite this high degree of attenuation, it induced substantial protection against challenge with wt HPIV1: challenge virus replication was restricted 200-fold in the URT and 15-fold in the LRT. rHPIV1-P(C−) was also evaluated in AGMs to determine its potential for use as a live attenuated pediatric vaccine against HPIV1; AGMs are a more-appropriate model, since they are evolutionarily and anatomically closer to humans than are hamsters. In AGMs, replication of rHPIV1-P(C−) was not detectable in the URT and was barely detectable in the LRT. Despite this very high level of attenuation, challenge virus titers were reduced 79- and 125-fold in the URT and LRT, respectively. It is possible that rHPIV1-P(C−) will replicate more efficiently (and thus be more immunogenic and protective) in humans than in AGMs since it is a human virus that is more permissive in its natural host. For example, while HPIV1 causes significant respiratory disease in humans, infection of AGMs is asymptomatic.
In order to obtain an independent assessment of attenuation prior to the initiation of clinical trials, the replication of rHPIV1-P(C−) was characterized in a HAE model, which uses primary HAE cells grown at an air-liquid interface to generate a differentiated, pseudostratified, ciliated epithelium that bears close structural and functional similarity to HAE in vivo (53, 72). A similar model has been used previously to evaluate the attenuation of RSV vaccines (70). We have also recently characterized the replication of wt HPIV1 and several HPIV1 vaccine candidates in the in vitro HAE model and observed a strong correlation between attenuation in AGMs and attenuation in HAE cells, indicating the potential usefulness of the HAE model for evaluating potential respiratory virus vaccine candidates (4). In our current HPIV1 study, the growth of rHPIV1-P(C−) in HAE cells was barely detectable, whereas wt rHPIV1 grew to high titers. Since rHPIV1-P(C−) is highly restricted in replication in AGMs and in HAE cells, previous observations that the level of replication in HAE cells mimics the level of replication in AGMs were confirmed, thus providing further evidence of the utility of the in vitro HAE model. This also suggests that this virus might be overattenuated in humans, potentially limiting its efficacy as a vaccine. Therefore, we are currently proceeding to clinical trials with a previously characterized virus, rHPIV1-CR84G/Δ170HNT553ALY942A (3), that is less attenuated than rHPIV1-P(C−) in AGMs. Only if rHPIV1-CR84G/Δ170HNT553ALY942A proves to be insufficiently attenuated will we advance rHPIV1-P(C−) into clinical trials.
The results of previous studies of rHPIV1 C mutants demonstrated that in vivo attenuation correlated with the ability to stimulate an effective type I IFN response, including IFN production and IFN signaling (1, 65). This was demonstrated in detail for rHPIV1-CF170S (65). The rHPIV1-P(C−) virus was indistinguishable from rHPIV1-CF170S with regard to the induction of type I IFN production and signaling to establish an antiviral state in vitro. However, rHPIV1-P(C−) was much more attenuated in AGMs than rHPIV1-CF170S. This increased level of attenuation might be due to the induction of apoptosis by rHPIV1-P(C−), a property not shared by rHPIV1-CF170S. We did not determine whether apoptosis was induced in vivo following rHPIV1-P(C−) infection; however, an SeV mutant containing the CF170S mutation was shown to induce apoptosis both in vitro and in vivo in the bronchial epithelium of mice, and this virus was also attenuated in its natural host (16, 29). It is possible that the in vitro apoptosis phenotype of rHPIV1-P(C−) can translate to in vivo effects. This would suggest that the greater level of in vivo attenuation of rHPIV1-P(C−) than of rHPIV1-CF170S is based on two additive effects: (i) the ability (of both viruses) to activate the type I IFN response and (ii) the ability of rHPIV1-P(C−) (but not PIV1-CF170S) to induce apoptosis. The results shown in Fig. 6 highlight this added degree of attenuation that could be due to the induction of apoptosis. However, it is also possible that a C protein function other than inhibition of the IFN response and apoptosis contributed to the high level of attenuation of rHPIV1-P(C−) in AGMs.
In summary, we have demonstrated that the HPIV1 C proteins are nonessential viral proteins that play an important role in viral replication in vitro and in vivo and act as antagonists of the type I IFN response and of apoptosis. Our HPIV1 C protein deletion mutant is highly attenuated in nonhuman primates and in primary HAE, yet this virus does confer significant protection against wt HPIV1 challenge in vivo. It remains to be determined whether such a highly attenuated virus would be useful as a vaccine candidate against PIV1 in humans.
Acknowledgments
We thank Brad Finneyfrock and Marisa St. Claire, Bioqual, Inc., for carrying out the primate studies. We are grateful to the bioimaging and flow cytometry groups, especially Lily Koo and David Stephany, within the Research Technology Branch at NIAID for their excellent support. We are also grateful to the directors and teams of the UNC Cystic Fibrosis Center Tissue Culture Core and to Susan Burkett for technical assistance. John Hiscott, McGill University, provided the VSV-GFP, and Yasuhiko Ito, Mie University School of Medicine, Mie, Japan, provided the mouse anti-HPIV1 HN (8.2.2A and 4.5) antibodies.
This project was funded as a part of the NIAID Intramural Program and National Institutes of Health (NIH) and by grant NIH R01 HL77844-1.
Footnotes
Published ahead of print on 9 July 2008.
REFERENCES
- 1.Bartlett, E. J., E. Amaro-Carambot, S. R. Surman, P. L. Collins, B. R. Murphy, and M. H. Skiadopoulos. 2006. Introducing point and deletion mutations into the P/C gene of human parainfluenza virus type 1 (HPIV1) by reverse genetics generates attenuated and efficacious vaccine candidates. Vaccine 242674-2684. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett, E. J., E. Amaro-Carambot, S. R. Surman, J. T. Newman, P. L. Collins, B. R. Murphy, and M. H. Skiadopoulos. 2005. Human parainfluenza virus type I (HPIV1) vaccine candidates designed by reverse genetics are attenuated and efficacious in African green monkeys. Vaccine 234631-4646. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett, E. J., A. Castano, S. R. Surman, P. L. Collins, M. H. Skiadopoulos, and B. R. Murphy. 2007. Attenuation and efficacy of human parainfluenza virus type 1 (HPIV1) vaccine candidates containing stabilized mutations in the P/C and L genes. Virol. J. 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartlett, E. J., M. Hennessey, M. H. Skiadopoulos, A. C. Schmidt, P. L. Collins, B. R. Murphy, and R. J. Pickles. 4 June 2008. The role of interferon in the replication of human parainfluenza virus type 1 wild type and mutant viruses in human ciliated airway epithelium. J. Virol. doi: 10.1128/JVI.02263-07. [DOI] [PMC free article] [PubMed]
- 5.Belshe, R. B., F. K. Newman, E. L. Anderson, P. F. Wright, R. A. Karron, S. Tollefson, F. W. Henderson, H. C. Meissner, S. Madhi, D. Roberton, H. Marshall, R. Loh, P. Sly, B. Murphy, J. M. Tatem, V. Randolph, J. Hackell, W. Gruber, and T. F. Tsai. 2004. Evaluation of combined live, attenuated respiratory syncytial virus and parainfluenza 3 virus vaccines in infants and young children. J. Infect. Dis. 1902096-2103. [DOI] [PubMed] [Google Scholar]
- 6.Bitko, V., O. Shulyayeva, B. Mazumder, A. Musiyenko, M. Ramaswamy, D. C. Look, and S. Barik. 2007. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-κB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 811786-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bitzer, M., S. Armeanu, F. Prinz, G. Ungerechts, W. Wybranietz, M. Spiegel, C. Bernlohr, F. Cecconi, M. Gregor, W. J. Neubert, K. Schulze-Osthoff, and U. M. Lauer. 2002. Caspase-8 and Apaf-1-independent caspase-9 activation in Sendai virus-infected cells. J. Biol. Chem. 27729817-29824. [DOI] [PubMed] [Google Scholar]
- 8.Bousse, T., R. L. Chambers, R. A. Scroggs, A. Portner, and T. Takimoto. 2006. Human parainfluenza virus type 1 but not Sendai virus replicates in human respiratory cells despite IFN treatment. Virus Res. 12123-32. [DOI] [PubMed] [Google Scholar]
- 9.Buchholz, U. J., S. Finke, and K. K. Conzelmann. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cadd, T., D. Garcin, C. Tapparel, M. Itoh, M. Homma, L. Roux, J. Curran, and D. Kolakofsky. 1996. The Sendai paramyxovirus accessory C proteins inhibit viral genome amplification in a promoter-specific fashion. J. Virol. 705067-5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colon-Ramos, D. A., P. M. Irusta, E. C. Gan, M. R. Olson, J. Song, R. I. Morimoto, R. M. Elliott, M. Lombard, R. Hollingsworth, J. M. Hardwick, G. K. Smith, and S. Kornbluth. 2003. Inhibition of translation and induction of apoptosis by Bunyaviral nonstructural proteins bearing sequence similarity to reaper. Mol. Biol. Cell 144162-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Counihan, M. E., D. K. Shay, R. C. Holman, S. A. Lowther, and L. J. Anderson. 2001. Human parainfluenza virus-associated hospitalizations among children less than five years of age in the United States. Pediatr. Infect. Dis. J. 20646-653. [DOI] [PubMed] [Google Scholar]
- 13.Curran, J., J. B. Marq, and D. Kolakofsky. 1992. The Sendai virus nonstructural C proteins specifically inhibit viral mRNA synthesis. Virology 189647-656. [DOI] [PubMed] [Google Scholar]
- 14.Elmore, S. 2007. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35495-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcin, D., J. Curran, M. Itoh, and D. Kolakofsky. 2001. Longer and shorter forms of Sendai virus C proteins play different roles in modulating the cellular antiviral response. J. Virol. 756800-6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcin, D., M. Itoh, and D. Kolakofsky. 1997. A point mutation in the Sendai virus accessory C proteins attenuates virulence for mice, but not virus growth in cell culture. Virology 238424-431. [DOI] [PubMed] [Google Scholar]
- 17.Garcin, D., J. B. Marq, S. Goodbourn, and D. Kolakofsky. 2003. The amino-terminal extensions of the longer Sendai virus C proteins modulate pY701-Stat1 and bulk Stat1 levels independently of interferon signaling. J. Virol. 772321-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodbourn, S., L. Didcock, and R. E. Randall. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 812341-2364. [DOI] [PubMed] [Google Scholar]
- 19.Gosselin-Grenet, A. S., J. B. Marq, L. Abrami, D. Garcin, and L. Roux. 2007. Sendai virus budding in the course of an infection does not require Alix and VPS4A host factors. Virology 365101-112. [DOI] [PubMed] [Google Scholar]
- 20.Gotoh, B., K. Takeuchi, T. Komatsu, and J. Yokoo. 2003. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J. Virol. 773360-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grandvaux, N., B. R. tenOever, M. J. Servant, and J. Hiscott. 2002. The interferon antiviral response: from viral invasion to evasion. Curr. Opin. Infect. Dis. 15259-267. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg, D. P., R. E. Walker, M. S. Lee, K. S. Reisinger, J. I. Ward, R. Yogev, M. M. Blatter, S. H. Yeh, R. A. Karron, C. Sangli, L. Eubank, K. L. Coelingh, J. M. Cordova, M. J. August, H. B. Mehta, W. Chen, and P. M. Mendelman. 2005. A bovine parainfluenza virus type 3 vaccine is safe and immunogenic in early infancy. J. Infect. Dis. 1911116-1122. [DOI] [PubMed] [Google Scholar]
- 23.Hasan, M. K., A. Kato, M. Muranaka, R. Yamaguchi, Y. Sakai, I. Hatano, M. Tashiro, and Y. Nagai. 2000. Versatility of the accessory C proteins of Sendai virus: contribution to virus assembly as an additional role. J. Virol. 745619-5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heikkinen, T., M. Thint, and T. Chonmaitree. 1999. Prevalence of various respiratory viruses in the middle ear during acute otitis media. N. Engl. J. Med. 340260-264. [DOI] [PubMed] [Google Scholar]
- 25.Henderson, F. W., A. M. Collier, M. A. Sanyal, J. M. Watkins, D. L. Fairclough, W. A. Clyde, Jr., and F. W. Denny. 1982. A longitudinal study of respiratory viruses and bacteria in the etiology of acute otitis media with effusion. N. Engl. J. Med. 3061377-1383. [DOI] [PubMed] [Google Scholar]
- 26.Heylbroeck, C., S. Balachandran, M. J. Servant, C. DeLuca, G. N. Barber, R. Lin, and J. Hiscott. 2000. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 743781-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horikami, S. M., R. E. Hector, S. Smallwood, and S. A. Moyer. 1997. The Sendai virus C protein binds the L polymerase protein to inhibit viral RNA synthesis. Virology 235261-270. [DOI] [PubMed] [Google Scholar]
- 28.Irie, T., N. Nagata, T. Yoshida, and T. Sakaguchi. 2008. Recruitment of Alix/AIP1 to the plasma membrane by Sendai virus C protein facilitates budding of virus-like particles. Virology 371108-120. [DOI] [PubMed] [Google Scholar]
- 29.Itoh, M., H. Hotta, and M. Homma. 1998. Increased induction of apoptosis by a Sendai virus mutant is associated with attenuation of mouse pathogenicity. J. Virol. 722927-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itoh, M., Y. Isegawa, H. Hotta, and M. Homma. 1997. Isolation of an avirulent mutant of Sendai virus with two amino acid mutations from a highly virulent field strain through adaptation to LLC- MK2 cells. J. Gen. Virol. 783207-3215. [DOI] [PubMed] [Google Scholar]
- 31.Iwane, M. K., K. M. Edwards, P. G. Szilagyi, F. J. Walker, M. R. Griffin, G. A. Weinberg, C. Coulen, K. A. Poehling, L. P. Shone, S. Balter, C. B. Hall, D. D. Erdman, K. Wooten, and B. Schwartz. 2004. Population-based surveillance for hospitalizations associated with respiratory syncytial virus, influenza virus, and parainfluenza viruses among young children. Pediatrics 1131758-1764. [DOI] [PubMed] [Google Scholar]
- 32.Karron, R. A., R. B. Belshe, P. F. Wright, B. Thumar, B. Burns, F. Newman, J. C. Cannon, J. Thompson, T. Tsai, M. Paschalis, S. L. Wu, Y. Mitcho, J. Hackell, B. R. Murphy, and J. M. Tatem. 2003. A live human parainfluenza type 3 virus vaccine is attenuated and immunogenic in young infants. Pediatr. Infect. Dis. J. 22394-405. [DOI] [PubMed] [Google Scholar]
- 33.Karron, R. A., and P. L. Collins. 2007. Parainfluenza viruses, p. 1497-1526. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 34.Karron, R. A., M. Makhene, K. Gay, M. H. Wilson, M. L. Clements, and B. R. Murphy. 1996. Evaluation of a live attenuated bovine parainfluenza type 3 vaccine in two- to six-month-old infants. Pediatr. Infect. Dis. J. 15650-654. [DOI] [PubMed] [Google Scholar]
- 35.Kohl, A., R. F. Clayton, F. Weber, A. Bridgen, R. E. Randall, and R. M. Elliott. 2003. Bunyamwera virus nonstructural protein NSs counteracts interferon regulatory factor 3-mediated induction of early cell death. J. Virol. 777999-8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolakofsky, D., T. Pelet, D. Garcin, S. Hausmann, J. Curran, and L. Roux. 1998. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J. Virol. 72891-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komada, H., S. Kusagawa, C. Orvell, M. Tsurudome, M. Nishio, H. Bando, M. Kawano, H. Matsumura, E. Norrby, and Y. Ito. 1992. Antigenic diversity of human parainfluenza virus type 1 isolates and their immunological relationship with Sendai virus revealed by using monoclonal antibodies. J. Gen. Virol. 73875-884. [DOI] [PubMed] [Google Scholar]
- 38.Komatsu, T., K. Takeuchi, J. Yokoo, and B. Gotoh. 2004. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 325137-148. [DOI] [PubMed] [Google Scholar]
- 39.Koyama, A. H., H. Irie, A. Kato, Y. Nagai, and A. Adachi. 2003. Virus multiplication and induction of apoptosis by Sendai virus: role of the C proteins. Microbes Infect. 5373-378. [DOI] [PubMed] [Google Scholar]
- 40.Kozak, M. 1987. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 196947-950. [DOI] [PubMed] [Google Scholar]
- 41.Kurotani, A., K. Kiyotani, A. Kato, T. Shioda, Y. Sakai, K. Mizumoto, T. Yoshida, and Y. Nagai. 1998. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells 3111-124. [DOI] [PubMed] [Google Scholar]
- 42.Latorre, P., T. Cadd, M. Itoh, J. Curran, and D. Kolakofsky. 1998. The various Sendai virus C proteins are not functionally equivalent and exert both positive and negative effects on viral RNA accumulation during the course of infection. J. Virol. 725984-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levy, D. E., and A. Garcia-Sastre. 2001. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 12143-156. [DOI] [PubMed] [Google Scholar]
- 44.Marx, A., T. J. Torok, R. C. Holman, M. J. Clarke, and L. J. Anderson. 1997. Pediatric hospitalizations for croup (laryngotracheobronchitis): biennial increases associated with human parainfluenza virus 1 epidemics. J. Infect. Dis. 1761423-1427. [DOI] [PubMed] [Google Scholar]
- 45.McLean, J. E., A. Ruck, A. Shirazian, F. Pooyaei-Mehr, and Z. F. Zakeri. 2008. Viral manipulation of cell death. Curr. Pharm. Des. 14198-220. [DOI] [PubMed] [Google Scholar]
- 46.Moeller, K., I. Duffy, P. Duprex, B. Rima, R. Beschorner, S. Fauser, R. Meyermann, S. Niewiesk, V. ter Meulen, and J. Schneider-Schaulies. 2001. Recombinant measles viruses expressing altered hemagglutinin (H) genes: functional separation of mutations determining H antibody escape from neurovirulence. J. Virol. 757612-7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy, B. R., G. A. Prince, P. L. Collins, K. Van Wyke Coelingh, R. A. Olmsted, M. K. Spriggs, R. H. Parrott, H. W. Kim, C. D. Brandt, and R. M. Chanock. 1988. Current approaches to the development of vaccines effective against parainfluenza and respiratory syncytial viruses. Virus Res. 111-15. [DOI] [PubMed] [Google Scholar]
- 48.Murphy, B. R., D. D. Richman, E. G. Chalhub, C. P. Uhlendorf, S. Baron, and R. M. Chanock. 1975. Failure of attenuated temperature-sensitive influenza A (H3N2) virus to induce heterologous interference in humans to parainfluenza type 1 virus. Infect. Immun. 1262-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newman, J. T., J. M. Riggs, S. R. Surman, J. M. McAuliffe, T. A. Mulaikal, P. L. Collins, B. R. Murphy, and M. H. Skiadopoulos. 2004. Generation of recombinant human parainfluenza virus type 1 vaccine candidates by importation of temperature-sensitive and attenuating mutations from heterologous paramyxoviruses. J. Virol. 782017-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newman, J. T., S. R. Surman, J. M. Riggs, C. T. Hansen, P. L. Collins, B. R. Murphy, and M. H. Skiadopoulos. 2002. Sequence analysis of the Washington/1964 strain of human parainfluenza virus type 1 (HPIV1) and recovery and characterization of wild-type recombinant HPIV1 produced by reverse genetics. Virus Genes 2477-92. [DOI] [PubMed] [Google Scholar]
- 51.Nolan, S. M., M. H. Skiadopoulos, K. Bradley, O. S. Kim, S. Bier, E. Amaro-Carambot, S. R. Surman, S. Davis, M. St. Claire, R. Elkins, P. L. Collins, B. R. Murphy, and A. Schaap-Nutt. 2007. Recombinant human parainfluenza virus type 2 vaccine candidates containing a 3′ genomic promoter mutation and L polymerase mutations are attenuated and protective in non-human primates. Vaccine 256409-6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peters, K., S. Chattopadhyay, and G. C. Sen. 2008. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J. Virol 823500-3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pickles, R. J., D. McCarty, H. Matsui, P. J. Hart, S. H. Randell, and R. C. Boucher. 1998. Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer. J. Virol. 726014-6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Power, U. F., K. W. Ryan, and A. Portner. 1992. The P genes of human parainfluenza virus type 1 clinical isolates are polycistronic and microheterogeneous. Virology 189340-343. [DOI] [PubMed] [Google Scholar]
- 55.Reed, G., P. H. Jewett, J. Thompson, S. Tollefson, and P. F. Wright. 1997. Epidemiology and clinical impact of parainfluenza virus infections in otherwise healthy infants and young children <5 years old. J. Infect. Dis. 175807-813. [DOI] [PubMed] [Google Scholar]
- 56.Sakaguchi, T., A. Kato, F. Sugahara, Y. Shimazu, M. Inoue, K. Kiyotani, Y. Nagai, and T. Yoshida. 2005. AIP1/Alix is a binding partner of Sendai virus C protein and facilitates virus budding. J. Virol. 798933-8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schultz-Cherry, S., N. Dybdahl-Sissoko, G. Neumann, Y. Kawaoka, and V. S. Hinshaw. 2001. Influenza virus ns1 protein induces apoptosis in cultured cells. J. Virol. 757875-7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skiadopoulos, M. H., T. Tao, S. R. Surman, P. L. Collins, and B. R. Murphy. 1999. Generation of a parainfluenza virus type 1 vaccine candidate by replacing the HN and F glycoproteins of the live-attenuated PIV3 cp45 vaccine virus with their PIV1 counterparts. Vaccine 18503-510. [DOI] [PubMed] [Google Scholar]
- 60.Spann, K. M., K. C. Tran, B. Chi, R. L. Rabin, and P. L. Collins. 2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J. Virol. 784363-4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stojdl, D. F., B. D. Lichty, B. R. tenOever, J. M. Paterson, A. T. Power, S. Knowles, R. Marius, J. Reynard, L. Poliquin, H. Atkins, E. G. Brown, R. K. Durbin, J. E. Durbin, J. Hiscott, and J. C. Bell. 2003. VSV strains with defects in their ability to shut down innate immunity are potent systemic anti-cancer agents. Cancer Cell 4263-275. [DOI] [PubMed] [Google Scholar]
- 62.Talon, J., C. M. Horvath, R. Polley, C. F. Basler, T. Muster, P. Palese, and A. Garcia-Sastre. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 747989-7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taniguchi, T., and A. Takaoka. 2002. The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 14111-116. [DOI] [PubMed] [Google Scholar]
- 64.Tapparel, C., S. Hausmann, T. Pelet, J. Curran, D. Kolakofsky, and L. Roux. 1997. Inhibition of Sendai virus genome replication due to promoter-increased selectivity: a possible role for the accessory C proteins. J. Virol. 719588-9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Cleve, W., E. Amaro-Carambot, S. R. Surman, J. Bekisz, P. L. Collins, K. C. Zoon, B. R. Murphy, M. H. Skiadopoulos, and E. J. Bartlett. 2006. Attenuating mutations in the P/C gene of human parainfluenza virus type 1 (HPIV1) vaccine candidates abrogate the inhibition of both induction and signaling of type I interferon (IFN) by wild-type HPIV1. Virology 35261-73. [DOI] [PubMed] [Google Scholar]
- 66.Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. Garcia-Sastre. 2000. Influenza A virus NS1 protein prevents activation of NF-κB and induction of alpha/beta interferon. J. Virol. 7411566-11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weber, F., A. Bridgen, J. K. Fazakerley, H. Streitenfeld, N. Kessler, R. E. Randall, and R. M. Elliott. 2002. Bunyamwera bunyavirus nonstructural protein NSs counteracts the induction of alpha/beta interferon. J. Virol. 767949-7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber, F., G. Kochs, and O. Haller. 2004. Inverse interference: how viruses fight the interferon system. Viral Immunol. 17498-515. [DOI] [PubMed] [Google Scholar]
- 69.Won, S., T. Ikegami, C. J. Peters, and S. Makino. 2007. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J. Virol. 8113335-13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wright, P. F., M. R. Ikizler, R. A. Gonzales, K. N. Carroll, J. E. Johnson, and J. A. Werkhaven. 2005. Growth of respiratory syncytial virus in primary epithelial cells from the human respiratory tract. J. Virol. 798651-8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang, M. R., S. R. Lee, W. Oh, E. W. Lee, J. Y. Yeh, J. J. Nah, Y. S. Joo, J. Shin, H. W. Lee, S. Pyo, and J. Song. 2008. West Nile virus capsid protein induces p53-mediated apoptosis via the sequestration of HDM2 to the nucleolus. Cell Microbiol. 10165-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang, L., M. E. Peeples, R. C. Boucher, P. L. Collins, and R. J. Pickles. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 765654-5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhirnov, O. P., T. E. Konakova, T. Wolff, and H. D. Klenk. 2002. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 761617-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]