

Abstract
We examined consecutive protease (PR) and reverse transcriptase (RT) sequences from human immunodeficiency virus (HIV) type 1—infected individuals, to distinguish changes resulting from sequence evolution due to possible superinfection. Between July 1997 and December 2001, ⩾2 PR and RT samples from 718 persons were sequenced at Stanford University Hospital. Thirty-seven persons had highly divergent sequence pairs characterized by a nucleotide distance of >4.5% in PR or >3.0% in RT. In 16 of 37 sequence pairs, divergence resulted from the loss of mutations during a treatment interruption or from the gain of mutations with reinstitution of treatment. tat and/or gag sequencing of HIV-1 from cryopreserved plasma samples could be performed on 15 of the 21 divergent isolate pairs from persons without a treatment interruption. The sequences of these genes, unaffected by selective drug pressure, were monophyletic. Although HIV-1 PR and RT genes from treated persons may become highly divergent, these changes usually are the result of sequence evolution, rather than superinfection.
Human immunodeficiency virus (HIV) type 1 genotypic drug-resistance testing performed by sequencing the protease (PR) and reverse transcriptase (RT) genes has, in many countries, become part of the routine care of infected individuals receiving antiretroviral therapy [1, 2]. Individuals experiencing persistent viremia may have ⩾2 of their isolates sequenced at different times. The extent to which different isolates from the same individual diverge has implications for sequence quality control and provides an opportunity to detect superinfection. We examined PR and RT sequences of viruses consecutively isolated from the same individuals, to determine whether sequence differences could have resulted from superinfection with a second HIV-1 isolate, rather than from the accumulation of changes in the original virus population.
METHODS
Individuals, isolates, and sequences
Between 1 July 1997 and 31 December 2001, the Stanford University Hospital (SUH) Diagnostic Virology Laboratory sequenced the PR and RT genes of 4366 HIV-1 isolates obtained from 3155 individuals in northern California at the request of their physicians. The present study is based on the HIV-1 isolates of those patients who had ⩾2 samples sent for sequencing at least 1 month apart. The study was approved by the Stanford University Panel of Human Subjects in Medical Research. Human experimentation guidelines of the US Department of Health and Human Services, the Stanford University institutional review board, and the University of California, San Francisco, institutional review board were followed in the conduct of this research.
The complete PR and RT positions 1–250 of plasma HIV-1 were sequenced as described elsewhere [3]. In brief, RNA was extracted from 0.2 mL of plasma using the guanidinethiocyanate lysis reagent in the AMPLICOR HIV Monitor test kit (Roche Diagnostic Systems). Reverse-strand cDNA was generated from viral RNA, and first-round polymerase chain reaction (PCR) was done with Superscript One-Step RT-PCR (Life Technologies). Direct PCR (population based) cycle sequencing was performed using AmpliTaq DNA fluorescent sequencing polymerase and dRhodamine terminators (Applied Biosystems). Electropherograms were generated using an Applied Biosystems Model 377 sequencer, and sequences were assembled using the manufacturer’s Factura and Auto Assembler sequence analysis software.
Quality control analyses performed at the time of sequencing
For quality control purposes, each sequence was examined manually and analyzed by software developed in the laboratory. This software compared each new sequence with all sequences generated within the preceding 2–3 months. Sample sequences having an uncorrected nucleotide distance of <2.0% different from a previous sequences were flagged and examined for the possibility of laboratory contamination. Each sequence was also compared with all previous sequences from the same individual. Sequences having an uncorrected nucleotide distance >3.5% different from a previous sequence were flagged and examined for the possibility of a sample mix-up. Approximately 1–2 confirmed sample mix-ups per year were identified in this manner. These sample mix-ups were excluded from the analysis described here.
Retrospective analyses performed to identify possible reinfection
We used 3 approaches to identify individuals whose viral sequences diverged the most over time (figure 1): measuring the nucleotide distance between consecutive isolates from the same individual; performing phylogenetic analyses of all the sequences produced by the laboratory, to determine whether sequences from the same individual were clustered with one another, rather than with the sequences of any other individual tested by the laboratory; and developing a genetic pattern similarity (GPS) score for determining whether 2 sequences shared uncommon amino acid substitutions. The 3 approaches were applied to PR and RT separately and were used to identify individuals whose treatment histories would be reviewed and whose isolates would undergo sequencing of tat and/or gag—genes that are not under direct antiretroviral selection pressure (figure 1).
FIGURE 1.
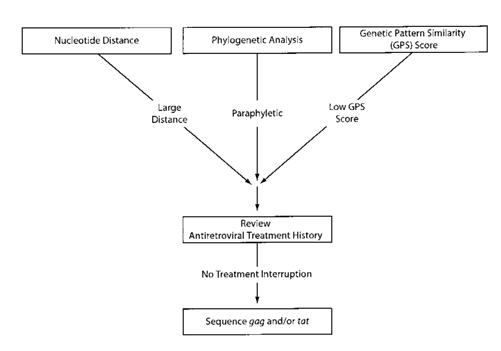
Approach to identifying divergent isolates and testing them for possible superinfection. Highly divergent isolates were identified by nucleotide distance measurement, phylogenetic analysis, and genetic pattern—similarity analysis. Highly divergent isolates that could not be explained on the basis of a treatment interruption were submitted for tat and/or gag gene sequencing.
Distance measures
Six different measures of nucleotide distance between 2 sequences were generated, depending on whether positions known to be associated with drug resistance were excluded and on how ambiguous nucleotides (indicative of mixtures) were handled. PR positions considered to be associated with drug resistance included codons 10, 20, 24, 30, 32, 33, 36, 46, 47, 48, 50, 53, 54, 63, 71, 73, 77, 82, 84, 88, 90, and 93. RT positions considered to be associated with drug resistance included codons 41, 44, 62, 65, 67, 69, 70, 74, 75, 77, 100, 101, 103, 106, 108, 115 116, 118, 151, 179, 181, 184, 188, 190, 210, 215, 219, 225, 227, and 230 [1].
Mixtures were handled in 1 of 3 ways: all differences between the aligned nucleotides in 2 sequences were counted as complete differences (Hamming distance), differences between the aligned nucleotides in 2 sequences were ignored if there was any overlap between nucleotides at a single position (i.e., if one sequence had a Y, indicating a mixture of C and T, and the second sequence had a C; unweighted distance), or differences were scored according to the extent of overlap between the 2 nucleotides being compared (mixture-weighted distance) [4]. For example, an R and an A mismatch was assigned a 0.5 difference, whereas a Y and an A mismatch was assigned a 1.0 difference.
Phylogenetic trees
Separate neighbor-joining trees of PR and RT were constructed from the matrices of mixture-weighted distances between all 4366 isolates [5]. Isolates from the same individual were considered to be monophyletic if they were the sole descendants of their most recent common ancestor. Isolates from the same individual were considered to be paraphyletic if they shared their most recent common ancestor with at least 1 other isolate.
GPS
We created position-specific profiles of the PR and the RT from the 4366 sequences performed by the SUH diagnostic laboratory. Each profile contained the proportion of sequences containing each amino acid at each position along both genes within the 4366 sequences. The profile, therefore, was based on sequences from both treated and untreated patients, as well as sequences from patients with different subtypes (although ∼99% of sequences belonged to subtype B). The GPS score at a position was defined as 0 when 2 sequences had different amino acids at the same position. Otherwise, the GPS score at a position was assigned a score of —log10(p), where p is the proportion of sequences containing the shared amino acid at that position. Thus, the GPS score at a position was also 0 if 2 sequences shared an absolutely conserved amino acid (p = 1; log10(1) = 0). The GPS score at a position was high if 2 sequences shared an uncommon amino acid (i.e., p = 0.001; —log10(0.001) = 3). The total GPS score between 2 sequences was calculated by adding the GPS scores at positions 1–99 in the PR and 1–250 in the RT.
To identify GPS scores that strongly suggest that 2 sequences were derived from isolates from the same individual rather than from different individuals, we compared the distribution of GPS scores of all pairs of sequences from different persons (3155 choose-2) using a bootstrap sample of 1,000,000 pairs with the pairs of sequences from those individuals with >1 sequence (718 individuals). On the basis of the distribution of GPS scores from different persons for the PR and RT, we chose a threshold that strongly suggested that 2 isolates were obtained from the same, rather than a different, individual.
Sequencing of gag and tat
The treatment histories of persons with sequences that had a high mixture-weighted distance and low GPS score or were paraphyletic were reviewed to determine whether a prolonged treatment interruption of either all PR and/or RT inhibitors took place that could explain a large sequence change. If no such treatment interruption occurred and if cryopreserved plasma samples were available for both isolates, we sequenced fragments of the gag and/or tat genes of virus from these samples (figure 1).
Viral RNA was extracted from plasma using the Qiagen viral RNA kit (Qiagen). Eluted RNA was converted to cDNA by incubation with 50 μg of random 6-nt-long oligomers, 1 μL of 10 mmol/L dNTP, 1 μL of 200 U/μL Moloney murine leukemia virus—RT (Life Technologies), and a master mix for 1 h at 37°C. First-round primers for tat were TatED1 5′-GCAGGAGTGGAAGCCATAATAAG-3′ (HXB2 position, 5721–5743) and TatED2 5′-TTCTATGAATACTATGGTCCACACAACTAT-3′ (HXB2 position, 6119–6148). Second-round primers were TatED3 5′-GAATTCTGCAACAACTGCTGTTTAT-3′ (HXB2 position, 5743–5767) and TatED4 5′-ATTGCTGCTACTACTAATGCTACTATTGC-3′ (HXB2 position, 6083–6111). First-round primers for gag were described elsewhere [6]. The second-round primers were p17EDHMA5 5′-GTGCGAGAGCGTCAGTATTAAGCG-3′ (HXB2 position, 794–817) and p17EDHMA3 5′-TTTCTTACTTTTGTTTTGCTCTTCC-3′ (HXB2 position, 1104–1128).
All RNA extractions, reverse transcription, and preparation of the first-round PCR tubes were performed in a preamplification room free of amplified HIV products. After purification, dideoxy terminator reactions of tat and gag PCR products were initiated using the second-round PCR antisense primer. Sequence products were resolved using an ABI 3100 capillary sequencer. Sequences of gag and tat genes were submitted to GenBank (accession nos. AY178912–AY178931 and AY178932–AY178961, respectively).
RESULTS
Nucleotide distances between paired sequences
Between 1 July 1997 and 31 December 2001, 4366 HIV-1 PR and RT isolates from 3155 individuals were sequenced. Seven hundred eighteen individuals submitted isolates for sequencing more than once. The mean number of sequences per individual was 2.5 (range, 2–6 sequences/individual), and a total of 1061 pairs of sequences were examined. The mean time between sequences was 12.2 months (range, 1–46 months), and the total time between sequence pairs was 1072 person-years.
Figure 2 shows the distribution of nucleotide distances between consecutive pairs of sequences from the same individual, by gene (PR or RT) and method for measuring nucleotide distance. Figures 2A and 2C show the distribution of unadjusted nucleotide distances between sequence pairs through PR positions 1–99 and RT positions 1–250, respectively. Figures 2B and 2D show the distribution of distances between sequence pairs of PR and RT, excluding codons associated with drug resistance (mutation-adjusted distance). Each of the graphs in figure 2 shows the distribution of distances using 3 approaches for handling mixtures: scoring mixtures as complete differences (Hamming distance), calculating a weighted distance on the basis of the components of a mixture (mixture-weighted distance), and ignoring mixtures.
FIGURE 2.
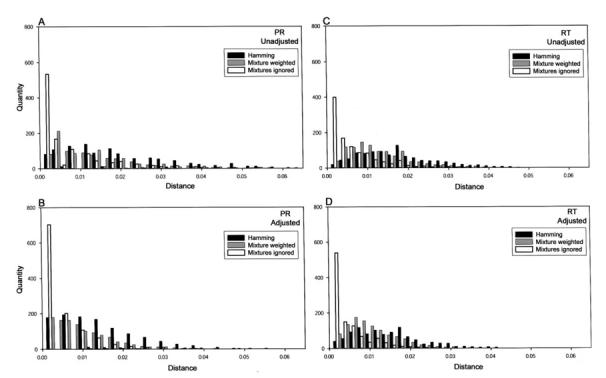
Distributions of nucleotide distances between consecutive pairs of sequences from the same individual, by gene (protease [PR] or reverse transcriptase [RT]) and method for measuring distance. A and C, Distribution of nucleotide distances between sequence pairs using PR positions 1–99 and RT distances 1–250, respectively. B and D, Distribution of distances between sequence pairs of PR and RT, excluding codons associated with drug resistance (mutation-adjusted distance). Each of the 4 graphs shows the distribution of distances using 3 approaches for handling mixtures: scoring mixtures as complete differences (Hamming distance; black bars), calculating a weighted distance based on the components of a mixture (mixture-weighted distance; gray bars), and ignoring mixtures (white bars).
The gene, the decision to include or exclude drug resistance positions (mutation adjustment), and the method for handling mixtures all influenced the distribution of nucleotide distance. Table 1 shows the various medians and the 95% and 99% quantiles for the greatest distances between the 1061 sequence-pairs, stratified by gene, mixture handling technique, and mutation adjustment. As expected, mutation-adjusted distances were lower than unadjusted distances, and mixture-weighted distances were lower than Hamming distances but higher than distances that ignored mixtures. For all methods, the median distance and 95% and 99% quantiles were higher for PR than for RT sequences.
Table 1.
Median and 95% and 99% quantiles of each distance method for protease (PR) and reverse transcriptase (RT).
| Gene, drug-resistance mutations,a distanceb |
Median | 95% quantile |
99% quantile |
|---|---|---|---|
| PR | |||
| Unadjusted | |||
| Hamming | 1.7 | 4.7 | 6.1 |
| Mixture-weighted | 1.0 | 3.7 | 5.4 |
| Unweighted | 0.3 | 3.0 | 4.7 |
| Adjusted | |||
| Hamming | 1.3 | 3.4 | 4.7 |
| Mixture-weighted | 0.6 | 2.4 | 3.4 |
| Unweighted | 0.0 | 1.7 | 3.0 |
| RT | |||
| Unadjusted | |||
| Hamming | 1.5 | 3.6 | 4.3 |
| Mixture-weighted | 0.9 | 2.7 | 3.5 |
| Unweighted | 0.3 | 1.9 | 2.9 |
| Adjusted | |||
| Hamming | 1.3 | 3.4 | 4.7 |
| Mixture-weighted | 0.7 | 2.1 | 2.8 |
| Unweighted | 0.1 | 1.3 | 2.2 |
NOTE. Data are percentage nucleotide distance.
The mutation-adjusted distance excludes codons associated with drug resistance. The unadjusted distance includes all 99 PR positions and 250 RT positions.
Three approaches were used to handle nucleotide mixtures. The Hamming distance weighed all nucleic acid differences between 2 sequences equally. The mixture-weighted distance weighed nucleotide distances according to the extent of overlap between 2 nucleotides (e.g., A and R, which represent a mixture of A/G overlap by 50%). The unweighted distance ignored positions that had nucleotide mixtures.
Phylogenetic analysis
In the PR neighbor-joining tree, 962 PR sequence-pairs were monophyletic, and 99 were paraphyletic. In the RT neighbor-joining tree, 1054 sequence-pairs were monophyletic, and 7 were paraphyletic. The median number of weeks between PR (59 vs. 43 weeks; P < .001) and RT sequences (60 vs. 46 weeks; P = .2) and the median nucleotide distance between PR (3.3% vs. 0.8%; P < .001) and RT (3.0% vs. 0.9%; P < .001) were higher for paraphyletic than for monophyletic sequence pairs.
GPS scores
Figure 3 shows the distribution of GPS scores obtained by comparing all PR and RT sequences from different individuals and all PR and RT sequences from the same individual. The PR GPS scores of sequence pairs from different individuals were 0.3–9.6, whereas the scores of sequence pairs from the same individual were 0.9–17.3. The RT GPS scores of sequence pairs from different individuals were 0.9–17.9, whereas the scores of sequence pairs from the same individual were 2.5–25.5. On the basis of the empirical distribution of the GPS interindividual scores, we determined that 2 PR sequences with a GPS score >7 or 2 RT sequences with a GPS score >9 were ∼10,000 times more likely to be from the same individual, rather than different individuals.
FIGURE 3.
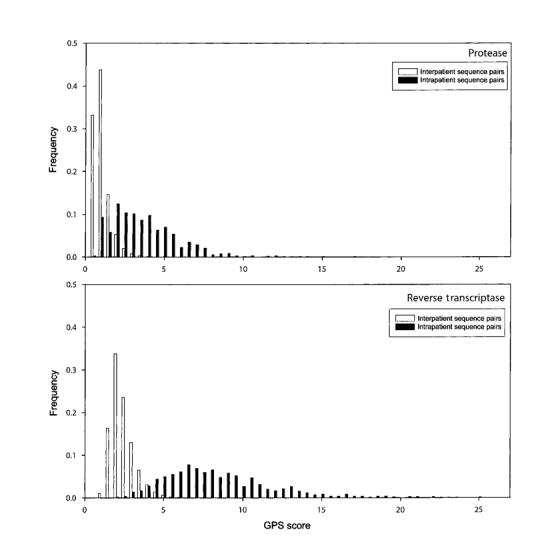
Distribution of genetic pattern similarity (GPS) scores obtained by comparing all intraperson and interperson protease (top) and reverse transcriptase (bottom) sequences.
Composite analysis
Figure 4 summarizes the nucleotide distances, phylogenetic analyses, and GPS scores for each pair of PR and RT sequences from the 718 individuals with >1 available sequence. Thirty-seven individuals had highly divergent sequence pairs (characterized by a nucleotide distance of at least 4.5% in PR or 3.0% in RT) and a high GPS score (>7 in PR or >9 in RT). Twenty-four of these individuals had sequence pairs that were paraphyletic (21 in the PR tree, 2 in the RT tree, and 1 in both trees). The remaining paraphyletic sequence pairs contained at least 1 sequence that was very close to the root of the tree (median, 2.5%), which makes the absence of clustering less meaningful.
FIGURE 4.
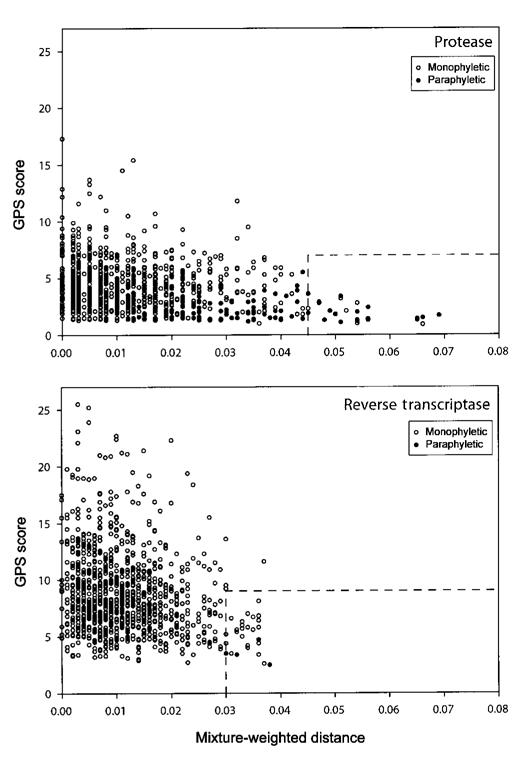
Graphic summary of the mixture-weighted nucleotide distances, genetic pattern similarity (GPS) scores, and phylogenetic clustering of consecutive of protease and reverse transcriptase isolates from 718 persons.
In 16 of the 37 individuals with divergent sequence pairs, the sequence change occurred during a treatment interruption (14 individuals) or followed the resumption of therapy after a treatment interruption (2 individuals). Of the remaining 21 individuals, 18 had a treatment change but no interruption, and 3 had treatment histories that were unavailable or considered to be unreliable.
gag and tat sequencing
We sequenced the tat and/or gag genes of 15 of 21 divergent sequence-pairs from individuals without a treatment interruption from whom cryopreserved samples were available. Phylogenetic trees created from the tat genes of 14 individuals and from the gag genes of 15 individuals (along with 20 additional San Francisco Bay Area control subjects) showed that all sequence pairs were monophyletic. Figure 5 shows the phylogenetic tree of the tat sequences. The median nucleotide distance between the 14 pairs of consecutive tat genes was 0.9% (range, 0%–2.0%). The median nucleotide distance between the 15 pairs of consecutive gag genes was 2% (range, 0%–5.0%). Table 2 summarizes the nucleotide distances, GPS scores, phylogenetic clustering results, and amino acid mutation changes for these 15 isolates.
FIGURE 5.
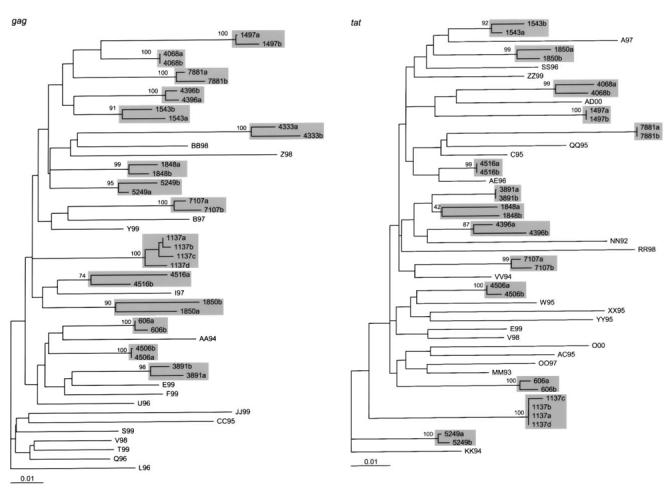
Neighbor-joining tree constructed from the gag (left) and tat (right) sequences of 20 northern California control subjects and the 15 pairs of highly divergent isolates for which cryopreserved plasma samples were available. The scale bar indicates percentage nucleotide distance.
Table 2.
Nucleotide distances, genetic pattern similarity (GPS) scores, phylogenetic clustering, and amino acid mutations of 15 highly divergent pairs of human immunodeficiency virus type 1 isolates from the same individual.
| Subject | Weeks | Nucleotide distance, % |
Paraphy letic |
GPS score |
Mutations |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PR | RT | gag | tat | PR | RT | PR | RT | PR | RT | ||
| 7107 | 57 | 2.7 | 3.8 | 1.2 | 2.2 | Yes | Yes | 1.3 | 2.5 |
L10I, L19LI, K20KR, M36MV, N37NS, K43KN, L63LP, I93L |
V35I, D113DE, K122E, D123E, I142T, A158S, E169D, D177E, V241VG, P243PS, V245VL, V245M |
| 4506 | 123 | 0.7 | 3.7 | 0.7 | 0.7 | No | No | 4.3 | 2.6 |
Q7QR, L10I, T12S, L63P, I72T, V77VI, I93L |
K20R, A33AG, T39A, M41L, V60I, D67N, K70R, V75M, K104T, V118I, T139TI, N175NH, Q207E, L210LW, R211RK, T215Y, D218DE, K219Q, L228H |
| 3891 | 69 | 4.4 | 3.6 | 2.3 | 0.4 | No | Yes | 2.3 | 4.7 |
L10V, L10I, V11VA, I15V, K20R, E35D, M36I, N37D, N37E, I54V, Q58E, L63T, L63P, I72V, V82A, L90LM |
M41L, V60I, D67DN, K103N, I135T, I142V, V179VG, M184V, L210W, R211K, T215Y, L234LH, V245K |
| 4068 | 40 | 5.4 | 3.6 | 0.0 | 1.8 | No | No | 2.8 | 8.1 |
L10V, L10LI, I13V, L24LF, M36MI, N37T, M46L, M46ML, F53L, F53FL, K55R, K55KR, L63P, C67F, A71V, A71AV, G73S, G73GXAST, V77I, V77VI, L90M, L90LM, C95L |
M41L, V60I, D67N, K70R, L74V, V75M, V75VM, K103T, L109M, V118I, V118VI, K122E, K122KE, I142V, I142IV, A158AS, K166KR, M184V, L187M, E203K, Q207E, Q207EK, H208Y, H208HY, L210W, R211K, T215Y, D218E, D218DE, K219Q, K219KQ, L228H, L228LH, M230L, K238T |
| 4396 | 59 | 6.6 | 3.6 | 0.7 | 2.5 | Yes | No | 1.5 | 5.8 |
R8RQ, L10I, K20IM, E35G, M36I, N37D, I54M, I62V, L63S, I64V, I72V, T74S, I85V, L90M, I93L |
D67N, K70R, L74I, K103N, K122E, D123N, I142IT, I142V, A158AS, A158S, S162A, E169D, T200A, E203EK, Q207E, R211K, K219E, L228H, L234I, K238KR |
| 1137 | 113 | 3.9 | 3.4 | 0.7 | 0.7 | Yes | No | 1.5 | 4.2 | L10V, M36MI, M46I, I54V, Q58E, L63P, A71V, L76V, V82T, V82VI, L90M, I93IL |
T39A, M41L, K43Q, E44D, L74V, R83K, V90I, K103N, Y115F, K122E, S162Y, S162A, V179VG, Y181C, M184V, I202IV, T215Y |
| 4516 | 47 | 6.5 | 3.1 | 5.0 | 0.0 | Yes | No | 1.4 | 6.6 |
L10I, V11VL, L24I, K43T, M46I, I54V, D60N, L63P, E65D, A71V, A71AV, I72IM, V77I, V82A, I93L |
L74V, A98S, K103N, V108I, D123E, I135L, I142V, M164MI, E169X, F171FS, I178L, V179I, Y181C, M184V, V189I, G196E, T200A, R211K, H221Y, L228LF |
| 1497 | 49 | 6.5 | 2.7 | 1.8 | 0.0 | Yes | No | 1.3 | 7.3 |
R8RQ, L10I, I13V, K14KR, I15IV, K20I, M36MI, M46I, F53Y, I54V, K55R, Q58E, L63P, L63LP, A71V, G73T, I84V, L90M |
K20R, V35I, M41L, K43E, E44D, D67N, R83RK, A98G, V118I, D123E, I135T, D177E, M184V, T200K, T200I, Q207E, H208Y, L210W, R211K, T215Y, T215TNSY, K223Q, E248D |
| 1543 | 50 | 5.2 | 2.7 | 2.5 | 1.8 | No | No | 2.1 | 9.9 |
L10F, M46I, L63P, I64V, I66IV, G73AT, V77I, I84V, I85V, L90M, I93L |
M41L, K43E, E44A, D67N, T69D, A98S, K103N, V118I, I135T, R143RT, M184V, H208Y, L210W, R211E, T215Y, K219R, L228H, Q242H, V245L |
| 7881 | 50 | 5.4 | 2.6 | 2.5 | 0.0 | Yes | No | 1.2 | 8.5 |
L10LI, L10I, I13V, K20KM, K20M, L33L_L, M36I, F53FL, I54V, Q58E, I62V, L63P, A71V, V82A, L89V, L90M, Q92E, Q92K |
V35T, D67N, T69TN, T69N, K70R, K103N, D121DH, K122E, D123DN, I142V, I167IV, D177DG, M184V, T200A, Q207K, R211A, T215TS, T215F, K219E |
| 606 | 18 | 5.0 | 0.5 | 0.7 | 1.1 | Yes | No | 1.8 | 6.9 |
L10I, K20MI, N37D, N37ND, M46I, I62V, L63P, I64V, K70R, A71AVIT, I72IL, G73S, N88E, L89M, L90M |
K20R, M41L, A98S, K102KR, K122E, A158S, M184V, Q197E, T215Y, L228LH |
| 1848 | 39 | 1.5 | 3.2 | 1.9 | 2.9 | No | No | 1.5 | 5.4 | D30N, L63P, A71V |
M41L, D67N, K103N, V118I, I135T, R143RS, E203EK, L205LR, H208Y, L210W, R211KQ, R211K, T215Y, K223KE, M230MV, V245M, D250E |
| 1850 | 43 | 4.0 | 3.4 | 4.8 | 1.4 | Yes | No | 1.4 | 5.8 |
L10I, V32I, M46I, F53L, L63P, I64IXLM, I64M, A71V, V82A, L89M, L90M, I93L |
E6D, D67N, K70R, V90I, A98S, K103N, K122P, I135V, K166R, D177E, I178V, I180L, T200V, I202V, R211K, V245M |
| 4333 | 109 | 4.9 | 2.7 | 2.6 | NA | Yes | No | 2.1 | 6.9 |
L10I, K20I, L24I, M36V, M36I, M46I, Q58E, L63P, I64V, A71V, V82T, I93L |
K20R, V21I, T39K, M41L, K43E, E44D, V60I, D67N, V75M, K104N, V118I, K122E, I135V, I135IV, K166R, M184V, I202V, L210W, R211K, T215Y, A272P, K277R, T286A, P294S, E297K |
| 5249 | 14 | 5.3 | 3.6 | 3.3 | 0.4 | No | No | 1.6 | 4.4 |
M46I, F53L, I54V, I62V, L63T, A71V, V77VI, V82A, L90M, I93L |
M41L, E79ED, K82KR, K103N, K122E, I142V, M184V, T215Y |
NOTE. Mutations in bold type were present in the first but not the second sequence. Mutations in italic type were present in the second but not the first sequence. Mutations in roman type were present in both the first and second sequence. NA, not available (unable to amplify); PR, protease; RT, reverse transcriptase.
DISCUSSION
Sequence divergence between PR or RT sequences obtained at different times from HIV-1—infected individuals receiving antiretroviral treatment may result from the acquisition or loss of mutations at positions associated with drug resistance [7–10], from genetic bottlenecks in which a new therapy selects for preexisting rare variants with differences at positions not associated with drug resistance [11, 12], from mix-ups with a sample from a different person [13], and from superinfection with a virus from a different person [14–16]. Of these possibilities, superinfection is the most interesting to clinicians and researchers because of its implications for protective immunity, viral interference, and the development of recombinant virus strains [17]. We have, therefore, focused our discussion on the ability of our data to detect superinfection. However, our study also provides new data on the extent to which PR and RT sequences of HIV-1 isolates from treated individuals may change over time. An understanding of these data is essential for maintaining the quality of PR and RT sequencing in laboratories performing genotypic resistance testing.
The present study shows that, although HIV-1 PR and/or RT genes from treated persons may become highly divergent, this divergence almost always results from intrahost sequence evolution, rather than from superinfection. We arrived at this conclusion by identifying 37 (5%) individuals with the most-divergent isolates from our sample of 718 individuals, reviewing their treatment histories, and then sequencing tat and/or gag—genes that are not under selective drug pressure—from these individuals. For 16 of these 37 individuals, the sequence divergence was consistent with a documented treatment interruption or with the resumption of therapy after a treatment interruption. For another 15 individuals, paired tat and/or gag genes were monophyletic, which argues against superinfection. Stored samples were not available for tat and gag sequencing for the remaining 7 individuals.
Although numerous cases of simultaneous infection with 2 distinct HIV-1 strains have been reported [17–20], there have been only 4 well-documented cases of superinfection, in which a second virus infected a person well after an initial infection [14–16]. All 4 cases occurred in persons who were monitored prospectively after the identification of primary infection. In 3 of the 4 cases, superinfection occurred with a virus belonging to a different subtype than the primary strain [14, 15].
Approximately 99% of isolates in our cohort belong to subtype B [21], which makes it more difficult to detect superinfection and requires the use of 2 new methods for assessing sequence divergence: the mixture-weighted distance allows the calculation of nucleotide distances between sequences containing nucleotide mixtures [4], whereas the GPS score allows the detection of signature polymorphisms within an infected individual that may be present even in the absence of phylogenetic clustering. The GPS method bears some similarity to the signature pattern analysis method of Korber et al. [22] but differs in that it evaluates 2 sequences at a time from a large set of sequences for which the amino acid profile (distribution of variants) at each position is known.
Each of the previous published cases of superinfection was initially identified by population-based sequencing. We cannot, however, exclude the possibility that superinfection occurred in our cohort but was undetected, because the superinfection virus remained a minor variant. To detect these cases, it would be necessary to sequence multiple clones from each of the plasma samples, rather than restricting the analysis to those samples with the most genetic divergence. Such a strategy would be optimally suited to a small cohort of persons at high risk of superinfection, rather than a large cohort like ours in which exposure history was not available.
Because HIV-1 is likely to undergo recombination when 2 viruses infect the same cell, we also examined sequences for the possibility that a second virus may have recombined with virus present in the first plasma sample. We divided each of the PR sequences into 2 segments of ∼150 nt and each of the RT sequences into 5 segments of 150 nt and calculated the divergence between matched gene segments from the same person. This analysis identified 61 additional persons with virus isolates having PR or RT segments with a mixture-weighted distance >4.5% (∼7 nt) between at least 1 pair of matched 150-nt segments. In nearly all these additional cases, divergence appeared to be caused by a major change in treatment (usually an interruption) or by a few unexplained nucleotide changes (data not shown), which suggests that, even if recombination with a superinfecting strain had occurred, it was extremely rare.
Surveillance data from the San Francisco Department of Health suggest that, among persons with the most common risk factors for HIV-1 in Northern California (e.g., male homosexuality and intravenous drug use), there is an ∼1%–2% annual incidence of new HIV-1 infection [23, 24]. Prospective studies in cohorts of individuals for whom risk behavior is documented have the potential to better define the incidence of superinfection. However, such studies are difficult to perform. Although we do not know the risk profile of our cohort, it is likely that ∼10–20 new cases of HIV-1 would have been expected during 1072 person-years of follow-up had the individuals in the cohort not already been infected.
Superinfection may be prevented as a result of partial immunity, the effect of antiretroviral drugs on superinfection with a drug-susceptible strain, or viral interference from the original virus strain. However, we cannot quantify the risk of superinfection because we do not know the extent of HIV-1 exposure within our cohort and because we cannot completely exclude the possibility that some cases of superinfection escaped detection. Therefore, infected individuals, even those receiving anti-retroviral therapy, should continue to avoid activities that could transmit HIV-1 to others or increase their risk of a second infection.
Acknowledgments
Financial support: National Institutes of Health (NIH; grant AI-46148 to M.J.G. and R.W.S.); Centers for Disease Control and Prevention (grant U64/CCU917889-01) and NIH (grant AI-447320) to E.D. and R.T.
References
- 1.Hirsch MS, Brun-Vezinet F, D’Aquila RT, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society—USA Panel. JAMA. 2000;283:2417–26. doi: 10.1001/jama.283.18.2417. [DOI] [PubMed] [Google Scholar]
- 2.US Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection A Guidelines for the use of antiretroviral agents in HIV-1—infected adults and adolescents 20024 FebruaryAvailable at:http://www.aidsinfo.nih.gov/guidelines/ [DOI] [PubMed]
- 3.Shafer RW, Hertogs K, Zolopa AR, et al. High degree of interlaboratory reproducibility of human immunodeficiency virus type 1 protease and reverse transcriptase sequencing of plasma samples from heavily treated patients. J Clin Microbiol. 2001;39:1522–9. doi: 10.1128/JCM.39.4.1522-1529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzales MJ, Dugan JM, Shafer RW. Synonymous—non-synonymous mutation rates between sequences containing ambiguous nucleotides (Syn-SCAN) Bioinformatics. 2002;18:886–7. doi: 10.1093/bioinformatics/18.6.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates; Sunderland, MA: 1998. [Google Scholar]
- 6.Leitner T, Escanilla D, Franzen C, Uhlen M, Albert J. Accurate reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proc Natl Acad Sci USA. 1996;93:10864–9. doi: 10.1073/pnas.93.20.10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devereux HL, Youle M, Johnson MA, Loveday C. Rapid decline in detectability of HIV-1 drug resistance mutations after stopping therapy. AIDS. 1999;13:F123–7. [PubMed] [Google Scholar]
- 8.Verhofstede C, Wanzeele FV, Van Der Gucht B, De Cabooter N, Plum J. Interruption of reverse transcriptase inhibitors or a switch from reverse transcriptase to protease inhibitors resulted in a fast reappearance of virus strains with a reverse transcriptase inhibitor—sensitive genotype. AIDS. 1999;13:2541–6. doi: 10.1097/00002030-199912240-00007. [DOI] [PubMed] [Google Scholar]
- 9.Kantor R, Fessel WJ, Zolopa AR, et al. Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy. Antimicrob Agents Chemother. 2002;46:1086–92. doi: 10.1128/AAC.46.4.1086-1092.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izopet J, Souyris C, Hance A, et al. Evolution of human immunodeficiency virus type 1 populations after resumption of therapy following treatment interruption and shift in resistance genotype. J Infect Dis. 2002;185:1506–10. doi: 10.1086/340215. [DOI] [PubMed] [Google Scholar]
- 11.Nijhuis M, Boucher CA, Schipper P, Leitner T, Schuurman R, Albert J. Stochastic processes strongly influence HIV-1 evolution during suboptimal protease-inhibitor therapy. Proc Natl Acad Sci USA. 1998;95:14441–6. doi: 10.1073/pnas.95.24.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibanez A, Clotet B, Martinez MA. Human immunodeficiency virus type 1 population bottleneck during indinavir therapy causes a genetic drift in the env quasispecies. J Gen Virol. 2000;81:85–95. doi: 10.1099/0022-1317-81-1-85. [DOI] [PubMed] [Google Scholar]
- 13.Learn GH, Jr, Korber BT, Foley B, Hahn BH, Wolinsky SM, Mullins JI. Maintaining the integrity of human immunodeficiency virus sequence databases. J Virol. 1996;70:5720–30. doi: 10.1128/jvi.70.8.5720-5730.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos A, Hu DJ, Nguyen L, et al. Intersubtype human immunodeficiency virus type 1 superinfection following seroconversion to primary infection in two injection drug users. J Virol. 2002;76:7444–52. doi: 10.1128/JVI.76.15.7444-7452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jost S, Bernard MC, Kaiser L, et al. A patient with HIV-1 superinfection. N Engl J Med. 2002;347:731–6. doi: 10.1056/NEJMoa020263. [DOI] [PubMed] [Google Scholar]
- 16.Altfeld M, Allen TM, Yu XG, et al. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature. 2002;420:434–9. doi: 10.1038/nature01200. [DOI] [PubMed] [Google Scholar]
- 17.Blackard JT, Cohen DE, Mayer KH. Human immunodeficiency virus superinfection and recombination: current state of knowledge and potential clinical consequences. Clin Infect Dis. 2002;34:1108–14. doi: 10.1086/339547. [DOI] [PubMed] [Google Scholar]
- 18.Zhu T, Wang N, Carr A, Wolinsky S, Ho DD. Evidence for coinfection by multiple strains of human immunodeficiency virus type 1 subtype B in an acute seroconvertor. J Virol. 1995;69:1324–7. doi: 10.1128/jvi.69.2.1324-1327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz RS, Sabino EC, Mayer A, Mosley JW, Busch MP. Dual human immunodeficiency virus type 1 infection and recombination in a dually exposed transfusion recipient. Transfusion Safety Study Group. J Virol. 1995;69:3273–81. doi: 10.1128/jvi.69.6.3273-3281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long EM, Martin HL, Jr, Kreiss JK, et al. Gender differences in HIV-1 diversity at time of infection. Nat Med. 2000;6:71–5. doi: 10.1038/71563. [DOI] [PubMed] [Google Scholar]
- 21.Gonzales MJ, Machekano RN, Shafer RW. Human immunodeficiency virus type 1 reverse-transcriptase and protease subtypes: classification, amino acid mutation patterns, and prevalence in a Northern California clinic—based population. J Infect Dis. 2001;184:998–1006. doi: 10.1086/323601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korber B, Myers G. Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retroviruses. 1992;8:1549–60. doi: 10.1089/aid.1992.8.1549. [DOI] [PubMed] [Google Scholar]
- 23.Waldo CR, McFarland W, Katz MH, MacKellar D, Valleroy LA. Very young gay and bisexual men are at risk for HIV infection: the San Francisco Bay Area Young Men’s Survey II. J Acquir Immune Defic Syndr. 2000;24:168–74. doi: 10.1097/00126334-200006010-00012. [DOI] [PubMed] [Google Scholar]
- 24.San Francisco Department of Public Health HIV/AIDS Epidemiology Annual Report 2001Available athttp://www.dph.sf.ca.us/Reports/ STD/HIVAIDSAnnlRpt2001.pdf