

Abstract
The reaction pathways of high-spin iron hydride complexes are relevant to the mechanism of N2 reduction by nitrogenase, which has been postulated to involve paramagnetic iron-hydride species. However, almost all known iron hydrides are low-spin, diamagnetic Fe(II) compounds. We have demonstrated that the first high-spin iron hydride complex, LtBuFeH (LtBu = bulky β-diketiminate), reacts with PhN=NPh to completely cleave the N-N double bond, giving LtBuFeNHPh. Here, we disclose a series of experiments that elucidate the mechanism of this reaction. Crossover and kinetic experiments rule out common non-radical mechanisms, and support a radical chain mechanism mediated by iron(I) species including a rare η2-azobenzene complex. Therefore, this high-spin iron(II) hydride can perform [1,2]-insertion through both non-radical and radical insertion mechanisms, a special feature that enables novel reactivity.
Introduction
Transition metal hydride complexes play a role in numerous bond transformations.1 One of their characteristic reactions is transfer of “H−” to substrates like alkenes and ketones, which results in an overall two-electron reduction of that substrate. Because nitrogenase substrates (e.g. N2, alkynes, CO2, CN−, N2O) are all reduced by multiples of two electrons, metal hydrides have been proposed as intermediates in the catalytic reduction of N2 to NH3 by nitrogenase enzymes.2,3 Iron is the only transition metal that is common to all nitrogenases,4 suggesting that an Fe-H intermediate could be an active species during nitrogenase catalysis. Consistent with the presence of a hydride intermediate, nitrogenase produces H2 from water at low substrate concentrations.5 Recent ENDOR and EPR evidence supports the presence of a paramagnetic iron-hydride species upon reduction of the α-70Ile mutant of A. vinelandii nitrogenase.6 Although this mutant is incapable of reducing N2, its close similarity to the wild-type nitrogenase indicates that Fe-H species should be considered as prospective intermediates during the nitrogenase catalytic cycle.
Our interest in synthetic analogues for nitrogenase7 motivated us to synthesize iron hydride complexes with coordination environments similar to those potentially present in nitrogenase. An ideal “model” complex would act as a functional model, reducing substrates like those reduced by nitrogenase (N2, alkynes, NO, CO2, HCN, diazenes, hydrazine) in a manner that is more amenable to detailed mechanistic inquiry than the enzyme.8 A hydride complex with perfect structural analogy to the “belt iron” sites implicated as active sites of the FeMoco (Figure 1)3,9,10 would have three weak-field sulfur donors, a high-spin electronic configuration, and a low coordination number (3, 4, 5) at iron. No complex with all of these properties is known. However, by using the bulky anionic β-diketiminate ligand LtBu (LR = 1,3-R2-1,3-bis(2,6-diisopropylphenyl)propyl, Figure 2), we were able to synthesize the first hydride complex of iron with a coordination number less than five, [LtBuFe([μ-H)]2.11,12 In solution, it is in equilibrium with a three-coordinate monomer, LtBuFeH, and it binds pyridine to give LtBuFe(H)(pyridine), which has a trigonal pyramidal geometry. The monomeric hydride complexes have the desired high-spin (S = 2) electronic configuration at the metal, by virtue of the low coordination number and weak ligand field at iron. Although they lack the characteristic three-sulfur coordination environment of the iron atoms in the FeMoco, diketiminate-supported iron hydrides show promise for giving hints into the relevant reactivity patterns of hydride ligands in a weak ligand field.13
Figure 1.
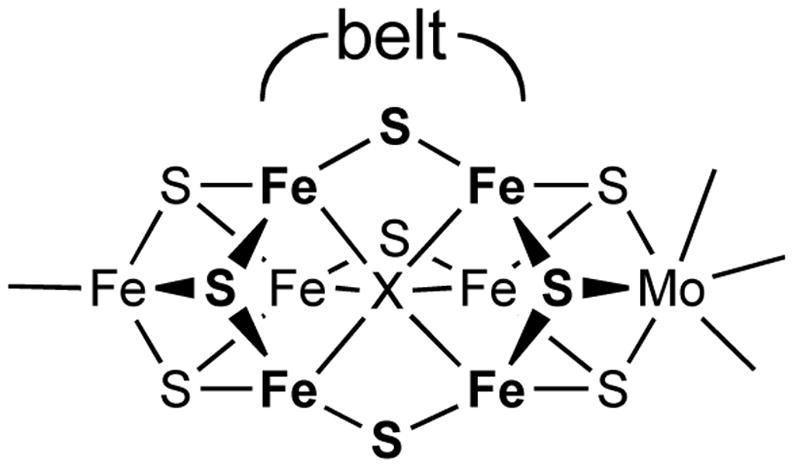
The active site “FeMoco” of nitrogenase. The “belt” region refers to the central Fe6S3X core, where X is an unidentified anion (probably C4−, N3−, or O2−).9
Figure 2.
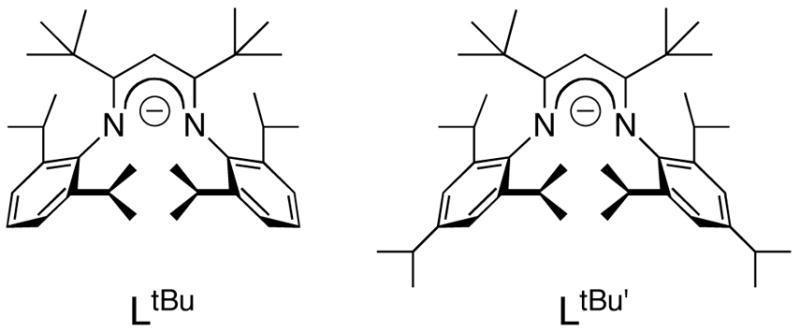
Bulky β-diketiminate ligands used in this work.
The main function of nitrogenase is the cleavage of N-N bonds, but in vitro it reduces numerous substrates. For example, A. vinelandii iron-molybdenum nitrogenase has been reported to reduce methyldiazene and hydrazine.14 N-N single bond cleavage is promoted by nitrogenase and model complexes,14,15,16 though rarely with iron.17 In a preliminary communication, we reported that [LtBuFeH]2 reacts with azobenzene (PhN=NPh) to afford the amido complex LtBuFeNHPh (boxed reaction in Scheme 1).11 This reaction was the first example of complete N=N cleavage by an iron-hydride complex, and one of the few by any iron complex.18,19,20,21 An unusual facet of the transformation in Scheme 1 is that the complete N=N bond cleavage of azobenzene requires no overall change in oxidation state at the iron. This contrasts with literature N=N cleavage reactions that result in four-electron oxidation of a metal (or multiple metals).22
Scheme 1.
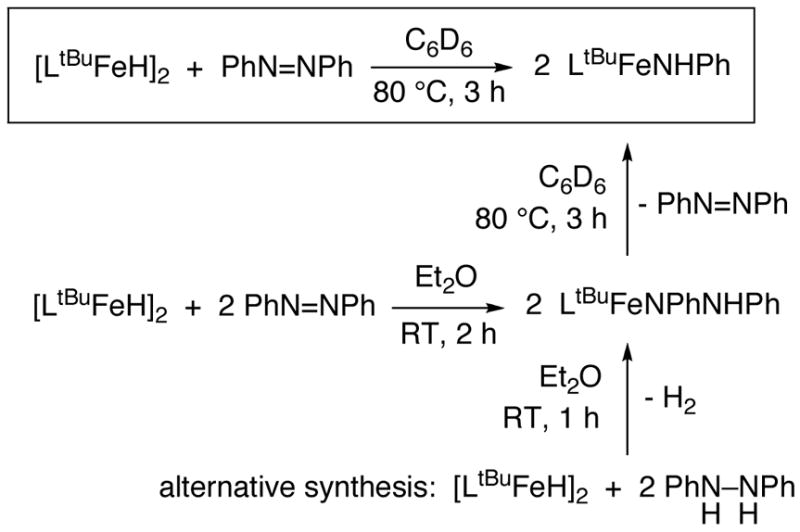
Given the importance of breaking nitrogen-nitrogen bonds in the formation of ammonia from N2 by nitrogenase, the ability of a low coordinate iron hydride to completely break an N=N bond is remarkable. This manuscript reports investigations aimed to determine the mechanism of this unusual transformation.
Results and Discussion
Synthesis of High-Spin Iron(II) Hydride Complexes
We have reported the synthesis of dimeric [LtBuFeH]2, and showed that it is in equilibrium with its monomeric, trigonal-planar form LtBuFeH.11 In order to provide an alternative ligand with comparable electronic properties and more steric bulk, we have prepared the new ligand abbreviated LtBu (Figure 2), which differs from LtBu by the presence of an additional isopropyl group at the para position of each aryl ring. Creation of LtBu’ necessitated the synthesis of 2,4,6-triisopropylaniline, which came from reduction of 2,4,6-triisopropylnitrobenzene. Incorporation of this aniline into the β-diketiminate ligand followed procedures analogous to those used in the synthesis of LtBuH (see the Experimental Section for details).
Hydride complexes were accessed by addition of KHBEt3 to the chloride complex LtBuFeCl or LtBuFeCl at room temperature in toluene. An immediate color change from red to brown was observed, with dissolution of the starting material (LtBu’ complexes tend to be somewhat more soluble than their LtBu analogues). In each case, it is important to remove the byproduct BEt3 immediately, because the borane reacts with the hydride complex. The details of the reactions between iron hydride complexes and boranes will be reported separately.23
The new hydride complex [LtBu’FeH]2 was characterized by solution methods and by X-ray crystallography. The solid state structure of [LtBu’FeH]2 (Figure 3) shows a dimer similar to [LtBuFeH]2.11 Each iron atom has a distorted tetrahedral geometry around the metal center, from binding of the diketiminate ligand in a η2 binding mode, and coordination of two bridging hydrides. These hydrides were located in a Fourier map, and their positional parameters were refined, but their positions must be regarded with uncertainty, given the small electron density at the hydrogen atom. The β-diketiminate ligands are twisted into a distorted boat conformation, with a substantial folding along the N…N axis (21.79(9)° and 27.39(7)°) in [LtBu’FeH]2; 24.3(3)° in [LtBuFeH]2). The Fe-Fe distance of [LtBu’FeH]2 is slightly shorter (2.5292(3) Å) than that of [LtBuFeH]2 (2.624(2) Å) despite the increased steric hindrance in the former. The nature of the iron-iron interaction is the subject of ongoing studies.
Figure 3.
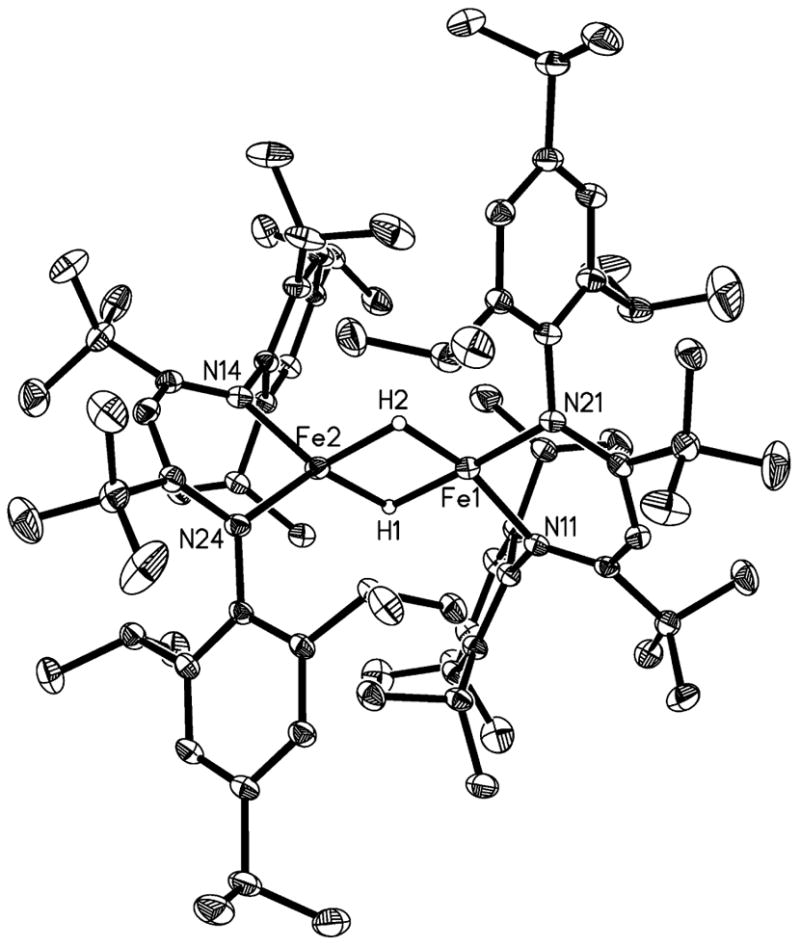
Solid-state structure of [LtBu’FeH]2, with 50% probability thermal ellipsoids. Hydrogen atoms on the diketiminate ligands are omitted for clarity. Fe1-N11 1.988(1) Å, Fe1-N21 1.978(1) Å, Fe2-N14 2.043(1) Å, Fe2-N24 2.010(1) Å, Fe1-Fe2 2.5292(3) Å, N11-Fe1-N21 96.41(5)°, N14-Fe2-N24 97.60(5)°.
The 1H NMR spectrum of each hydride dimer is unusually complicated, with at least 17 relatively sharp but partially overlapped peaks present over a range of 80 to −130 ppm. The complicated room-temperature 1H NMR spectrum is attributed to hindered C-C and C-N bond rotations in the dimer, because the X-ray crystal structures show that the complexes are exceptionally crowded. Despite the complex 1H NMR spectra, these materials are analytically pure, react with 3-hexyne to give high yields of vinyl products,11 and convert to monomeric forms with simple 1H NMR spectra at elevated temperatures. The ratio of monomer to dimer in a 17 mM solution in C6D6 at room temperature is slightly larger for the LtBu’ compound (ca. 0.5) than the LtBu compound (ca. 0.3). Therefore, a majority of the hydride complex is present as dimer at room temperature. However, our previous studies have shown that monomer is formed rapidly (the reported activation parameters for addition of alkyne to monomer extrapolate to a half-life of several min at room temperature and a few seconds at 80 °C),11 so in the following discussion the dimeric hydride complexes are treated as being functionally equivalent to the monomers.
Reaction with Azobenzene
Reaction of [LtBuFeH]2 or [LtBu’FeH]2 with 1 equiv of PhN=NPh in diethyl ether gave the isolable hydrazido complexes LtBuFeNPhNHPh and LtBu’FeNPhNHPh, respectively. The reactions are complete in less than 30 min at room temperature. Although the swiftness of this reaction has prevented kinetic studies, it is possible to propose a mechanism based on analogous reactions we have reported previously. We envision the first step of the reaction as dissociation of the hydride dimer into monomeric LtBuFeH or LtBu’FeH, which is rapid at room temperature.11 This is followed by [1,2]-insertion of the Fe-H bond across the N=N bond.24 We have observed [LRFeH]2 to add across C=C, C≡C, C=N, and C=O bonds, and we have used kinetic isotope effects, kinetics, and computations to support a [1,2]-insertion mechanism.25 Therefore, it is reasonable to presume that reduction of the N=N bond of diazene to the N-N bond of the hydrazido ligand follows an analogous pathway. Note that this reaction requires 1 equiv of azobenzene per iron, twice the amount needed for the overall conversion of [LtBuFeH]2 and PhN=NPh to LtBuFeNHPh.
The spectroscopic characteristics of the hydrazido complexes are unremarkable, and the characterization of LtBuFeNPhNHPh has been presented previously.11 As an alternative to the preparation of the hydrazido complexes from azobenzene, they can also be accessed by addition of two equiv of PhNHNHPh to [LtBuFeH]2, releasing H2. Samples of LtBuFeNPhNHPh from the two preparatory methods gave similar rate constants in subsequent reactions (see below), but the rate constants were more reproducible when generated from the latter method.
Kinetic and Crossover Studies on the N–N Cleavage Reaction
Benzene and toluene solutions of purified LtBuFeNPhNHPh (36–48 mM) decompose at 80 °C to form 1 equiv of LtBuFeNHPh and 0.5 equiv of PhN=NPh (Scheme 1).11,26 We followed the progress of this reaction using 1H NMR spectroscopy in C6D6 and toluene-d8. The reaction was monitored at a number of temperatures, and rate constants were derived from exponential fits to the integrations of peaks at 32 and −72 ppm for LtBuFeNPhNHPh and 28 ppm for LtBuFeNHPh. Integrations were calibrated to an internal capillary containing Tp*2Co in toluene-d8. No intermediates or other products were observed under these conditions. At most temperatures (see below), [LtBuFeNPhNHPh] followed an exponential decay with a rate constant near that calculated for growth of [LtBuFeNHPh]. The exponential fit implies a first-order rate dependence on [LtBuFeNPhNHPh], and plots of ln[LtBuFeNPhNHPh] vs. time were linear (Figure 4).27
Figure 4.
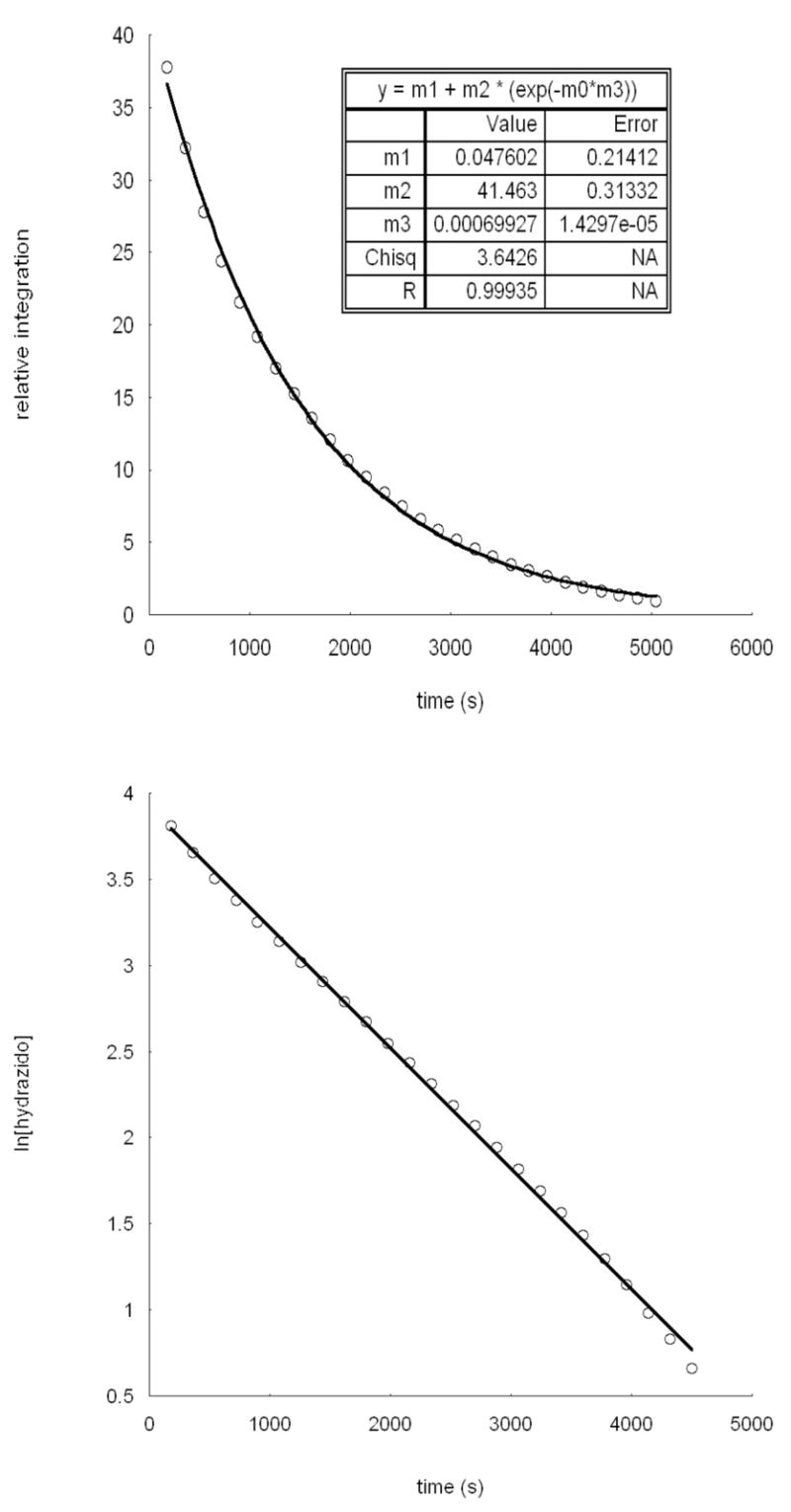
(a) Top: Plot of [LtBuFeNPhNHPh] vs time at 358 K. (b) Bottom: Plot of ln[LtBuFeNPhNHPh] vs time at 358 K.
Interestingly, the plots of [LtBuFeNPhNHPh] and [LtBuFeNHPh] vs. time at the lowest temperature used (42 °C) show an induction period before the beginning of the exponential decay or growth (Figure 5). This behavior, evident in three different trials, suggests an initiation step in the reaction pathway. The length of the induction period varied from 45 to 90 min in different trials. The implications of this observation will be described in more detail below.
Figure 5.
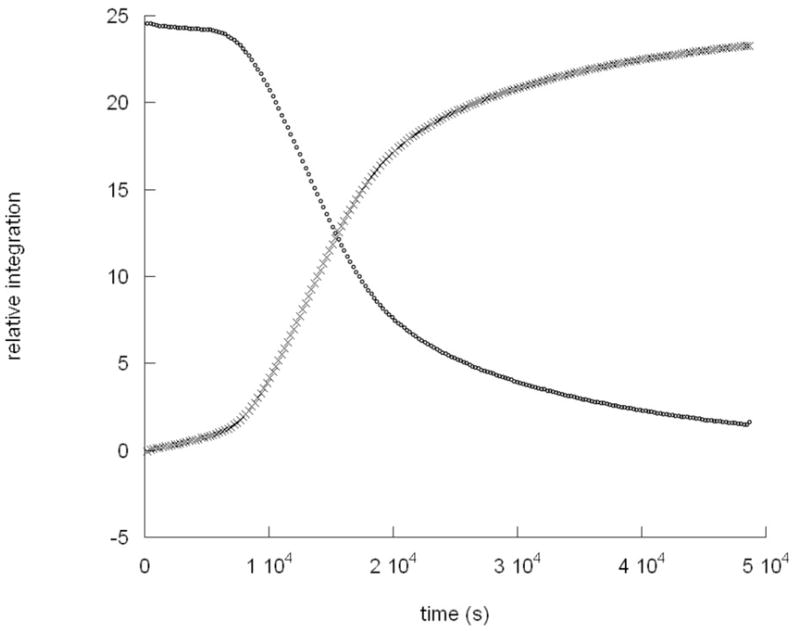
Kinetic data for the conversion of LtBuFeNPhNHPh to LtBuFeNHPh at 315 K: (°), [LtBuFeNPhNHPh]; (×), [LtBuFeNHPh].
The reaction path to the products was studied using a crossover experiment. A mixture of two different hydrazido compounds, LtBuFeNPhNHPh and LtBuFeNTolNHTol (Tol = m-tolyl), was heated to 80 °C for 2 h and passed through an activated alumina column to remove iron salts. Analysis of the organic byproducts by mass spectrometry shows the formation of PhN=NPh (m/z = 182) and TolN=NTol (m/z = 210), but no mixed diazene PhN=NTol (m/z = 196). This result implies that diazene is formed before N-N bond cleavage.
Finally, we used a double crossover experiment with two different diketiminate ligands and two different hydrazido ligands. A mixture of LtBuFeNPhNHPh and LtBu’FeNTolNHTol was heated to 80 °C for 2 h and the products were analyzed by mass spectrometry. LtBuFeNHPh (m/z = 649), LtBuFeNHTol (m/z = 663), LtBu’FeNHPh (m/z = 733), and LtBu’FeNHTol (m/z = 747) were observed in roughly equal amounts. Because the amido compounds had statistically scrambled between (diketiminate)Fe units, we conclude that all Fe-N bonds are broken at some point in the mechanism.
We also attempted to study the kinetic isotope effect for the reaction. For this purpose, deuterium labeled hydrazido compound, LtBuFeNPhNDPh, was synthesized from addition of PhNDNDPh to [LtBuFeH]2. This compound was heated under similar conditions as used with LtBuFeNPhNHPh. Unfortunately, the IR spectrum of the product shows the presence of LtBuFeNHPh (νN-H = 3319 cm−1) as well as LtBuFeNDPh (νN-D = 2499 cm−1), suggesting that some of the deuterium label was lost through an unknown mechanism. While the fate of these deuterons remains a mystery, the complication of the proton exchange makes it impossible to interpret the relative rates as a genuine kinetic isotope effect for the reaction.28
Exploring the possible mechanisms
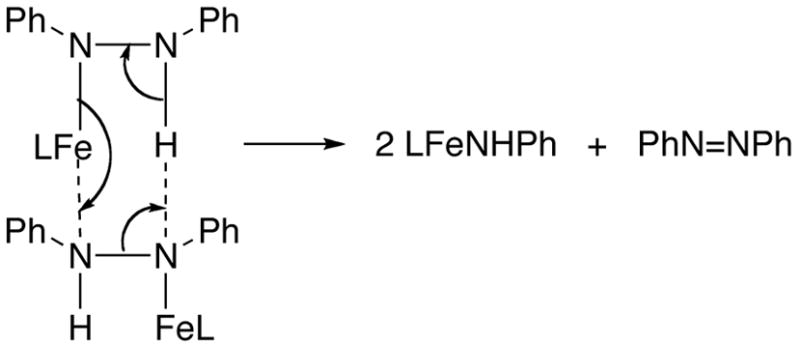
1. Disproportionation mechanism
One possible mechanism for the formation of amido product is shown in Scheme 2. A pericyclic mechanism of this type was proposed for the disproportionation of N,N′-diphenylhydrazine to azobenzene and aniline at high temperature.29 However, later kinetic studies showed a first-order dependence on [PhNHNHPh], ruling out this mechanism.30 According to this mechanism, the products are formed through a bimolecular transition state as shown in Scheme 2. This requires the rate of our reaction to have a second order dependence on [LtBuFeNPhNHPh]. The observed first-order dependence on iron concentration, and the induction period, are inconsistent with the disproportionation mechanism.
Scheme 2.
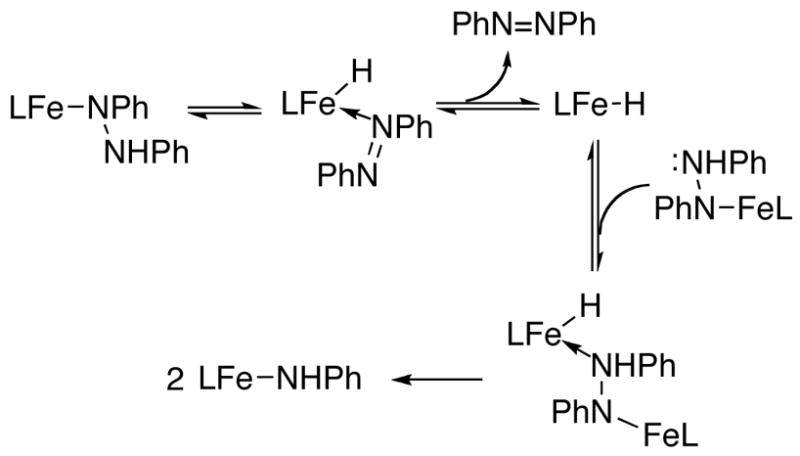
2. β-Hydride elimination mechanism
Next, we considered (β-hydride elimination (Scheme 3) as a potential rate-limiting step in the mechanism. This pathway has some attractive features: it would be expected to give a first-order dependence on [Fe]; the azobenzene is generated without N-N bond cleavage; and, the (β-hydride elimination step is the reverse of the [1,2]-insertion of the Fe-H bond across the N=N bond, which forms the hydrazido complex. If the reaction goes by this mechanism, LtBuFeH should be formed as an intermediate of the reaction. However, no intermediate species were detected by 1H NMR spectroscopy during the course of the reaction and the rate of the reaction was not affected by the addition of excess azobenzene. To account for these observations the (β-hydride elimination would have to be the rate limiting step: in other words, the intermediate LtBuFeH has to react quickly with LtBuFeNPhNHPh.
Scheme 3.
To query the intermediacy of the hydride species, the hydrazido complex was heated in the presence of 3-hexyne in varying amounts. The reaction of LtBuFeH with 3-hexyne to afford the vinyl compound, LtBuFeCEtCHEt, is very fast and irreversible at room temperature,11,31 so it is expected to be an effective trap. However, only a small amount (< 20%) of LtBuFeCEtCHEt was formed, and the ratio of vinyl product and amido product was independent of the concentration of 3-hexyne (see Supporting Information for details). This result is inconsistent with the intermediacy of LtBuFeH in the reaction.
It is instructive to further consider the possibility that the hydride and hydrazido react with each other so quickly that trapping is not possible. This possibility was experimentally tested by adding 1 equiv of [LtBuFeH]2 to LtBuFeNPhNHPh in C6D6. This mixture does not react under ambient conditions, even though [LtBuFeH]2 is expected to form monomer within a few minutes at room temperature.11 When this mixture was heated to 358 K, LtBuFeNHPh is formed with a rate constant of 5.3 ± 0.7 × 10−4 s−1. Because the rate constant for this reaction is roughly the same as that in the reaction without added hydride complex (7.1 ± 1.3 × 10−4 s−1) it is unlikely that the hydride is an intermediate in the transformation.
One final piece of evidence argues against the mechanism in Scheme 3: if LtBuFeH reacted rapidly with LtBuFeNPhNHPh, as required in Scheme 3, then it would not have been possible to isolate LtBuFeNPhNHPh during its synthesis from [LtBuFeH]2 and PhN=NPh (some LtBuFeH is present in the reaction mixture at room temperature). These combined observations suggest that the N-N cleavage reaction is unlikely to follow a β-hydride elimination pathway.
3. Ion-pair and acid-catalyzed mechanisms
The formation of LtBuFeNHPh, LtBuFeNHTol, LtBu’FeNHPh, and LtBu’FeNHTol from heating LtBuFeNPhNHPh and LtBu’FeNTolNHTol (see above) suggests that Fe-N bonds are disrupted as part of the mechanism. If the reaction proceeds through heterolytic Fe-N bond cleavage in an ion-pair mechanism as shown in equation 1, the rate of the reaction should depend on the polarity of the solvent in which the reaction is carried out. However, the rate constant determined in THF-d8 (k = 4.6 ± 0.2 × 10−4 s−1) at 352 K was not faster than that in toluene-d8 (k = 6.9 ± 0.2 × 10−4 s−1) or benzene-d6 (k = 5.8 ± 0.2 × 10−4 s−1) under similar conditions, arguing against an heterolytic cleavage in the rate-limiting step of the mechanism.
| (1) |
The possibility that trace acid catalyzes the reaction was tested by adding a non-nucleophilic base under the same reaction conditions. When the reaction was monitored at 359 K in the presence of lutidine (35 mM), the rate constant was 6.5 ± 0.2 × 10−4 s−1, again showing no effect on the reaction rate.
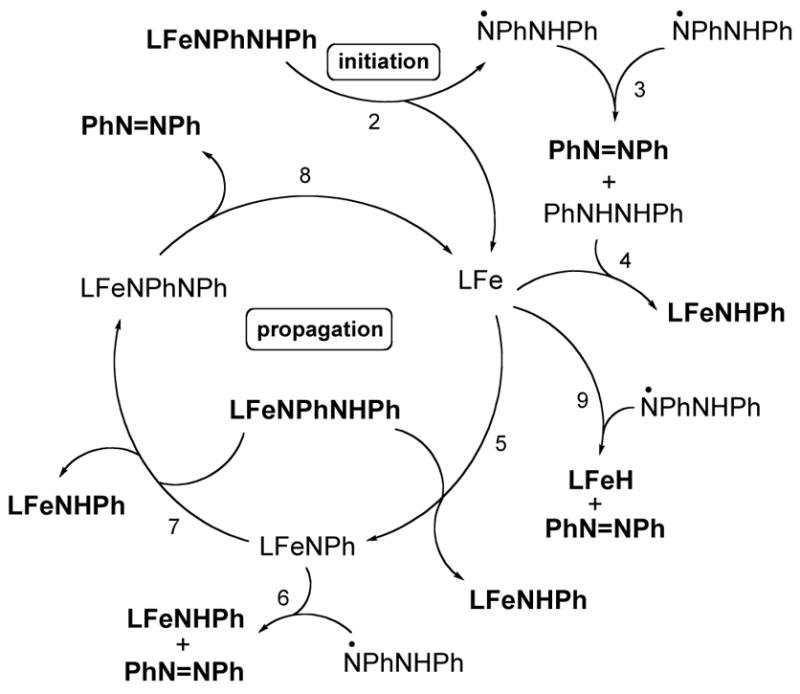
4. Radical Mechanisms
The results discussed so far are consistent with a pathway initiated by a homolytic cleavage to yield radicals that react through a chain mechanism. The crossover experiment shows that the mechanism cannot involve N-N bond homolysis along the way to PhN=NPh, and so Fe-N bond homolysis is most strongly implicated as an initiation reaction (eq 2).32 Light does not initiate the reaction, because all kinetic studies were done in the dark (inside an NMR probe).
| (2) |
Homolytic Fe-N bond cleavage would give the N,N′-diphenylhydrazinyl radical, which could undergo spontaneous disproportionation to PhN=NPh and PhNHNHPh (eq 3).33 The two sides of the double bond in the PhN=NPh molecule derive from the same hydrazido complex, assuming a pericyclic mechanism.33 Therefore, this initial step is consistent with the crossover experiments given above.34,35
| (3) |
What would happen to the iron(I) LtBuFe fragment formed in eq 2? First, it could react with PhNHNHPh (eq 4). This hypothesis was tested by reacting a genuine iron(I) complex, LtBuFeClK(solvent)n,36 with N,N′-diphenylhydrazine (1 equiv) in C6D6. The amido compound LtBuFeNHPh was formed within a few minutes at ambient temperature. Therefore, this step is a reasonable route to the observed product of the overall reaction.
| (4) |
The iron(I) fragment could also abstract an NHPh radical from LtBuFeNPhNHPh to give LtBuFeNHPh and LtBuFeNPh, an iron(III) imido species (eq 5). We have spectroscopically characterized a similar complex, LMeFeNAd (where Ad = 1-adamantyl).37,38 One characteristic reaction of LMeFe=NAd is to abstract hydrogen atoms from hydrocarbons (e.g. 9,10-dihydroanthracene and indene), giving an iron(II) amido complex. While we have been unsuccessful in preparing LtBuFeNPh,39 the analogy to LMeFeNAd suggests strongly that the transient formation of LtBuFeNPh is feasible.
| (5) |
| (6) |
| (7) |
LtBuFeNPh should be capable of abstracting a hydrogen atom from the hydrazinyl radical (which has a very weak N-H bond) to form LtBuFeNHPh and PhN=NPh (eq 6). This would represent a chain termination step.34 Alternatively, the transient imido species could abstract a hydrogen atom from LtBuFeNPhNHPh to give LtBuFeNPhNPh (eq 7), which may be formulated either as an iron(II) complex with a radical ligand, or an iron(I) complex with coordinated PhN=NPh.
LtBuFeNPhNPh was prepared independently by treating LtBuFeNNFeLtBu with PhN=NPh at room temperature, and the solid-state structure was determined using X-ray crystallography. Figure 6 shows that the azobenzene ligand is coordinated through the N=N π-bond, and backbonding weakens the N=N bond considerably (N-N = 1.398(2) Å) compared to free trans-azobenzene (1.247(2) Å).40 This feature indicates that the formal azobenzene-iron(I) formulation does not account for substantial backbonding into the azobenzene ligand. There are strong analogies to diketiminate-iron(I) complexes of alkenes and alkynes recently reported by our group, in which crystallographic, vibrational, Mössbauer, and theoretical evidence support substantial backbonding into the unsaturated ligand.41 Therefore, it is reasonable to think of LtBuFeNPhNPh as an iron(II)-radical complex LtBuFe2+(N2Ph2)−.
Figure 6.
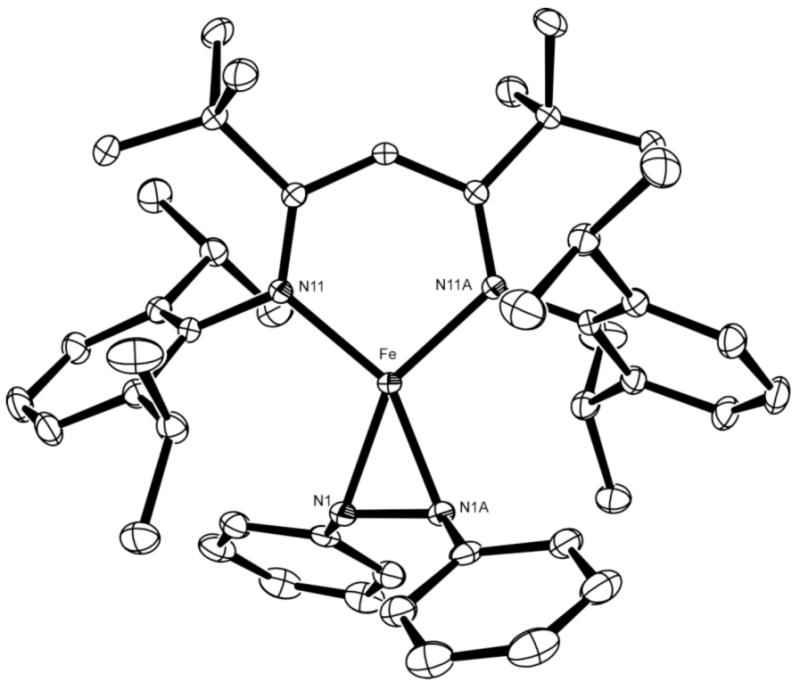
Crystal structure of LtBuFeNPhNPh. Thermal ellipsoids shown at 50% probability. Hydrogen atoms have been omitted for clarity. Selected metrical parameters: Fe-N11 1.987(1) Å, Fe-N1 1.953(1) Å, N1-N1A 1.398(2) Å, N11-Fe-N11A 97.52(5)°, N1-Fe-N1A 41.94(5)°.
In turn, LtBuFeNPhNPh can lose azobenzene to regenerate the active iron(I) fragment (eq 8), which continues the radical chain (Scheme 4). Both sides of the azobenzene produced through eq 8 have come from a single molecule of LtBuFeNPhNHPh, so this mechanism is consistent with the crossover experiment described above.
Scheme 4.
| (8) |
Effect of radical traps on the rate of the reaction
Kinetic studies on the decomposition of LtBuFeNPhNHPh in C6D6 at 358 K were repeated with large concentrations of the radical traps triphenylmethane (Ph3CH) (0.46 M) and dihydroanthracene (DHA) (0.35 M). The rate constant for the reaction was not affected (9.8 ± 0.3 × 10−4 s−1 and 9.3 ± 0.2 × 10−4 s−1, respectively). The bond dissociation energy (BDE) of the C-H bonds in Ph3CH (81 kcal/mol) and DHA (78 kcal/mol) is higher than the BDE of the N-H bond in N,N′-diphenylhydrazine (69 kcal/mol),42 and lower than the BDE of N-H in aniline (92 kcal/mol), so this observation is compatible with the presence of hydrazinyl radicals and argues against amido radicals. Because LtBuFeNPhNHPh is decomposed by TEMPO and nitrosobenzene, these radical traps are not informative.
Which species are chain carriers?
The decomposition of solutions of LtBuFeNPhNHPh at 42 °C in C7D8 shows an induction period, as described above (Figure 5). This reaction was repeated with different reagents added (12–14 mM), to learn whether they eliminate the induction period. We used LtBuFeCl as a representative of iron(II) compounds, LtBuFeClK(solvent)n as a proxy for the “LtBuFe” intermediate in the catalytic cycle, and the azobenzene complex LtBuFeNPhNPh, in separate experiments. Addition of LtBuFeCl did not eliminate the induction period, while addition of LtBuFeClK(solvent)n or LtBuFeNPhNPh resulted in formation of LtBuFeNHPh without an induction period at 42 °C. These observations support the chain mechanism including LtBuFe and LtBuFeNPhNPh as chain carriers. In a different experiment, we added benzophenone (2 equiv) to LtBuFeNPhNHPh in order to trap transient iron(I).43 Heating this mixture afforded unidentified paramagnetic complexes and no LtBuFeNHPh was formed. Other traps for iron(I) gave ambiguous results.43
Which is the rate-limiting step after initiation?
This is a subject that must be approached with caution. As stated by Huyser, “Rate laws for free-radical chain reactions are of limited value in determining the general mechanism of the reaction.”46 This is especially true when considering Scheme 4, in which many of the propagation and termination steps give the same products. However, the following arguments may be advanced. The post-initiation kinetics follow a first-order rate dependence on [Fe], which suggests that eq 8 (top arrow in the catalytic cycle of Scheme 4) may be rate-limiting. When LtBuFeNPhNHPh was heated in the presence of added PhN=NPh (45 mM) in C6D6 at 358 K, there was no effect on the rate constant (7.0 ± 0.1 × 10−4 s−1). This result suggests that eq 8 is effectively irreversible: every time LtBuFe is formed it is trapped by PhNHNHPh or LtBuFeNPhNHPh rather than by PhN=NPh. In order to test this idea, 1 equiv of PhN=NPh was added to a C6D6 solution of LtBuFeClK(solvent)n with and without 1 equiv of LtBuFeNPhNHPh. Without LtBuFeNPhNHPh, only LtBuFeNPhNPh was observed in the 1H NMR spectrum, but in the presence of the hydrazido species no LtBuFeNPhNPh was evident (instead, there was slow conversion to LtBuFeNHPh). Therefore, our experiments suggest that loss of PhN=NPh from the formally iron(I) center in LtBuFeNPhNPh is the slow step of the chain.44
Although the details are tentative, the amassed experimental data are most supportive of a chain mechanism of the type shown in Scheme 4. It remains only to explain how addition of 3-hexyne to the hydrazido complex could yield a small, concentration-independent amount of LtBuFeCEtCHEt (see above). To rationalize this observation, we note that LtBuFeH could be formed by abstraction of a hydrogen atom from the weak N-H bond in the hydrazinyl radical •NPhNHPh by “LtBuFe” (equation 9; Scheme 4). Because the hydride complex is off the main reaction pathway, the amount of vinyl complex formed is independent of [3-hexyne].
| (9) |
| (10) |
Conclusions
The reaction of LtBuFeH with 0.5 equiv of PhN=NPh cleanly leads to LtBuFeNHPh, formally cleaving the diazene into “NPh” units that insert into the Fe-H bond. Although the reaction stoichiometry is simple, the mechanism is complex. Fortunately, a combination of mechanistic experiments can be used to rule out most mechanisms, and to support a chain mechanism. The first step of this chain process consists of adding the Fe-H bond across the N=N bond to give a hydrazido complex LtBuFeNPhNHPh. This intermediate is susceptible to Fe-N bond homolysis to give iron(I) fragments that are capable of breaking N-N bonds. The picture that emerges is a radical chain mechanism that has odd-electron iron species as chain carriers.
One lesson to be learned from this mechanism is that iron complexes with weak ligand fields are capable of [1,2]-additions through both non-radical (in the addition of Fe-H across the N=N bond) and radical (in the N-N cleaving chain reaction) pathways.45,46 Although there are clear differences between the coordination sphere of the diketiminate-iron complexes here and the important iron sites in the FeMoco of nitrogenase, one can use these conclusions to speculate about the numerous N-N cleavage and N-H bond forming steps that lie along the path from N2 to ammonia. Previous workers have understandably favored simpler non-radical pathways as in the Chatt cycle,47 and these are representative of the mechanisms of reactions at molybdenum and other second- and third-row metal ions. However, given the recent evidence supporting high-spin iron as the reactive site on the FeMoco of nitrogenase,10 it is important to consider one-electron reactivity in the enzymatic system.
Experimental Section
General procedures
All manipulations were performed under a nitrogen atmosphere by standard Schlenk techniques or in an M. Braun glove box maintained at or below 1 ppm of O2 and H2O. Glassware was dried at 150 °C overnight. NMR data were recorded on a Bruker Avance 400 (400 MHz) or Bruker Avance 500 spectrometer (500 MHz). All peaks in the NMR spectra are referenced to residual C6D5H at 7.16 ppm. In parentheses are listed: integrations, T2 values in ms calculated as (πΔν1/2)−1 (paramagnetic complexes only), and assignments. In some cases, it was not possible to determine integrations and T2 values because of peak overlap. UV-vis spectra were measured on a Cary 50 spectrophotometer, using screw-cap cuvettes. Solution magnetic susceptibilities were determined by the Evans method,48 monitoring the shift in the residual solvent peak relative to a capillary of solvent. Elemental analyses were determined by Desert Analytics, Tucson, AZ.
Pentane, diethyl ether and toluene were purified by passage through activated alumina and “deoxygenizer” columns from Glass Contour Co. (Laguna Beach, CA). Deuterated benzene and toluene were first dried over CaH2, then over Na/benzophenone, and then vacuum transferred into a storage container. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF solution. Celite was dried overnight at 200 °C under vacuum. The compounds nitro-2,4,6-triisopropylbenzene,49 LtBuFeCl,50 KBEt3H,51 [LtBuFeH]2,11 LtBuFeNPhNHPh,11 LtBuFeCEtCHEt,11 LtBuFeNNFeLtBu,36 LtBuFe(Cl)K(solvent)x36 were prepared by published procedures. 3-Hexyne, N,N′-diphenylhydrazine, and azobenzene were obtained from Aldrich and m-azotoluene was obtained from TCI America. 3-Hexyne was degassed, vacuum transferred to a new container, and stored in the glovebox at −35 °C. Azobenzene and m-azotoluene were dissolved in pentane, dried over molecular sieves, filtered through Celite then dried under vacuum. 1,3,5-Triisopropylbenzene, lutidine, n-butyllithium, methyllithium, TMEDA, Pd/C, phosphorus pentachloride, pivaloyl chloride, and triphenylmethane were purchased from Aldrich Chemical Co. and used as received. 9,10-Dihydroanthracene and Ph3CH were crystallized from ethanol and the crystals were vacuum dried. D2O was purchased from Cambridge Isotope Laboratories, Inc.
1. Preparation of LtBu’H
A 200 mL Paar bomb was charged with nitro-2,4,6-triisopropylbenzene (20.0 g, 80.2 mmol) dissolved in isopropanol (80 mL) and Pd/C (3.0 g). H2 gas (~ 600 psi) was introduced, and the mixture was stirred at 60 °C for 18 h. The contents were filtered in air after cooling to room temperature. Isopropanol was removed by distillation and the product was distilled at 80 °C (3 mbar) to afford 2,4,6-triisopropylaniline (16.5 g, 94%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 1.1 (12, d, J = 8 Hz, o-iPr-CH3), 1.3 (6, d, J = 8 Hz, p-iPr-CH3), 2.9 (1, septet, p-iPr-CH), 3.0 (2, septet, o-iPr-CH), 3.8 (2, br, NH), 6.9 (2, s, m-H).
A round bottom flask was charged with 2,4,6-triisopropylaniline (16.5 g, 75.2 mmol) and dichloromethane (50 mL). To the flask was added triethylamine (7.7 g, 75 mmol) and then a solution of pivaloyl chloride (9.3 mL, 75 mmol) in 50 ml of dichloromethane slowly (~5 drops/sec). The reaction mixture was refluxed for 2 h at 40 °C and the solvent was removed using a rotovap. The resultant pink solid was washed with water and diethyl ether to yield N-(2,4,6-triisopropylphenyl)-2,2-dimethylpropionamide as a white microcrystalline powder which was dried under vacuum (21.8 g, 96%). 1H NMR (400 MHz, CDCl3) δ 1.17 (12, d, J = 8 Hz, o-iPr-CH3), 1.22 (6, d, J = 8 Hz, p-iPr-CH3), 1.33 (9, s, tBu), 2.86 (1, septet, p-iPr-CH), 2.97 (2, septet, o-iPr-CH), 6.73 (1, s, NH), 6.98 (2, s, m-H)
Phosphorus pentachloride (16 g, 73 mmol) was added in small portions to a slurry of N-(2,4,6-triisopropylphenyl)-2,2-dimethylpropionamide (21.8 g, 71.8 mmol) in benzene (33 mL) under N2 flow. During the course of the addition, HCl gas was liberated and it was passed through aqueous Na2CO3. The resultant yellow reaction mixture was stirred for 18 h under the continuous flow of N2. The solvent and byproducts were distilled off and the product was distilled at 110 °C (3 mbar) to yield 1-chloro-l-(2,4,6-triisopropylphenylimino)-2,2-di(methylpropane) (17.8 g, 77%) as a colorless gel. This product was handled under N2. 1H NMR (400 MHz, CD2Cl2) δ 1.16 (12, d, J = 8 Hz, o-iPr-CH3), 1.18 (6, d, J = 8 Hz, p-iPr-CH3), 1.42 (9, s, tBu), 2.73 (2, septet, p-iPr-CH), 2.95 (1, septet, o-iPr-CH), 7.0 (2, s, m-H).
A round bottom flask was charged with l-chloro-l-(2,4,6-triisopropylphenylimino)-2,2-dimethylpropane (14.8 g, 46 mmol) and pentane (80 mL) inside a glovebox and cooled in a cold well (−60 °C). To this solution was added 1.6 M methyllithium (29 mL, 46 mmol) in diethyl ether. The turbid yellow reaction mixture was stirred at room temperature for 18 h, and then brought out of the glove box. Water (50 mL) was added to the reaction mixture slowly and very carefully. The aqueous layer was separated and extracted with diethyl ether. The organic fractions were combined and dried with anhydrous magnesium sulfate, filtered, and solvent was removed. The product was dried under vacuum to yield 2-(2,4,6-triisopropylphenylimino)-3,3-dimethylbutane (10.7 g, 77%) as a pale yellow solid. 1H NMR (400 MHz, CD2Cl2) δ 1.2 (12, br, o-iPr-CH3), 1.3 (24, br, p-iPr-CH3, tBu), 1.7 (3, br, CH3), 2.6 (2, br, o-iPr-CH), 2.8 (1, br, p-iPr-CH), 6.9 (2, s, m-H).
A round bottom flask was charged with 2-(2,4,6-triisopropylphenylimino)-3,3-dimethylbutane (10.7 g, 35 mmol) and diethyl ether (60 mL). To this was added TMEDA (5.5 mL, 36 mmol) and the reaction mixture was cooled in a cold well (−60 °C). n-Butyllithium (14.6 mL, 2.5 M in hexanes, 36 mmol) was added slowly while stirring. The resultant dark orange reaction mixture was stirred in the cold well for 1 h and then stirred at room temperature for 18 h. The dark orange reaction mixture with yellow precipitate was cooled in the cold well and then l-chloro-l-(2,4,6-triisopropylphenylimino)-2,2-dimethylpropane (11.4 g, 35 mmol) in 20 mL of pentane was added dropwise. Once the addition was complete the reaction mixture was stirred at 40 °C for 6 h. Water (50 mL) was added; the aqueous layer was separated and extracted with diethyl ether. All the organic fractions were combined and dried with magnesium sulfate, filtered, and the volatile components were removed under vacuum. The pale yellow solid was dissolved in hot pentane, filtered, and stored at −25 °C to afford LtBu’H (15.3 g, 72%) as pale yellow crystals. 1H NMR (400 MHz, CDCl3) δ 1.1 (24, br, o-iPr-CH3), 1.3 (18, br, tBu), 1.7 (12, br, p-iPr-CH3), 2.7 (1, br, p-iPr-CH), 2.9 (2, br, o-iPr-CH), 3.3 (2, br, backbone CH), 6.9 (4, s, m-H); LCMS: m/z = 587. Anal. Cald. for C41H66N2 C 83.89, H 11.33, N 4.77. Found C, 82.62, H 11.33, N 4.65.
2. Preparation of LtBu’Li(THF)
A Schlenk flask was charged with LtBu’H (12.1 g, 20.6 mmol) and THF (100 mL) and cooled to −60 °C. A solution of n-butyllithium (9.0 mL, 2.5 M in hexanes, 22.5 mmol) was added slowly while stirring. The reaction mixture was warmed to room temperature and heated at 60 °C for 2 h. The volatile components were removed under vacuum and the product was dissolved in warm pentane and stored at −35 °C to afford pale yellow crystals of LtBu’ Li(THF) (9.6 g, 70%). 1H NMR (400 MHz, C6D6) δ 0.9 (4, br, THF), 1.14 (12, d, iPr-CH3), 1.20 (12, d, iPr-CH3), 1.39 (18, s, tBu), 1.42 (12, d, iPr-CH3), 2.3 (4, br, THF), 2.8 (2, septet, iPr-CH), 3.5 (4, septet, iPr-CH), 5.24 (1, s, backbone-CH), 6.96 (4, s, m-H).
3. Preparation of LtBu’FeCl
A bomb flask was charged with FeCl2(THF)1.5 (1.3 g, 5.6 mmol), LtBu’Li(THF) (3.7 g, 56 mmol), and toluene (70 mL). The red reaction mixture was heated at 100 °C for 18 h. The volatile components were removed under vacuum, and the red residue was transferred to a glass thimble, which was placed in a Soxhlet extractor. The residue was extracted continuously with boiling diethyl ether until the extracting solvent was clear (24 h). The red extract was concentrated under vacuum and the resultant red solid was isolated (2.04 g, 54%). The mother liquor was stored at −35 °C to afford LtBu’FeCl as red crystals (1.2 g, 32%). Total yield 3.2 g (86%). 1H NMR (400 MHz, C6D6) δ 105 (1, backbone-CH), 42 (18, 0.3, tBu), 35 (2, 0.7, iPr-CH), 2.1 (4, m-H), −5 (12, 0.7, iPr-CH3), −28 (12, 0.5, iPr-CH3), −111 (12, 0.1, iPr-CH3), −115 (4, iPr-CH) LCMS: m/z = 676. Anal. Cald. for C41H65N2FeCl C 71.71, H 9.67, N 4.14. Found C, 70.81, H 10.25, N 3.99.
4. Preparation of [LtBu’FeH]2
A Schlenk flask was charged with LtBu’FeCl (0.884 g, 1.30 mmol) and toluene (30 mL). To this red solution was added a clear solution of KBEt3H (0.180 g, 1.30 mmol) in toluene (10 mL). The dark red reaction mixture was stirred at room temperature for 45 min, and the volatile materials were removed under vacuum. The residue was extracted with pentane and filtered through Celite to give a dark red solution. This solution was concentrated, warmed to dissolve the product, and then cooled to −35 °C to afford very dark red crystals (501 mg, 78%). It is critical that the reaction is not stirred for too long because [LtBu’ FeH]2 reacts with the BEt3 byproduct. Unlike its LtBu analogue, [LtBu’FeH]2 is soluble in pentane. 1H NMR ([LtBu’FeH], 22°C, C6D6) 72, 42, 22, 21, 14, 13, 1.2, −1.3, −5, −8, −10, −19, −26, −29, −42, −51, −120. 1H NMR (LtBu’FeH, 80°C, C6D6) δ 45 (1, backbone-CH), 31 (2, p-iPr-CH), 30 (18, tBu), 9 (4, m-H), −3 (12, iPr-CH3), −17 (12, iPr-CH3), −81 (4, o-iPr-CH), −82 (12, iPr-CH3). Anal. Cald. for C82H132N4Fe C 76.61, H 10.35, N 4.36. Found C, 76.59, H 10.42, N 4.22.
5. Preparation of LtBu’FeNPhNHPh
From [LtBu’FeH]2: An orange solution of azobenzene (14 mg, 77 μmol) in Et2O (2 mL) was added to a dark red slurry of [LtBu’FeH]2 (50 mg, 38 μmol) in Et2O (8 mL). A red solution was formed over the course of an hour. The volatile materials were removed under vacuum. The residue was extracted with pentane, filtered through Celite, concentrated, and stored at −35 °C to give LtBu’FeNPhNHPh as red crystals. From LtBu’FeCl: A 20 mL scintillation vial was charged with LtBu’FeCl (172 mg, 253 μmol), azobenzene (46 mg, 253 μmol) and Et2O (10 mL). A solution of KBEt3H (35 mg, 253 μmol) in Et2O (2 mL) was added to give a dark red solution. After stirring at room temperature for 2 h, the volatile components were removed under reduced pressure. The resultant residue was extracted with pentane, and filtered through Celite to give a red solution. The solution was concentrated and stored at −35 °C to afford LtBu’FeNPhNHPh as red crystals (126 mg, 60%).
1H NMR (400 MHz, C6D6) δ 111 (1, backbone-CH), 43 (18, 0.4, tBu), 23 (2, 1.2), −7.8 (12, 0.8, p-iPr-CH3), −8.6 (4, 1.2, diketiminate m-H), −23 (6, 0.6, o-iPr-CH3)), −29 (6, 0.6, o-iPr-CH3),), −45 (1, 0.7), −82 (6, 0.09, o-iPr-CH3),), −170 (6, 0.09, o-iPr-CH3),), −181 (br). Due to the thermal instability of the solid, we have not been able to obtain successful elemental analysis.
6. Preparation of LtBuFeNTolNHTol
A 20 mL scintillation vial was charged with LtBuFeCl (102 mg, 172 μmol), azo-m-toluene (36 mg, 172 μmol) and Et2O (10 mL). A solution of KBEt3H (24 mg, 172 μmol) in Et2O (2 mL) was added to yield a dark red solution. After stirring at room temperature for 2 h, the volatile components were removed under reduced pressure. The resultant residue was extracted with pentane, filtered through Celite to give a red solution. The solution was concentrated and stored at −35 °C to afford LtBuFeNTolNHTol as red crystals (82 mg, 62%). 1H NMR (400 MHz, C6D6) δ 112 (1, backbone-CH), 43 (18, tBu), −7 (2, diketiminate m-H), −8 (2, diketiminate m-H), −23 (6, iPr-CH3), −28 (6, iPr-CH3), −44 (1, hydrazido p-H), −83 (6, iPr-CH3), −91 (2, diketiminate p-H), −172 (8, iPr-CH3 and iPr-CH).
7. Preparation of LtBu’FeNTolNHTol
An orange solution of azo-m-toluene (16 mg, 76 μmol) in Et2O (2 mL) was added to a dark red slurry of [LtBu’FeH]2 (49 mg, 38 μmol) in Et2O (8 mL). A red solution was formed over the course of an hour. The volatile materials were removed under vacuum. The residue was extracted with pentane, filtered through Celite, concentrated, and stored at −35 °C to give LtBu’FeNTolNHTol as red crystals (39 mg, 60%).
8. Preparation of PhNDNDPh
A Schlenk tube was charged with N,N′-diphenylhydrazine (215 mg, 1.17 mmol) and pentane (10 mL). A solution of n-butyllithium (0.95 mL, 2.5 M in hexanes) was added while stirring. The thick yellow suspension was stirred for 18 h at room temperature. The volatile components were removed under reduced pressure. The contents were cooled in an ice bath and D2O (10 mL) was added under nitrogen flow to give a white precipitate. The slurry was stirred for 2 h and the reaction mixture was allowed to settle. The yellow solution was decanted using a cannula and the white solid was dried under reduced pressure. The solid was extracted with pentane/diethyl ether solvent mixture, filtered through Celite, concentrated, and stored at −35 °C to afford PhNDNDPh as colorless flaky crystals (~98% deuteration by NMR). 1H NMR (400 MHz, C6D6) δ 6.7 (4, o-H), 6.8 (2, p-H), 7.2 (4, m-H).
9. Preparation of LtBuFeNPhNDPh
A solution of PhNDNDPh (8.3 mg, 44 μmol) in Et2O (2 mL) was added to a dark red slurry of [LtBuFeH]2 (25 mg, 22 μmol) in Et2O (8 mL). A red solution was formed over the course of an hour. The volatile materials were removed under vacuum. The residue was extracted with pentane, filtered through Celite, concentrated, and stored at −35 °C to give LtBuFeNPhNDPh as red crystals (56 mg, 60%). 1H NMR (400 MHz, C6D6) δ 112(1, backbone-CH), 43 (18, tBu), −7 (2, diketiminate m-H), −8 (2, diketiminate m-H), −23 (6, iPr-CH3), −28 (6, iPr-CH3), −45 (1, hydrazido p-H), −87 (6, iPr-CH3), −94 (2, diketiminate p-H), −171 (8, iPr-CH3 and iPr-CH).
10. Preparation of LtBuFeNPhNPh
A 20 mL scintillation vial was charged with LtBuFeN=NFeLtBu (98 mg, 86 μmol) and Et2O (10 mL). A solution of azobenzene (31 mg, 170 μmol) in Et2O (2 mL) was added to yield a brown solution, which turned dark green upon stirring for 30 minutes. After stirring at room temperature for 2 h, the volatile components were removed under reduced pressure. The resultant residue was extracted with toluene, and filtered through Celite to give a dark green solution. The solution was concentrated and stored at −35 °C to afford LtBuFeNPhNPh as dark green crystals (51 mg, 40%). 1H NMR (400 MHz, C6D6) δ 119 (4, br, Ph o-H), 72 (1, Ph p-H), 42 (1, Ph p-H), 24 (18, tBu), −11 (12, iPr-CH3), −15 (4, diketiminate m-H), −17 (2, diketiminate p-H), −22 (2, Ph m-H), −50 (12, iPr-CH3), −70 (2, br, iPr-CH). Anal. Calcd. for C47H63N4Fe C 76.30, H 8.58, N 7.57. Found C, 76.51, H 8.42, N 8.00.
Crystallography
Crystals were placed onto the tip of a 0.1 mm diameter glass capillary tube or fiber and mounted on a Bruker SMART APEX II CCD Platform diffractometer52 for a data collection at 100.0(1) K using MoKα radiation (graphite monochromator). A randomly oriented region of reciprocal space was surveyed: four major sections of frames were collected with 0.30° steps in ω at four different φ settings and a detector position of −28° in 2θ. The intensity data were corrected for absorption.53 Final cell constants were calculated from the xyz centroids of >3500 strong reflections from the actual data collections.54 The structures were solved using direct methods and refined using SHELXL-97.55 The space groups (C2/c in each case) were determined based on systematic absences and intensity statistics. All non-hydrogen atoms were refined with anisotropic displacement parameters. The bridging hydrogen atoms in [LtBu’FeH]2 were found from the difference Fourier map and their positions were refined independently from the iron atoms, but with relative (based on Fe1) isotropic displacement parameters. It is understood that these positions are approximate. All other hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. The final full matrix least squares refinement for [LtBu’FeH]2 converged to R1 = 0.0467 (F2, I > 2σ(I)) and wR2 = 0.1328 (F2, all data). The final full matrix least squares refinement for LtBuFe(N2Ph2) converged to R1 = 0.0384 (F2, I > 2σ(I)) and wR2 = 0.1035 (F2, all data).
Supplementary Material
Eyring plot and NMR data (pdf) and crystallographic data (cif). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This paper is dedicated to the memory of Ian Rothwell, who taught the chemical community about transition-metal-mediated N=N bond breaking, and who provided useful discussions in the early part of this work. We also thank Joseph Dinnocenzo, William Jones, and Richard Finke for helpful discussions. Financial support was provided by the National Institutes of Health (GM-065313).
References and Notes
- 1.(a) Collman JP, Hegedus L. Principles and Applications of Organotransition Metal Chemistry. 1980 [Google Scholar]; (b) Peruzzini M, Poli R. Recent Advances in Hydride Chemistry. Elsevier; New York: 2001. [Google Scholar]
- 2.Thorneley RNF, Eady RR, Lowe DJ. Nature. 1978;272:557–8. [Google Scholar]
- 3.Theoretical evaluation of hydrides on belt irons: Dance IJ. Am Chem Soc. 2005;127:10925–10942. doi: 10.1021/ja0504946.Dance I. Biochemistry. 2006;45:6328–6340. doi: 10.1021/bi052217h.
- 4.(a) Eady RR. Chem Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]; (b) Seefeldt LC, Dance IG, Dean DR. Biochemistry. 2004;43:1401–1409. doi: 10.1021/bi036038g. [DOI] [PubMed] [Google Scholar]
- 5.Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 6.(a) Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]; (b) Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Proc Natl Acad Sci USA. 2007;104:1451–1455. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holland PL. Can J Chem. 2005;83:296–301. [Google Scholar]
- 8.Lee SC, Holm RH. Proc Natl Acad Sci USA. 2003;100:3595–3600. doi: 10.1073/pnas.0630028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Structure: Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877.
- 10.Evidence that substrate binding takes place at belt irons: Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC. J Biol Chem. 2004;279:53621–53624. doi: 10.1074/jbc.M410247200.Dos Santos PC, Igarashi RY, Lee H-I, Hoffman BM, Seefeldt LC, Dean DRAce. Chem Res. 2005;38:208–214. doi: 10.1021/ar040050z.
- 11.Smith JM, Lachicotte RJ, Holland PL. J Am Chem Soc. 2003;125:15752–15753. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]
- 12.A recent paper postulates a transient four-coordinate iron-hydride complex that reacts with benzene: Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122.
- 13.Studies of other promising iron-hydride complexes: Brown SD, Mehn MP, Peters JC. J Am Chem Soc. 2005;127:13146–13147. doi: 10.1021/ja0544509.Franke O, Wiesler BE, Lehnert N, Peters G, Burger P, Tuczek FZ. Anorg Allg Chem. 2006;632:1247–1256.Gilbertson JD, Szymczak NK, Crossland JL, Miller WK, Lyon DK, Foxman BM, Davis J, Tyler DR. Inorg Chem. 2007;46:1205–1214. doi: 10.1021/ic061570o.
- 14.(a) Burgess BK, Wherland S, Newton WE, Stiefel EI. Biochemistry. 1981;20:5140–5146. doi: 10.1021/bi00521a007. [DOI] [PubMed] [Google Scholar]; (b) Barney BM, Laryukhin M, Igarashi RY, Lee H-I, Dos Santos PC, Yang T-C, Hoffman BM, Dean DR, Seefeldt LC. Biochemistry. 2005;44:8030–8037. doi: 10.1021/bi0504409. [DOI] [PubMed] [Google Scholar]; (c) Barney BM, Yang T-C, Igarashi RY, Dos Santos PC, Laryukhin M, Lee H-I, Hoffman BM, Dean DR, Seefeldt LC. J Am Chem Soc. 2005;127:14960–14961. doi: 10.1021/ja0539342. [DOI] [PubMed] [Google Scholar]
- 15.Reduction of hydrazine by Mo/S-based nitrogenase model complexes: Block E, Ofori-Okai G, Kang H, Zubieta J. J Am Chem Soc. 1992;114:758–759.DeBord JRD, George TA, Chang Y, Chen Q, Zubieta J. Inorg Chem. 1993;32:785–786.Coucouvanis D, Mosier PE, Demadis KD, Patton S, Malinak SM, Kim CG, Tyson MA. J Am Chem Soc. 1993;115:12193–12194.Demadis KD, Coucouvanis D. Inorg Chem. 1994;33:4195–4197.Malinak SM, Demadis KD, Coucouvanis D. J Am Chem Soc. 1995;117:3126–3133.Demadis KD, Malinak SM, Coucouvanis D. Inorg Chem. 1996;35:4038–4046. doi: 10.1021/ic960098b.Schollhammer P, Petition FY, Poder-Guillou S, Saillard JY, Talarmin J, Muir KW. Chem Commun. 1996:2633–2634.Petition FY, Schollhammer P, Talarmin J, Muir KW. Inorg Chem. 1999;38:1954–1955. doi: 10.1021/ic9813359.Le Grand N, Muir KW, Petition FY, Pickett CJ, Schollhammer P, Talarmin J. Chem Eur J. 2002;8:3115–3127. doi: 10.1002/1521-3765(20020715)8:14<3115::AID-CHEM3115>3.0.CO;2-Z.
- 16.Hydrazine reduction by ruthenium complexes: Chatterjee D. J Mol Catal A. 2000;154:1–3.Prakash R, Ramachandraiah G. J Chem Soc, Dalton Trans. 2000:85–92.Nakajima Y, Suzuki H. Organometallics. 2003;22:959–969.Nakajima Y, Inagaki A, Suzuki H. Organometallics. 2004;23:4040–4046.Nakajima Y, Suzuki H. Organometallics. 2005;24:1860–1866.Nakajima Y, Kameo H, Suzuki H. Angew Chem, Int Ed. 2006;45:950–952. doi: 10.1002/anie.200503222.
- 17.Hydrazine reduction by iron complexes: Verma AK, Lee SC. J Am Chem Soc. 1999;121:10838–10839.
- 18.(a) Hansert B, Vahrenkamp HJ. Organomet Chem. 1993;459:265–269. [Google Scholar]; (b) Bazhenova TA, Emelyanova NS, Shestakov AF, Shilov AE, Antipin MY, Lyssenko KA. Inorg Chim Acta. 1998;280:288–294. [Google Scholar]
- 19.Ohki Y, Takikawa Y, Hatanaka T, Tatsumi K. Organometallics. 2006;25:3111–3113. [Google Scholar]
- 20.In a related reaction, azides are catalytically reduced by H2 using a sterically hindered iron catalyst: Bart SC, Lobkovsky E, Bill E, Chirik PJ. J Am Chem Soc. 2006;128:5302–5303. doi: 10.1021/ja057165y.
- 21.Some synthetic Fe/Mo clusters reduce diazenes, and the available evidence indicates that the reaction takes place at molybdenum: Malinak SM, Simeonov A, Mosier PE, McKenna CE, Coucouvanis D. J Am Chem Soc. 1997;119:1662–1667.
- 22.(a) Gambarotta S, Floriani C, Chiesi-Villa A, Guastini C. J Chem Soc, Chem Commun. 1982:1015. [Google Scholar]; (b) Cotton FA, Duraj S, Roth W. J Am Chem Soc. 1984;106:4749. [Google Scholar]; (c) Lahiri GK, Goswami S, Falvello L, Chakravorty A. Inorg Chem. 1987;26:3365. [Google Scholar]; (d) Hill IE, Profilet RD, Fanwick PE, Rothwell IP. Angew Chem Int Ed Engl. 1990;29:664. [Google Scholar]; (e) Hill JE, Fanwick PE, Rothwell IP. Inorg Chem. 1991;30:1143. [Google Scholar]; (f) Arney DSJ, Burns C. J J Am Chem Soc. 1992;115:10068. [Google Scholar]; (g) Peters RG, Warner BP, Burns CJ. J Am Chem Soc. 1999;121:5585. [Google Scholar]; (h) Arney DSJ, Burns CJ. J Am Chem Soc. 1995;117:9448–9460. [Google Scholar]; (i) Schrock RR, Glassman TE, Vale MG, Kol M. J Am Chem Soc. 1993;115:1760. [Google Scholar]; (j) Zambrano CH, Fanwick PE, Rothwell IP. Organometallics. 1994;13:1174. [Google Scholar]; (k) Lockwood MA, Fanwick PE, Eisenstein O, Rothwell IP. J Am Chem Soc. 1996;118:2762. [Google Scholar]; (l) Gray SD, Thorman JL, Adamian VA, Kadish KM, Woo LK. Inorg Chem. 1998;37:1. doi: 10.1021/ic970952e. [DOI] [PubMed] [Google Scholar]; (m) Warner BP, Scott BL, Burns CJ. Angew Chem Int Ed. 1998;37:959. doi: 10.1002/(SICI)1521-3773(19980420)37:7<959::AID-ANIE959>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]; (n) Maseras F, Lockwood MA, Eisenstein O, Rothwell IP. J Am Chem Soc. 1998;120:6598. [Google Scholar]; (o) Aubart MA, Bergman RG. Organometallics. 1999;18:811. [Google Scholar]; (p) Pétillon FY, Schollhammer P, Talarmin J, Muir KW. Inorg Chem. 1999;38:1954–1955. doi: 10.1021/ic9813359. [DOI] [PubMed] [Google Scholar]; (q) Diaconescu PL, Arnold PL, Baker TA, Mindiola DJ, Cummins CC. J Am Chem Soc. 2000;122:6108. [Google Scholar]; (r) Guillemot G, Solari E, Scoppelliti R, Floriani C. Organometallics. 2001;20:2446–2448. [Google Scholar]; (s) Lentz MR, Vilardo JS, Lockwood MA, Fanwick PE, Rothwell IP. Organometallics. 2004;23:329–343. [Google Scholar]; (t) Evans WJ, Kozimor SA, Ziller JW. Chem Commun. 2005:4681–4683. doi: 10.1039/b508612d. [DOI] [PubMed] [Google Scholar]
- 23.Yu Y, Brennessel WW, Holland PL. submitted. [Google Scholar]
- 24.This step might be preceded by coordination of the azobenzene in an η1 binding mode; see ref 22o.
- 25.Vela J, Vaddadi S, Cundari TR, Smith JM, Gregory EA, Lachicotte RJ, Flaschenriem CJ, Holland PL. Organometallics. 2004;23:5226–5239. [Google Scholar]
- 26.When [LtBuFeH]2 is reacted with 1 equiv of PhN=NPh (i.e., 0.5 equiv of PhN=NPh per iron atom), then all of the PhN=NPh is consumed. In this case, the PhN=NPh formed in the decomposition of LtBuFeNPhNHPh immediately reacts with the excess iron-hydride complex to form more LtBuFeNPhNHPh, eventually giving only LtBuFeNHPh as a product.
- 27.A linear Eyring plot (Figure S-l) derived from the rate constants at different temperatures gives apparent activation parameters of ΔH± =13 ± 1 kcal·mol−1 and ΔS± = −36 ± 4 eu. However, the induction period suggests a chain mechanism (discussed at length below), in which the rate constants do not reflect elementary steps. Therefore, these apparent activation parameters should be interpreted only with the greatest caution.
- 28.The apparent kH/kD is 0.95 ± 0.23, and the value near unity is consistent with loss of deuterium label.
- 29.Holt PF, Hughes BP. J Chem Soc. 1953:1666–1669. [Google Scholar]
- 30.Heesing A, Schinke U. Chem Ber. 1977;110:2867–2871. [Google Scholar]
- 31.The reaction of LtBuFeCEtCHEt and PhN=NPh at room temperature does not form LtBuFeNPhNHPh. Many other control experiments were also performed to verify that the products do not react with each other or with byproducts (see Supporting Information).
- 32.We cannot rule out that the initiation step is N-N bond homolysis, which would give LtBuFeNPh (a potential chain carrier, as discussed below) and PhNH•, which would give a small amount of PhNH2 (presumably undetected). After initiation, the chain would proceed as in Scheme 4.
- 33.(a) Shizuka H, Kayoiji H, Morita T. Mol Photochem. 1970;2:165–176. [Google Scholar]; (b) Van Beek HCA, Heertjes PM, Houtepen C, Retzloff D. J Soc Dyers Colourists. 1971;87:87–92. [Google Scholar]; (c) Patai, editor. The Chemistry of the Hydrazo, Azo, and Azoxy Groups. Wiley; New York: 1975. [Google Scholar]
- 34.However, second-order reactions between two transient radicals are relatively unlikely, a phenomenon known as the “persistent radical effect.”35 The implication is that the transient radical species will react primarily with the major species in solution, LtBuFeNPhNHPh.
- 35.(a) Fischer H. Chem Rev. 2001;101:3581–3610. doi: 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]; (b) Daikh BE, Finke RG. J Am Chem Soc. 1992;114:2938–43. [Google Scholar]
- 36.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 37.Eckert NA, Vaddadi S, Stoian S, Lachicotte RJ, Cundari TR, Holland PL. Angew Chem, Int Ed Engl. 2006;45:6868–6871. doi: 10.1002/anie.200601927. [DOI] [PubMed] [Google Scholar]
- 38.Other recently isolated iron(III) species with a terminal imido ligand: Brown SD, Betley TA, Peters JC. J Am Chem Soc. 2003;125:322–323. doi: 10.1021/ja028448i.Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122.Brown SD, Peters JC. J Am Chem Soc. 2005;127:1913–1923. doi: 10.1021/ja0453073.Mehn MP, Peters JC. J Inorg Biochem. 2006;100:634–643. doi: 10.1016/j.jinorgbio.2006.01.023.Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4956–4957. doi: 10.1021/ja0604358.
- 39.All attempts to create (diketirninate)FeIII=N(aryl) species have given (diketiminate)FeII–NH(aryl) products, consistent with powerful H-atom abstraction ability of the putative imido. Eckert, N. A. Ph. D. Thesis, University of Rochester, 2005.
- 40.Bouwstra JA, Schouten A, Kroon J Acta Crystallogr C. 1983;39:1121–1123. [Google Scholar]
- 41.(a) Stoian SA, Yu Y, Smith JM, Holland PL, Bominaar EL, Münck E. Inorg Chem. 2005;44:4915–4922. doi: 10.1021/ic050321h. [DOI] [PubMed] [Google Scholar]; (b) Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Inorg Chem. 2006;45:5742–5751. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Bordwell FG, Cheng JP, Wang D. J Am Chem Soc. 1997;119:9125–9129. [Google Scholar]
- 43.Diketiminate-iron(I) complexes react with ketones to give pinacol coupling products: see ref 36. We have obtained ambiguous results when using other traps for iron(I) in the conversion of LtBuFeNPhNHPh to LtBuFeNHPh, as follows. PPh3 and PEt3 bind weakly to the LtBuFe fragment (see ref 36 and 41b) and do not affect the rate of conversion. CO does bind to LtBuFe (see ref 36), but does not affect the decomposition of LtBuFeNPhNHPh: this is consistent with the observation that addition of a small amount of LtBuFe(CO)2 to the LtBuFeNPhNHPh abolishes the induction period for its decomposition. Therefore, it is likely that CO binds reversibly to the LtBuFe fragment, and does not substantially affect the chain reaction.
- 44.Azobenzene loss may require an isomerization of the η2-bound ground state (Figure 6) to an η1 isomer, a process that can be slow: see ref 22o. We are unable to reconcile this rate-limiting step with the apparent negative value of ΔS± in footnote 27.
- 45.Recent review on radical organometallic reactions in diketiminate chemistry: Smith KM. Organometallics. 2005;24:778–784.
- 46.Despite the conceptual complexity of radical processes, they can lead to high yields and clean products. See, for example: Huyser ES. Free-Radical Chain Reactions. Wiley; New York: 1970.
- 47.(a) Chatt J, Pearman AJ, Richards RL. Nature. 1975;253:39–40. doi: 10.1038/259204a0. [DOI] [PubMed] [Google Scholar]; (b) Yandulov DV, Schrock RR. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]; (c) Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]; (d) Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252–6254. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]
- 48.(a) Baker MV, Field LD, Hambley TW. Inorg Chem. 1988;27:2872. [Google Scholar]; (b) Schubert EM. J Chem Ed. 1992;69:62. [Google Scholar]
- 49.Newton A. J Am Chem Soc. 1943;65:2434–2439. [Google Scholar]
- 50.(a) Smith JM, Lachicotte RJ, Holland PL. Chem Commun. 2001:1542–1543. doi: 10.1039/b103635c. [DOI] [PubMed] [Google Scholar]; (b) Smith JM, Lachicotte RJ, Holland PL. Organometallics. 2002;21:4808–4814. [Google Scholar]
- 51.Fryzuk MD, Lloyd BR, Clentsmith GKB, Rettig SJ. J Am Chem Soc. 1994;116:3804–3812. [Google Scholar]
- 52.APEX2 V10-22. Bruker Analytical X-ray Systems; Madison, WI: 2004. [Google Scholar]
- 53.SADABS V210: Blessing R. Acta Cryst. 1995;A51:33–38. doi: 10.1107/s0108767394005726.
- 54.SAINT V706A. Bruker Analytical X-ray Systems; Madison, WI: 2003. [Google Scholar]
- 55.SHELXTL V6.14. Bruker Analytical X-ray Systems; Madison, WI: 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Eyring plot and NMR data (pdf) and crystallographic data (cif). This material is available free of charge via the Internet at http://pubs.acs.org.