

Abstract
Act3p/Arp4, an essential actin-related protein of Saccharomyces cerevisiae located within the nucleus, is, according to genetic data, involved in transcriptional regulation. In addition to the basal core structure of the actin family members, which is responsible for ATPase activity, Act3p possesses two insertions, insertions I and II, the latter of which is predicted to form a loop-like structure protruding from beyond the surface of the molecule. Because Act3p is a constituent of chromatin but itself does not bind to DNA, we hypothesized that insertion II might be responsible for an Act3p-specific function through its interaction with some other chromatin protein. Far Western blot and two-hybrid analyses revealed the ability of insertion II to bind to each of the core histones, although with somewhat different affinities. Together with our finding of coimmunoprecipitation of Act3p with histone H2A, this suggests the in vivo existence of a protein complex required for correct expression of particular genes. We also show that a conditional act3 mutation affects chromatin structure of an episomal DNA molecule, indicating that the putative Act3p complex may be involved in the establishment, remodeling, or maintenance of chromatin structures.
INTRODUCTION
Actin-related proteins (Arps), a group of protein families that exhibit moderate sequence similarity among each other and to conventional actin (i.e., muscle actin), have been found in a variety of eukaryotic organisms (Frankel and Mooseker, 1996; Frankel, 1998). According to the known three-dimensional structure of rabbit muscle actin and sequence comparisons, Arps and conventional actin compose the actin branch within a superfamily of proteins that possess ATPase activity, and this superfamily includes the 70-kDa heat shock cognate protein and hexokinase (Holmes et al., 1993). Although actin and Arps most probably possess a common tertiary structure centered around an ATP/ADP-binding pocket, named the actin fold, they exhibit divergent surface features caused by insertions and point mutations (Mullins et al., 1996). The actin fold undergoes major conformational shifts in response to the 5′ phosphorylation–hydrolysis state of the adenine nucleotide, and such shifts are thought to be crucial to the functions of the various actin family members (Frankel, 1998). Additionally, the divergent nature of molecular surfaces for each protein is thought to be involved in unique functions specific for each family member.
Poch and Winsor (1997) proposed a classification of Arps in which the Arp classes are numbered according to their similarity to conventional actin: class 1 is the most and class 10 is the least similar to muscle actin. Functional analyses on Arps of classes 1–3 revealed their distinct functions in the cytoplasm, and recently it was shown that two related chromatin-remodeling complexes of Saccharomyces cerevisiae, RSC (remodel the structure of chromatin) and SWI/SNF (switching/sucrose nonfermenting), share Arp7 and Arp9 (Cairns et al., 1998; Peterson et al., 1998). However, functional information of distantly related Arps is still limited. Among the 10 Arps of S. cerevisiae, Act3p, a class 4 Arp (Arp4), is characterized by its nuclear localization confirmed by immunofluorescence (Weber et al., 1995; Jiang and Stillman, 1996). ACT3 is an essential gene coding for a polypeptide of 489 amino acids with a calculated molecular mass of 54.8 kDa (Harata et al., 1994). Subfractionation of nuclear proteins revealed association of Act3p with chromatin (Weber et al., 1995), and genetic studies indicated that Act3p is involved in transcriptional regulation (Jiang and Stillman, 1996) as described below.
S. cerevisiae contains a retrovirus-like transposable element, Ty1, which possesses two domains with strong promoters, the δ element. Insertion of Ty1 or a δ element into the 5′ region of genes often causes inactivation of the adjacent gene because of interference or competition between transcriptional signals in the δ element and the native gene promoter (Winston and Carlson, 1992). Selections for extragenic suppressors of Ty1- or δ element-inactivated genes have identified numerous SPT (suppressor of Ty) genes, and many of them were shown to be involved in transcriptional regulations via effects on chromatin structure (Winston and Carlson, 1992). For example, SPT11 and SPT12 encode histones (Clark-Adams et al., 1988), SPT15 encodes a TATA-binding factor (Eisenmann et al., 1989), the gene products of SPT3, SPT7, and SPT20 are components of the Spt-Ada-Gcn5-acetyltransferase complex (Grant et al., 1997), and SPT6 encodes a protein that binds to core histones (Bortvin and Winston, 1996). Because certain nonlethal mutations in the ACT3 gene suppress the transcriptional defect caused by the insertion of a δ element into the yeast HIS4 promoter (his4-912δ), ACT3 belongs to the group of SPT genes.
Interestingly, act3 mutations cause variegated suppression of the δ element-inactivated HIS4 gene (the his4-912δ allele) in cells of identical genetic background (Jiang and Stillman, 1995), suggesting an epigenetic effect of the mutated Act3p proteins on transcription. On and off states of transcription of the his4-912δ gene in the act3 mutants were found heritable with a low degree of reversibility. This situation seems to be comparable with the position effect variegation originally observed for genes adjacent to heterochromatin in Drosophila (Elgin, 1996). Because position effect variegation in Drosophila is associated with an altered chromatin structure (Wallrath and Elgin, 1995), it is likely that the act3 mutations cause a defect in chromatin structure that affects gene regulation.
Here we report the ability of Act3p to bind to core histones in vitro and in vivo. We also show that an act3 mutation affects chromatin structure by changes in DNA-linking number indicating altered chromatin. Our findings suggest that Act3p is involved in the establishment or the maintenance of chromatin structure through its association with core histones.
MATERIALS AND METHODS
Yeast Strains, Media, and General Methods
Strain MZ3 (MATa pep4-3 trp1 leu2-Δ1 ura3-Δ1) was derived from 20B12 (Wintersberger et al., 1995). The yeast reporter strain Y190 (MATa ura3-52 his3-200 lys2-801 ade2-101 trp1-901 leu2-3 112 gal4Δ gal80Δ cyhr2 LYS2::GAL1UAS-HIS3TATA-HIS3 URA3::GAL1UAS-GAL1TATA-lacZ) (Clontech Laboratories, Palo Alto, CA) was used in the two-hybrid analysis. Yeast strains DY3462 (MATa his4-912δ-ADE2 his4-912δ lys2-128δ can1 leu2 ura3 ACT3) and DY3189 (MATa his4-912δ-ADE2 his4-912δ lys2-128δ can1 leu2 trp1 ura3 act3-3) were used for analyses of chromatin structure of plasmid YCp50. YPD growth medium was used as standard medium for the culture of yeast cells. Synthetic complete medium was prepared as described by Sherman (1991). Protease inhibitors (0.5 μg/ml antipain, chymostatin, elastatinal, leupeptin, and pepstatin A) were added to the buffers described below.
Preparation of Nuclei, Chromatin Fraction, and Core Histones from Yeast and Chicken Cells
Yeast nuclei were prepared as described by Weber et al. (1995). To prepare yeast chromatin fractions, the isolated nuclei were suspended in 10 mM Tris-HCl, pH 7.5, 20 mM KCl, 5 mM MgCl2, 3 mM DTT, 1 mM EDTA, and 10% glycerol, and protease inhibitors as well as 0.25 mg/ml DNaseI were added. After incubation for 2 h on ice, the soluble chromatin fraction was obtained by centrifugation. Purification of core histones from isolated yeast nuclei was performed according to the method of Smith et al. (1996).
To prepare chicken core histones, female chicken MSB-1 cells (Akiyama and Kato, 1974) were cultured in suspension in 1 l of RPMI 1640 medium (Sigma, St. Louis, MO) containing 1% (vol/vol) FBS (Irvine Scientific, Santa Ana, CA) and 4% (vol/vol) chicken serum (Sigma) at 41°C under 5% CO2/95% air, and ∼2 × 109 cells were harvested. The following steps were all carried out at 0–4°C. Nuclei were prepared as described by Suka et al. (1993). The nuclear pellet was washed with solution E (300 mM sucrose, 50 mM triethanolamine, 25 mM KCl, 5 mM sodium butyrate, 4 mM MgCl2, 1 mM CaCl2, 1% thiodiglycol, and 1 mM PMSF) and suspended in the same buffer at a concentration of 5 mg DNA/ml. Micrococcal nuclease (Worthington Biochemical, Freehold, NJ) was added to the suspension at a concentration of 40 U/mg DNA, and the mixture was incubated at 37°C for 3.5 min. Thereafter the nuclei were sedimented by centrifugation for 10 min at 13,000 × g, and the pellet was extracted twice with solution G (2 mM EDTA and 0.73 mM tetrapropylammonium hydroxide, pH 7.4) by resuspension and centrifugation as above to yield the combined supernatants. The supernatants were dialyzed against 0.1 M K-phosphate buffer, pH 6.8, containing 0.63 M NaCl and protease inhibitors and applied to a hydroxylapatite column (40 ml). After washing the column with the dialysis buffer, histones H2A/H2B and histones H3/H4 were eluted with 0.1 M K-phosphate buffer, pH 6.8, containing 0.93 and 2 M NaCl, respectively.
Gel Electrophoresis and Western Blot Analysis
Discontinuous SDS-PAGE was performed with gels containing 12.5% acrylamide. Proteins were blotted to nitrocellulose membranes as described previously (Harata et al., 1994). Act3p and histone H2A on the blots were detected with the combination of an affinity-purified polyclonal rabbit antibody against Act3p (Weber et al., 1995), HRP-conjugated anti-rabbit immunoglobulin G antibody, and a chemiluminescence developing reagent (ECL system; Amersham, Buckinghamshire, United Kingdom), as well as with the combination of a monoclonal mouse antibody against histone H2A (Weber et al., 1995), alkaline phosphatase-conjugated anti-mouse immunogolublin G antibody, and the ProtoBlot alkaline phosphatase detection system (Promega, Madison, WI), respectively.
Far Western Blot Analysis
To construct a plasmid for the GST-Act3p(Ins II) fusion protein, a DNA fragment encoding amino acids 269S–387D of Act3p (nucleotides 803-1162 in Harata et al., 1994), which contains the insertion II of Act3p, was isolated by Sau3A I digestion and inserted into the BamHI site of the vector pGEX-2TK (Pharmacia Biotech, Uppsala, Sweden). To produce control fusion proteins, DNA fragments encoding amino acids 1M–295P of Act3p [Act3p(N-ter)], 380N–489R of Act3p [Act3p(C-ter)], and 192I–310A of S. cerevisiae Act1p [Act1p(flank)] were amplified with the following primers and with yeast genomic clones containing ACT3 or ACT1: Act3p(N-ter), sense primer N-terF (5′-ATAGGATCCATGTCCAATGCTGCTTTGCAA-3′) and antisense primer N-terR (5′-GAAGGCCTGGAAGGAAAAGCTCTTCAG-3′); Act3p(C-ter), sense primer C-terF (5′-CGGGATCCAATGAATTGATTGGTCTAGCG-3′) and antisense primer C-terR (5′-CGCGAATTCCTATCTAAACCTATCGTTAAG-3′); and Act1p(flank), sense primer Act1F (5′-CGGGATCCATCTTGAGTGAACGTGGT-3′) and antisense primer (5′-CGGAATTCGGCAATACCTGGGAACAT-3′). The PCR products were cleaved at primer-derived BamHI and EcoRI sites or BamHI and StuI sites, and the DNA fragments were cloned into BamHI- and EcoRI-digested or BamHI- and SmaI-digested pGEX-2TK. Escherichia coli (XL-1 blue) was transformed with each of the constructed plasmids or with a control pGEX-2TK, and expression of the fusion proteins was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside. After further incubation at 30°C for 3 h, cells were harvested by centrifugation, and the cell pellets were resuspended in 10 ml of PBS containing 0.03% SDS and protease inhibitors. Cells lysed by sonication were centrifuged, and the supernatants were collected. After addition of two volumes of PBS (containing protease inhibitors) and 500 μl of a slurry of 50% glutathione-Sepharose 4B (Pharmacia Biotech) in the same buffer to the supernatants, the fusion proteins were bound to the matrices at 4°C for 12 h with shaking. Subsequently the matrices were sedimented by centrifugation and washed five times with PBS and twice with 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 12 mM MgCl2. The fusion proteins contain a cAMP-dependent kinase recognition site following the GST region, and the matrix-bound fusion proteins were labeled each with 100 U of the catalytic subunit of bovine heart kinase (Sigma), 100 μCi of [γ-32P]ATP (3000 Ci/mmol; Amersham), and 1.3 mM DTT at 4°C for 30 min. The reactions were stopped by addition of 2 ml of the stop solution (10 mM sodium phosphate, pH 8.0, 10 mM sodium pyrophosphate, 10 mM EDTA, and 1 mg/ml BSA). After washing the matrices five times with PBS, the labeled fusion proteins were recovered by addition of 500 μl of elution buffer (50 mM Tris-HCl, pH 9.5, and 20 mM reduced glutathione) and centrifugation. To prepare unlabeled fusion proteins, the labeling procedure was omitted, and the fusion proteins were recovered as described above.
Proteins to be tested for binding to Act3p(Ins II) were separated by SDS-PAGE and blotted to a nitrocellulose membrane in 25 mM Tris base and 192 mM glycine. After incubation in binding buffer (20 mM HEPES, pH 7.7, 75 mM KCl, 75 mM NaCl, 1 mM EDTA, 0.25 mM MgCl2, 0.5 mM DTT, 0.05% NP-40, 0.4% BSA, 0.4% Ficoll 400, and 0.4% polyvinylpyrrolidone) at room temperature for 30 min, the membrane was incubated for 16 h at 4°C in the binding buffer (0.2 ml/lane on the membrane) containing the 32P-labeled Act3p(Ins II) (∼5 μg [∼200,000 cpm]/ml). Thereafter the membrane was washed three times with the binding buffer for 5 min each, and the binding of GST-Act3p(Ins II) to the membrane-bound proteins was analyzed with BAS2000 bio-image analyzer (Fuji Photo Film, Tokyo, Japan). As controls membranes with bound proteins were incubated with 32P-labeled GST not fused to Act3p(Ins II), GST-Act3p(N-ter), GST-Act3p(C-ter), and Act1p(flank).
Two-Hybrid Analysis
From the cloned ACT3 gene the region encoding amino acids 269S–413G of Act3p was excised with BglII and RsaI and subcloned into the BamHI- and HincII-digested pUC119 vector. Thereafter the fragment was excised with EcoRI and PstI and subcloned into the EcoRI- and PstI-digested pAS2–1 vector (Clontech). To produce a control construct, a DNA fragment encoding 192I–310A of S. cerevisiae Act1p [Act1p(flank)] was amplified from a yeast genomic clone containing ACT1 using the sense primer Act1FII (5′-CATGCCATGGAGATCTTGAGTGAACGTG-3′) and Act1R (see above). The PCR product was cleaved at primer-derived NcoI and EcoRI sites and subcloned into the NcoI- and EcoRI-digested pAS2–1 vector. DNA sequencing confirmed that the resulting plasmids, GAL4-DBD-Act3p and GAL4-DBD-Act1p, express the GAL4 activation domain (AD) fused in frame to amino acids 269S-413G of Act3p and 192I-310A of Act1p, respectively.
The ORFs of the yeast core histones were amplified with the following primers and with yeast genomic DNA as a template: HTA1 (histone H2A), sense primer H2AF (5′-AAAGGATCCGAATGTCCGGTGGTAAAGG-3′) and antisense primer H2AR (5′-TTTCTCGAGCCTTCCGCCTTCTTTAA-3′); HTB1 (histone H2B), sense primer H2BF (5′-CCCGGATCCGAATGTCTGCTAAAGC-3′) and antisense primer H2BR (5′-TATCTCGAGCAAAGGAAGTGATTTCA-3′); HHT1 (histone H3), sense primer H3F (5′-AAAGGATCCGAATGGCCAGAACAAAGC-3′) and antisense primer H3R (5′-AATCTCGAGCTGATGACAATCAACAA-3′); and HHF2 (histone H4), sense primer H4F (5′-AAAGGATCCGAATGTCCGGTAGAGG-3′) and antisense primer H4R (5′-AAACTCGAGAAATTAACCACCGAAACC-3′). The PCR products were cleaved at primer-derived BamHI and XhoI sites, and the DNA fragments were cloned into the BamHI- and XhoI-digested pACT2 vector (Clontech), respectively. The resulting plasmids expected to contain in-frame fusions of each of the yeast core histones with the AD of GAL4 were confirmed by DNA sequencing. The yeast reporter strain, Y190, was cotransformed with various combination of the two-hybrid vectors, plasmids containing the constructs or control constructs, and were plated on synthetic complete media lacking the appropriate amino acids.
For quantitation of the β-galactosidase activity, β-galactosidase liquid assays were done by using o-nitrophenyl-β-d-galactopyranoside as a substrate. Yeast transformants were grown overnight in appropriate synthetic media. Then 1.5-ml aliquots were inoculated into 6 ml of YPD and grown to an OD600 of 0.5–0.8. Cells were harvested and permeabilized by freezing–thawing cycles (three times) with liquid nitrogen. β-Galactosidase activity was assayed (Kaiser et al., 1994), and the unit of the activity was defined according to the method of Miller (1972).
Fractionation of Complexes Containing His6-tagged Act3p
The ORF coding for the peptide 320V–489R of Act3p together with a continuous His6-tag was amplified by PCR with the sense primer ActHisF (5′-GCCCAAGCTTCGATCGACGTGCCATTAAAAAGAACCAAGC-3′) and the antisense primer ActHisR (5′-GACTAGTGGTGATGATGATGATGTCTAAACCTATCGTTAAGCAATC-3′), using the cloned ACT3 gene (Harata et al., 1994) as template. By taking advantage of the primer-derived HindIII and SpeI sites of the amplified PCR product, the fragment was subcloned into the pRS406 vector (Stratagene, La Jolla, CA), and its presence was confirmed by DNA sequence analysis. For introduction of the His6 tag at the terminus of the genomic ACT3 gene by homologous recombination, strain MZ3 was transformed with the plasmid linearized at the internal BsaBI site within the insert. Among Ura+ transformants, strains expressing the Act3p-His6 fusion protein were identified by Southern blot analysis of the genomic locus of ACT3 and Western blot analysis for expression of Act3p-His6 with an anti-Act3p antibody as well as an anti-His6 tag antibody.
The Act3-His6-expressing strain, MZ3ACT3H, was grown in 1 l of YPD, harvested, washed with cold water, and resuspended in 10 ml of disruption buffer (20 mM HEPES, pH 7.4, 4 mM MgCl2, 100 mM NaCl, 50 mM KCl, 5% glycerol, 25 mM imidazole, and protease inhibitors). After addition of DNaseI at 350 U/ml, cells were disrupted with glass beads. The cell extract was clarified by centrifugation and subsequently filtered through a 0.22-μm pore filter. After addition of NP-40 at a concentration of 0.05% to the extract (∼10 ml), 2 ml of the 50% Ni2+-nitrilotriacetic acid (NTA) slurry (Qiagen, Santa Clarita, CA), equilibrated with the disruption buffer containing 0.05% NP-40, were added to the extract. The suspension was mixed gently for 1 h at room temperature and loaded into an empty column. The column filled with Ni2+-NTA was washed with the disruption buffer containing 0.05% NP-40. Bound proteins were then eluted with a linear gradient of imidazole (0.025–0.2 M in 9 ml in the same buffer), and 0.5-ml fractions were collected. Aliquots of 10 μl from each fraction were subjected to Western blot analysis.
To check the specificty of the anti-histone H2A antibody, yeast histone H2A was further purified from the acid-precipitated yeast core histones. The fraction was separated by SDS-PAGE and subjected to imidazole-zinc reverse staining (Fernandez-Patron et al., 1995). The band of histone H2A was cut out and crushed in the same amount of SDS sample buffer (50 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, and 2% 2-mercaptoethanol). After heating in boiling water for 5 min, the supernatant was subjected to Western blot analysis with the anti-histone H2A antibody.
Immunoprecipitation of Protein Complex Containing Act3p
Cell extract from strain MZ3ACT3H (containing Act3p-His6) was prepared and applied to a Ni2+-NTA column. Bound protein complexes were eluted with the disruption buffer containing 0.05% NP-40 and 0.3 M imidazole. To separate proteins that might bind nonspecifically to protein A-Sepharose beads (Pharmacia Biotech) from the protein fraction containing the Act3p-His6, the preparation (200 μl) was preincubated with 40 μl of a slurry of the beads (50% in the disruption buffer containing 0.05% NP-40) for 1 h at 4°C. After removal of the beads by centrifugation, the protein fraction was reacted with 0.1 μl of anti-histone H2A antibody (Weber et al., 1995) or 0.1 μl of a serum from a nonimmunized mouse and incubated at 4°C for 50 min. Then 20 μl of a slurry of protein A-Sepharose beads was added to the mixture and incubated at 4°C for 1 h. The beads that were expected to be associated with the protein complexes via the specific antibody were sedimented and washed five times with 0.5 ml each of the disruption buffer containing 0.05% NP-40. Bound proteins were eluted with 20 μl of the SDS sample buffer without 2-mercaptoethanol, and the elute was loaded onto gels for Western blot analysis without reduction.
Chloroquine Gel Electrophoresis
Yeast DNAs prepared from strains containing the YCp50 plasmid were subjected to the two-dimensional electrophoresis as descried previously (Jiang and Stillman, 1992). Briefly, electrophoresis in the first dimension was done in 0.7% agarose in the presence of 0.8 μg/ml chloroquine. Then in the second dimension, electrophoresis was done in the presence of 4.0 μg/ml chloroquine. The DNA in the gel was transferred to a membrane and detected with a 32P-labeled YCp50 probe.
RESULTS
Structural Feature of Act3p
Although Act3p shows obvious similarity to conventional actins over nearly the whole sequence, it is a unique member of the actin protein family in terms of the presence of two insertions (Harata et al., 1994; insertions I and II in Figure 1). Insertion I, comprising 24 amino acids, contains a putative nuclear targeting signal (Weber et al., 1995) and seems to actually be responsible for the nuclear localization of Act3p in vivo (Wintersberger, manuscript in preparation). Recently, it was reported that conventional actins have two nuclear export signals, which contain four hydrophobic residues with characteristic spacing (Wada et al., 1998). In the regions of Act3p (175T–186A and 241E–252T) corresponding to the nuclear export signals of conventional actin, two of the four hydrophobic residues in each region are substituted by amino acids not belonging to the hydrophobic group (180T and 183N and 242C and 251P, respectively), suggesting that regulated nuclear export of Act3p does not occur in vivo (for review, see Nigg, 1997). Insertion II, consisting of 83 amino acids, is relatively abundant in charged amino acids (15 acidic and 16 basic residues) and forms one of the most hydrophilic regions of Act3p. We used the algorithm of Janin (1979) to predict the surface of Act3p that would be accessible to interaction with other molecules, and found that insertion II represents the most accessible domain of the Act3p molecule (Figure 1). In addition the 3D–1D profile predicts that insertion II forms a loop-like structure, which protrudes from the surface of the Act3p molecule (our unpublished results). Thus, although various surface parts of Act3p may contribute to Act3p-specific functions, insertion II, for which homologous regions exist neither in actin nor in any other Arps of S. cerevisiae, seems to be the most characteristic surface feature of the molecule; therefore we started our investigations with a study of possible interactions of insertion II with other molecules in vitro. Considering the nuclear location of Act3p (Weber et al., 1995), we first tested the DNA-binding ability of insertion II of Act3p. Using a fusion protein containing insertion II, GST-Act3p(Ins II) (see below), we performed gel shift assays and South-Western blot analyses with DNA fragments prepared from plasmids as probes; however, we did not detect any DNA-binding ability of Act3p(Ins II) (our unpublished results). In addition, chromatography of a yeast cell extract, free of nucleic acids, on a DNA cellulose column in presence of 0.1 M NaCl resulted in Act3p elution with the flowthrough fraction, whereas known DNA-binding proteins had to be eluted with a buffer containing 10–20 times higher salt concentrations (our unpublished results). Therefore, we supposed that insertion II of Act3p might associate with chromatin proteins.
Figure 1.

Accessibility plot of Act3p. The accessible surface area of Act3p was calculated according to the method of Janin (1979) using a computation program available on the Internet (http://expasy.hcuge.ch/cgi-bin/protascale; window size 21). Numbers under the horizontal axis represent those of the amino acids of Act3p. Scores beside the vertical axis show the probability to be accessible on the surface of the molecule for each region of Act3p. Roman numerals (I and II) represent the positions of the insertions I and II of Act3p, respectively.
Far Western Blot Analysis with Insertion II of Act3p
To screen for chromatin proteins that might interact with insertion II of Act3p, we performed far Western blot experiments. This method uses a 32P-labeled protein to probe for interaction with proteins present on a nitrocellulose membrane. Our 32P-labeled probe was a fusion protein, GST-Act3p(Ins II), which consisted of insertion II (amino acids 269–387) of Act3p fused to GST. When chromatin proteins prepared from isolated yeast nuclei were subjected to far Western blot analysis (Figure 2B), GST-Act3p(Ins II) recognized a 17-kDa protein (lane 4, arrow) and to a lesser extent a 30-kDa protein (lane 4, arrowhead) but neither any other chromatin nor marker protein (Figure 2B, lanes 4 and 1). 32P-labeled GST alone did not bind to any protein under the test conditions (our unpublished results).
Figure 2.

Far Western blot analysis with 32P-labeled GST-Act3p(Ins II). Marker proteins (lane 1), chicken histone fraction enriched in H3 and H4 (lane 2), purified chicken histones H2A and H2B (lane 3), and the chromatin fraction prepared from isolated yeast nuclei (lane 4) were electrophoresed on SDS-polyacrylamide gels. The gels were subjected to staining with Coomassie brilliant blue R-250 (A) and to far Western blot analysis with 32P-labeled GST-Act3p(Ins II) (B). Radioactivity of the 32P-labeled peptide bound to proteins on the membrane were detected with BAS2000 bio-image analyzer. An arrow and an arrowhead to the right of B mark the positions of a 17-kDa band and a 30-kDa band in lane 4, respectively.
We concentrated on the 17-kDa protein of the yeast chromatin fraction because it showed the strongest binding to GST-Act3p(Ins II), and because its migration on SDS-PAGE seemed to coincide with that of chicken histone H3 (Figure 2, A and B, lanes 2 and 4). Binding of GST-Act3p(Ins II) to all chicken core histones, which were purified by hydroxylapatite column chromatography, was detected after extended autoradiographic exposure (Figures 2B, lanes 2 and 3, and 3B). With shorter exposure time, it can be seen that GST-Act3p(Ins II) binds preferentially to chicken histones H3 and H2B (Figure 3B). In the experiment with acid-precipitated yeast core histones, GST-Act3p(Ins II) binding to histone H3 was visible after short exposure, and after extended exposure binding to all four core histones was seen (Figure 3C). This in vitro binding specificity of GST-Act3p(Ins II) might explain why histone H3 was selectively detected in the fraction of total yeast chromatin (Figure 2B, lane 4).
Figure 3.

Binding of GST-Act3p(Ins II) to purified core histones. (A) Fragments used for the construction of the GST fusions. Numbers correspond to the N- and C-terminal amino acids retained of each fragment. Chicken histones H3 and H4 (H3/4 in B), chicken histones H2A and H2B (H2A/B in B), and acid-precipitated yeast core histones (H2A/B/3/4 in C) were electrophoresed on SDS-polyacrylamide gels. The gels were subjected to staining with Coomassie brilliant blue R-250 or to far Western blot analyses with 32P-GST-Act3p(Ins II), 32P-GST-Act3p(N-ter), 32P-GST-Act3p(C-ter), and 32P-GST-Act1p(flank). Panels for the far Western blot analyses with 32P-GST-Act3p(Ins II) differ in the time of autoradiographic exposure.
In addition to insertion II, GST-Act3p(Ins II) contains its flanking amino acid residues, a part of which is conserved in other members of actin-related proteins. To determine whether the flanking residues without insertion II are responsible for the interaction, we performed far Western blot analyses with 32P-labeled fusion proteins containing the flanking residues of Act3p without insertion II or the equivalent segment of Act1p of S. cerevisiae, consisting of the residues corresponding to the flanking sequences of insertion II: GST-Act3p(N-ter) containing amino acids 1M–295P of Act3p, GST-Act3p(C-ter) containing 380N–489R of Act3p, and GST-Act1p(flank) containing amino acids 192I–310A of Act1p. The fusion proteins were subjected to far Western blot analyses with purified core histones (Figure 3). None of the fusion proteins without insertion II bound to core histones even after the same extended exposure time during which GST-Act3p(Ins II) bound to all of the core histones. This result shows that insertion II itself, but not its flanking residues, is responsible for the interaction with core histones.
Two-Hybrid Analysis of Act3p
We used the two-hybrid system to confirm the interaction between the insertion II of Act3p and core histones. Fusion constructs were prepared by cloning the ACT3 gene and the yeast core histone genes into plasmids encoding the DNA-binding domain (DBD) or the AD of the Gal4p protein, respectively. Interactions between insertion II of Act3p and core histones were expected to result in the activation of HIS3 transcription and lacZ transcription within the transformants. Yeast cells coexpressing GAL4-DBD-Act3p(269–413), which contains insertion II, and one of the GAL4-AD-histone fusions grew on plates lacking histidine (Figure 4A). The activity of β-galactosidase was measured by the liquid assay (Figure 4B). GAL4-DBD-Act3p(269–413) together with each of the GAL4-AD-histones activated the transcription of lacZ compared with the combination with GAL4-AD alone. The activation was not observed when GAL4-DBD-Act1p(192–310) was coexpressed with each of the GAL4-AD-histones. The strength of the activation was different between expressed core histones and did not depend on the strength of their basic charge. These results demonstrate that Act3p is capable of binding to each of the core histones within yeast cells as observed in the far Western blot analysis. The preferential binding of insertion II of Act3p to yeast histone H3 as seen in the far Western blot analysis (Figure 3C) was not observed in the two-hybrid analysis, where insertion II interacted preferentially with histone H2A. The construct used for the two-hybrid analysis for technical reasons contained more flanking residues of insertion II than the one used for the far Western blot analysis; however, as we have shown that the flanking residues did not interact with core histones by the far Western blot analysis, and that the segment of Act1p containing corresponding flanking regions did not interact with histones by the two-hybrid analysis, the difference of the binding preference probably reflects the difference of the principle of these two methods, but not the composition of the flanking residues (also see DISCUSSION).
Figure 4.

Two-hybrid analyses for the detection of a possible interaction between Act3p and yeast core histones. Plasmids encoding the listed sequences were introduced into yeast strain Y190. The growth of the transformant on the dropout media containing 25 mM 3-amino-1,2,4-triazole and no histidine after 4 d of incubation (A) and the activities of β-galactosidase (B) were examined: GAL4-DBD (control bait vector), plasmid pAS2-1 containing the GAL4 DNA-binding domain alone; GAL4-DBD-Act3p, pAS2-1 containing an additional peptide 269S–413G of Act3p, including insertion II; GAL4-DBD-Act1p, pAS2-1 containing an additional peptide 192I–310A of Act1p; GAL4-AD (control prey vector), plasmid pACT2 containing the GAL4 AD alone; H2A, H2B, H3, and H4: plasmids containing each of the yeast histone genes fused with GAL4-AD. Positive controls were plasmids containing the fused genes of the following proteins: GAL4-DBD-p53, the murine p53 gene fused with GAL4-DBD; GAL4-AD-SVT, the SV40 large T-antigen gene fused with GAL4-AD; and in B, the transformants were inoculated from media containing histidine (solid bar) or media lacking histidine (hatched bar). The data shown are the averages and SDs (error bars) of β-galactosidase activities represented by Miller units (Miller, 1972).
In Vivo Interaction between Act3p and Core Histones
To get information about a possible interaction between Act3p and core histones in vivo, we investigated whether in yeast nuclei there existed a protein complex containing both Act3p and core histones. As a control we tested for the presence of histone H2A with the appropriate antibody.
First, we searched for a putative protein complex using the strong interaction between a Ni2+-NTA resin and the His6 tag introduced at the C-terminal part of the Act3 protein molecule. Nuclear extracts prepared from strain MZ3ACT3H, which contains the His6-tagged Act3p (Act3p-His6), or from the control strain, MZ3, were subjected to Ni2+-NTA column chromatography. Western blot analysis was carried out with antiserum against Act3p and anti-histone H2A monoclonal antibody (Weber et al., 1995). Because anti-Act3p antiserum was raised selectively against insertion II of Act3p (Weber et al., 1995), cross-reactions with other nuclear proteins were not to be expected. The anti-histone H2A monoclonal antibody recognized purified yeast histone H2A (Figure 5B, right panel). The Western blot analysis revealed the presence of both proteins in fractions obtained from the chromatography of the nuclear extract from strain MZ3ACT3H (Figure 5B). Act3p without His6 tag, as it is present in the control strain, did not bind to the Ni2+-NTA column (Figure 5A). During chromatography of the control extract a small amount of histone H2A itself or a protein with which it associated was bound to the Ni2+ resin and was eluted from the column as a faint smear throughout all the fractions. In contrast, histone H2A from strain MZ3ACT3H eluted at a specific imidazole concentration together with Act3p-His6. Because in vivo histone H2A is present almost exclusively in histone octamers together with the other core histones, and because the histone octamers should be stable under the condition we used for the Ni2+-NTA column chromatography, we consider it likely that also core histones other than H2A were included in the protein complex eluted from the column. This is also supported by our observation that in vitro–reconstituted nucleosomes are precipitated with GST-Act3p fusion proteins (our unpublished data). Remarkably, whereas Act3p appeared to be present exclusively in the eluted fractions compared with the column load (Figure 5B, top panel), less histone H2A (and thus protein complex) was detected in the eluted fraction than in the column load (Figure 5B, lower panel). This result may indicate that Act3p associates only with a small fraction of either histone H2A or histone octamers.
Figure 5.

Association of Act3p-His6 with histone H2A during chromatography on a Ni2+-NTA column. Nuclear extracts prepared from strains MZ3 (A) and MZ3ACT3H (B) were applied to a Ni2+-NTA column and bound protein complex(es) were eluted with buffer containing increasing concentrations of imidazole (0.025–0.2 M). Aliquots of the applied extracts (AP), of the column flow through (FT), and of each fraction eluted from the columns were subjected to Western blot analyses with antiserum against Act3p (α-Act3p, upper panels) and anti-histone H2A monoclonal antibody (α-H2A, lower panels). Binding of the anti-histone H2A antibody to purified histone H2A is shown in B, right panel. Positions of Act3p and histone H2A on the blots are marked.
To corroborate the data reported above, we performed an immunoprecipitation of the putative complex containing Act3p and core histones. The extract from strain MZ3ACT3H containing the complex(es) was partially purified by Ni2+-NTA column chromatography, and the eluted fractions were immunoprecipitated with the anti-histone H2A antibody. A possible coprecipitation of Act3p with histone H2A was examined by Western blot analyses with the anti-Act3p antiserum (Figure 6). When we analyzed the immunoprecipitated fractions, Act3p was not detected in the fraction precipitated with preimmune serum (Figure 6, lane 3); however, Act3p was indeed coprecipitated with histone H2A by the anti-histone H2A antibody (Figure 6, lane 2), proving the presence of complex(es) containing both Act3p and histone H2A. When we compared the intensity of Act3p-signals, ∼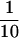 of Act3p was detected as precipitate relative to the input fraction (Figure 6, compare lanes 1 and 2). This suggests that only a portion of Act3p forms a stable complex with core histones in vivo or that the complex is not stable enough during the procedure of the immunoprecipitation.
of Act3p was detected as precipitate relative to the input fraction (Figure 6, compare lanes 1 and 2). This suggests that only a portion of Act3p forms a stable complex with core histones in vivo or that the complex is not stable enough during the procedure of the immunoprecipitation.
Figure 6.

Immunoprecipitation of protein complex(es) with anti-histone H2A monoclonal antibody. Protein complex(es) expected to contain Act3p-His6 in the extract from strain MZ3ACT3H were enriched with a Ni2+-NTA column. Aliquots of the fraction eluted with a buffer containing 0.3 M imidazole were subjected to immunoprecipitation using anti-histone H2A antibody. The presence of Act3p in the input fraction (lane 1) and in the fraction immunoprecipitated with anti-histone H2A antibody (lane 2) or with a serum from a nonimmunized mouse (lane 3) was examined by Western blot analysis with anti-Act3p antiserum. Positions for Act3p are marked on the left.
Taken together with the binding ability of insertion II of Act3p to core histones in vitro, the presence of a complex containing both Act3p and histone H2A suggests that Act3p directly interacts with core histones in vivo and thus is a constituent part of chromatin.
Mutations in ACT3 Affect Chromatin Structure
Because Act3p associates with core histones in vitro and in vivo, and act3 mutants cause variegated suppression of transcription (Jiang and Stillman, 1996), we hypothesized that act3 mutations might have an epigenetic effect on transcription through the contribution of Act3p on chromatin structure. We therefore decided to use a biochemical assay to investigate the effect of act3 mutations on chromatin structure.
The effect of an act3 mutation on plasmid DNA linking number was checked, because previous studies have correlated alterations in chromatin structure with changes in linking number of DNA (Worcel et al., 1981). In these experiments, the linking number of circular DNA molecules is measured by gel electrophoresis in the presence of the intercalating dye chloroquine. Each nucleosome changes the linking number by one, and thus linking number reflects the density of nucleosomes in vivo. Repression of histone gene transcription leads to a loss of nucleosomes, detected by altered DNA linking number, and to alterations in transcriptional regulation (Han et al., 1987). Mutations in the SIR regulatory genes, in addition to causing a loss of silencing at the silent mating type loci, lead to a change in the linking number of plasmids containing silent mating type loci (Abraham et al., 1983; Bi and Broach, 1997; Cheng et al., 1998).
Figure 7 shows the results of an experiment designed to measure linking numbers of circular DNAs. DNA was isolated from isogenic wild-type and act3-3 strains, each carrying the single copy YCp50 plasmid. The act3-3 allele has a single amino acid replacement (155C to Y), and the mutation causes an epigenetic effect on transcriptional regulation (Jiang and Stillman, 1996). The DNA was separated by two-dimensional electrophoresis in the presence of chloroquine, transferred to nitrocellulose, and hybridized with a YCp50 probe. The two-dimensional gel resolves topoisomers of DNA in a broad arc, from the lower left, up to the center, and then down to the right, with linking number increasing clockwise through the arc. The act3-3 mutation causes a counterclockwise shift in topoisomer distribution, indicating a decrease in linking number. In contrast, the distribution of topoisomers in a sin4 mutant is shifted in the opposite direction, indicating increased linking number, as described previously (Jiang and Stillman, 1992; Jiang et al., 1995). In sin4 mutants, changes in linking number were seen with other plasmids, and thus the effect of sin4 is not specific to YCp50. We speculate that this decreased linking number in an act3 mutant could be due to an increased density of nucleosomes or to an increase in stability of nucleosome–DNA complexes, compared with wild type, during the isolation procedure. However, it has been shown that transcription by RNA polymerase II can cause a decrease in DNA linking number (Brill and Sternglanz, 1988; Osborne and Guarente, 1988). Thus, although cause and effect are not clear, the linking number experiment is consistent with the idea that an act3 mutation causes a change in the structure of chromatin.
Figure 7.

Linking number of episomal DNA molecules is altered by an act3 mutation. DNA was isolated from logarithmically growing yeast cells containing the YCp50 plasmid, electrophoresed in two-dimensional chloroquine-agarose gels, and subjected to Southern blot analysis with 32P-labeled URA3 DNA fragment. Left panel, DY3462 (wild-type); right panel, DY3189 (act3-3). The directions of the first and second dimensions are indicated.
DISCUSSION
Act3p Binds to Core Histones In Vitro and In Vivo
If compared with classes 1–3 of the Arps, a characteristic feature of the amino acid sequence of the yeast class 4 Arp, Act3p, is the presence of two inserted domains. Insertion I is involved in nuclear targeting of the Act3p molecule (Wintersberger, manuscript in preparation) and insertion II might, because of its predicted loop-like structure and its high accessibility on the surface of Act3p (Figure 1), be involved in intermolecular interactions. Because Act3p most probably is a component of chromatin but does not directly bind to DNA, we supposed that insertion II might interacted with other component(s) of chromatin.
Far Western blot analysis of a chromatin fraction from yeast with a labeled Act3p-insertion II fusion polypeptide pointed to a possible interaction with histone H3 (Fig, 2B, lane 4). This finding was substantiated by showing an interaction with purified yeast histones, although in this experiment binding also to the other core histones was detected. In the chromatin fraction the smaller amount of the histones and the presence of many proteins besides histones could inhibit the ability of Act3p to interact with all of the histones.
Because histones are basic proteins, and the insertion II of Act3p contains a number of acidic amino acids, it might be assumed that ionic strength between the histone molecules and the Act3p-peptide might be the major force of binding. However, GST-Act3p(Ins II) in vitro interacts only weakly with histone H4, which is the most basic core histone, in the far Western blot analysis. In addition, the in vitro binding of the GST-Act3p(Ins II) to core histones was detected even after washing with a buffer containing 2 M NaCl (our unpublished results), suggesting that ionic charge alone cannot explain the particularly strong binding of yeast histone H3 to GST-Act3p(Ins II). In the experiments with chicken histones, GST-Act3p(Ins II) showed high-affinity binding to both histone H3 and H2B (Figure 3A), whereas the affinity to the yeast histone H2B was only moderate. The reason might be the difference in the primary sequences of the two histone molecules or the different methods used for preparation of core histones from chicken and yeast. A similar result was observed for the Spt6p histone-binding protein of budding yeast, which binds strongly to human, but not yeast, histone H2B (Bortvin and Winston, 1996).
The two-hybrid experiments, which take place within living yeast cells, showed interaction between the insertion II of Act3p and all of the core histones, similar to the far Western result. However, the strongest interaction was observed with histone H2A, but not with H3 (Figure 4B). These results may just reflect the different principles of the two methods, the far Western blot analysis and two-hybrid system, which both do not exactly reproduce the in vivo situation. On the other hand, possible modifications of the histones (e.g., acetylation within certain parts of chromatin) may influence the binding of Act3p and thus may be responsible for the subtle differences in results from the two methods. However, additional results strongly support our hypothesis of Act3p being a component of chromatin that directly interacts with core histones in vivo: complex(es) containing both Act3p and histone H2A (and probably other proteins), could be isolated from living cells (Figures 5 and 6). Furthermore, genetic data from the Winston laboratory support our biochemical results, because they have demonstrated genetic interactions between ACT3 and particular mutations in an H2A gene (Hirschhorn et al., 1995; Pinto and Winston, personal communication).
Conditional Mutations in ACT3 Affect Both Transcription Regulation and Chromatin Structure
As mentioned above, Spt6p of budding yeast is similar in some respects to Act3p. In far Western blot analysis, Spt6p recognized all four human core histones to varying degrees, and within yeast cells it interacted most strongly with histone H3. Importantly, there are also genetic similarities between SPT6 and ACT3, because point mutations in either of these essential genes show an Spt− phenotype, changing transcription start sites at the his4-912δ promoter. Furthermore, these spt6 mutations caused changes of the chromatin structure in the promoter region of the SUC2 gene (Bortvin and Winston, 1996). Genetic studies suggested that Spt6p, in addition to its role in transcriptional initiation (Swanson and Winston, 1992), is required for transcriptional elongation (Hartzog et al., 1998) and normal recombination (Malagon and Aguilera, 1996). Thus we asked whether Act3p, like Spt6p, might also be involved in diverse regulatory processes concerning chromatin function.
A biochemical experiment showed that a nonlethal act3 mutation affects chromatin structure. The act3 mutation changed the linking number of a plasmid DNA molecule, possibly by changing the density or stability of nucleosomes on DNA. Although this result suggests a general effect of the act3 mutation on chromatin structure, it has, on the other hand, been shown that act3 mutations influence transcription of some, but not all, genes (Jiang and Stillman, 1996). Together these data might reflect a strong effect of chromatin structure on the transcription of particular genes, whereas transcription of other genes (at least of those as yet tested) does not respond to a change in chromatin structure caused by the act3 mutation. Alternatively, considering our observation that only a fraction of Act3p associated with core histones in vivo (Figure 6), Act3p might interact with nucleosomes in specific regions of chromatin. If this were the case, a transcriptional unit of one of the selective markers on plasmid YCp50 could have responded to the act3 mutation. The histidine-tagged Act3p was recovered from cell extracts together not only with histones but also with other as yet unidentified proteins (our unpublished results). We suggest that Act3p may be a part of a multiprotein complex that affects chromatin structure such as SWI/SNF or RSC that contains Arp7 and Arp9 (Cairns et al., 1998; Peterson et al., 1998), or the Spt-Ada-Gcn5-acetyltransferase complex (Grant et al., 1997).
Nuclear Actin-related Proteins Are Found in Organisms Other Than S. cerevisiae
Like Act3p, it has been shown that Arp5 (Grava and Winsor, unpublished results) of S. cerevisiae and Arp4/ArpX of Drosophila (Frankel et al., 1997) are each located in the nucleus (Frankel, 1998). Arp4/ArpX of Drosophila colocalizes with heterochromatin protein 1 within central heterochromatin and probably contributes to heterochromatin functions (Frankel et al., 1997). In addition, Arp7 and Arp9 of S. cerevisiae were recently shown to be components of two chromatin remodeling complex, RSC and SWI/SNF (Cairns et al., 1998; Peterson et al., 1998). Furthermore, we recently showed that there are two isoforms of a human Arp, and that both are localized in the nucleus (Harata et al., 1999). These isoforms are expressed in a mutually exclusive manner, either in the brain or other tissues, with one of the isoforms identical to the recently described BAF53 subunit of the human SWI/SNF-related nucleosome remodeling complex (Zhao et al., 1998). Although these “nuclear” Arps show less similarity to conventional actin than the Arps in groups 1–3, it is interesting to note that they seem to have similar functions; they are involved in structural modulation of chromatin. Recently the Schizosaccharomyces pombe genome project revealed the existence of a putative Act3p homologue of S. pombe, Arp23D3 (Poch and Winsor, 1997), and it would be interesting to know whether this protein is also a constituent of chromatin.
A common structural feature of the actin family proteins is the ATP/ADP-binding pocket. Hydrolytic removal of a phosphate group from ATP within the pocket induces a conformational shift of the protein folding, and such shifts are thought to be crucial for the functions of the actin family members (Frankel, 1998). Actually, Act3p binds ATP in vitro (Wintersberger, manuscript in preparation), and it will be of interest whether this binding or even hydrolysis of ATP might be important for the function of Act3p.
Another characteristic feature of all well studied members of the actin family is the ability to participate in multiprotein complexes, the assembly of which is reversible and highly regulated (Frankel and Mooseker, 1996). Also in this respect the essential yeast protein Act3p represents a typical actin family member, and we suggest that it fulfills a role in the establishment or maintenance of a specific chromatin structure by stabilizing or destabilizing the protein complex, of which it is an intrinsic part.
ACKNOWLEDGMENTS
We thank Dr. Fred Winston and Dr. Barbara Winsor for communicating results in advance of publication. We also thank Dr. Kosuke Morikawa for advice regarding the structure of insertion II. This research was supported by International Scientific Research Program grant-in-aid 08044188 from the Ministry of Education, Science, Sports, and Culture, Japan (to M.H.) and grants from the National Institutes of Health (to D.J.S.).
REFERENCES
- Abraham J, Feldman J, Nasmyth KA, Strathern JN, Klar AJS, Broach JR, Hicks JB. Sites required for position-effect regulation of mating-type information in yeast. Cold Spring Harbor Symp Quant Biol. 1983;47:989–998. doi: 10.1101/sqb.1983.047.01.113. [DOI] [PubMed] [Google Scholar]
- Akiyama Y, Kato S. Two cell lines from lymphomas of Marek’s disease. Biken J. 1974;17:105–116. [PubMed] [Google Scholar]
- Bi X, Broach JR. DNA in transcriptionally silent chromatin assumes a distinct topology that is sensitive to cell cycle progression. Mol Cell Biol. 1997;17:7077–7087. doi: 10.1128/mcb.17.12.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortvin A, Winston F. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996;272:1473–1476. doi: 10.1126/science.272.5267.1473. [DOI] [PubMed] [Google Scholar]
- Brill SJ, Sternglanz R. Transcription-dependent DNA supercoiling in yeast DNA topoisomerase mutants. Cell. 1988;54:403–411. doi: 10.1016/0092-8674(88)90203-6. [DOI] [PubMed] [Google Scholar]
- Cairns BR, Erdjument-Bromage H, Tempst P, Winston F, Kornberg RD. Two actin-related proteins are shared functional components of the chromatin-remodeling complexes RSC and SWI/SNF. Mol Cell. 1998;2:639–651. doi: 10.1016/s1097-2765(00)80162-8. [DOI] [PubMed] [Google Scholar]
- Cheng TH, Li YC, Gartenberg MR. Persistence of an alternate chromatin structure at silenced loci in the absence of silencers. Proc Natl Acad Sci USA. 1998;95:5521–5526. doi: 10.1073/pnas.95.10.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Adams CD, Norris D, Osley MA, Fassler JS, Winston F. Changes in histone gene dosage alter transcription in yeast. Genes & Dev. 1988;2:150–159. doi: 10.1101/gad.2.2.150. [DOI] [PubMed] [Google Scholar]
- Eisenmann DM, Dollard C, Winston F. SPT15, the gene encoding the yeast TATA binding factor TFIID, is required for normal transcription initiation in vivo. Cell. 1989;58:1183–1191. doi: 10.1016/0092-8674(89)90516-3. [DOI] [PubMed] [Google Scholar]
- Elgin SC. Heterochromatin and gene regulation in Drosophila. Curr Opin Genet Dev. 1996;6:193–202. doi: 10.1016/s0959-437x(96)80050-5. [DOI] [PubMed] [Google Scholar]
- Fernandez-Patron C, Hardy E, Sosa A, Seoane J, Castellanos L. Double staining of Coomassie blue-stained polyacrylamide gels by imidazole-SDS-zinc reverse staining: sensitive detection of Coomassie blue-undetected proteins. Anal Biochem. 1995;224:263–269. doi: 10.1006/abio.1995.1039. [DOI] [PubMed] [Google Scholar]
- Frankel S. The more distantly related of the actin-related proteins. (1998) In: Vale R, Kreis T, editors. Guidebook to the Cytoskeletal and Motor Proteins. Oxford: Oxford University Press; 1998. (in press). [Google Scholar]
- Frankel S, Mooseker MS. The actin-related proteins. Curr Opin Cell Biol. 1996;8:30–37. doi: 10.1016/s0955-0674(96)80045-7. [DOI] [PubMed] [Google Scholar]
- Frankel S, Sigel EA, Craig C, Elgin SC, Mooseker MS, Artavanis-Tsakonas S. An actin-related protein in Drosophila colocalizes with heterochromatin protein 1 in pericentric heterochromatin. J Cell Sci. 1997;110:1999–2012. doi: 10.1242/jcs.110.17.1999. [DOI] [PubMed] [Google Scholar]
- Grant PA, et al. Yeast Gcn5 functions in two multisubunit complexes: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes & Dev. 1997;11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- Han M, Chang M, Kim UJ, Grunstein M. Histone H2B repression causes cell-cycle-specific arrest in yeast: effects on chromosomal segregation, replication, and transcription. Cell. 1987;48:589–597. doi: 10.1016/0092-8674(87)90237-6. [DOI] [PubMed] [Google Scholar]
- Harata M, Karwan A, Wintersberger U. An essential gene of Saccharomyces cerevisiae coding for an actin-related protein. Proc Natl Acad Sci USA. 1994;91:8258–8262. doi: 10.1073/pnas.91.17.8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harata M, Mochizuki R, Mizuno S. Two isoforms of a human actin-related protein show nuclear localization and mutually selective expression between brain and other tissues. Biosci Biotechnol Biochem. 1999;63:917–923. doi: 10.1271/bbb.63.917. [DOI] [PubMed] [Google Scholar]
- Hartzog GA, Wada T, Handa H, Winston F. Evidence that Spt4, Spt5 and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes & Dev. 1998;12:357–369. doi: 10.1101/gad.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Bortvin AL, Ricupero-Hovasse SL, Winston F. A new class of histone H2A mutations in Saccharomyces cerevisiae causes specific transcriptional defects in vivo. Mol Cell Biol. 1995;15:1999–2009. doi: 10.1128/mcb.15.4.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KC, Sander C, Valencia A. A new ATP-binding fold in actin, hexokinase and Hsc70. Trends Cell Biol. 1993;3:53–59. doi: 10.1016/0962-8924(93)90161-s. [DOI] [PubMed] [Google Scholar]
- Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277:491–492. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- Jiang YW, Dohrmann PR, Stillman DJ. Genetic and physical interactions between yeast RGR1 and SIN4 in chromatin organization and transcriptional regulation. Genetics. 1995;140:47–54. doi: 10.1093/genetics/140.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YW, Stillman DJ. Involvement of the SIN4 global transcriptional regulator in the chromatin structure of Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:4503–4514. doi: 10.1128/mcb.12.10.4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YW, Stillman DJ. Epigenetic effects on yeast transcription caused by mutations in an actin-related protein present in the nucleus. Genes & Dev. 1996;10:604–619. doi: 10.1101/gad.10.5.604. [DOI] [PubMed] [Google Scholar]
- Kaiser C, Michaelis S, Mitchell A. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1994. [Google Scholar]
- Malagon F, Aguilera A. Differential intrachromosomal hyper-recombination phenotype of spt4 and spt6 mutants of S. cerevisiae. Curr Genet. 1996;30:101–106. doi: 10.1007/s002940050107. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Mullins RD, Kelleher JF, Pollard TD. Actin’ like actin? Trends Cell Biol. 1996;6:208–212. doi: 10.1016/0962-8924(96)20017-0. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- Osborne BI, Guarente L. Transcription by RNA polymerase II induces changes of DNA topology in yeast. Genes & Dev. 1988;2:766–772. doi: 10.1101/gad.2.6.766. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Zhao Y, Chait BT. Subunits of the yeast SWI/SNF complex are members of the actin-related protein (ARP) family. J Biol Chem. 1998;273:23641–23644. doi: 10.1074/jbc.273.37.23641. [DOI] [PubMed] [Google Scholar]
- Poch O, Winsor B. Who’s who among the Saccharomyces cerevisiae actin-related proteins? A classification and nomenclature proposal for a large family. Yeast. 1997;13:1053–1058. doi: 10.1002/(SICI)1097-0061(19970915)13:11<1053::AID-YEA164>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. In: Guthrie C, Fink GR, editors. Guide to Yeast Genetics And Molecular Biology. New York: Academic Press; 1991. pp. 3–21. [Google Scholar]
- Smith MM, Yang P, Santisteban MS, Boone PW, Goldstein AT, Megee PC. A novel histone H4 mutant defective in nuclear division and mitotic chromosome transmission. Mol Cell Biol. 1996;16:1017–1026. doi: 10.1128/mcb.16.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suka N, Shinohara Y, Saitoh Y, Saitoh H, Ohtomo K, Harata M, Shpigelman E, Mizuno S. W-heterochromatin of chicken: its unusual DNA components, late replication, and chromatin structure. Genetica. 1993;88:93–105. doi: 10.1007/BF02424466. [DOI] [PubMed] [Google Scholar]
- Swanson MS, Winston F. SPT4, SPT5, and SPT6 interactions: Effects on transcription and viability in Saccharomyces cerevisiae. Genetics. 1992;132:325–336. doi: 10.1093/genetics/132.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Fukuda M, Mishima M, Nishida E. Nuclear export of actin: a novel mechanism regulating the subcellular localization of a major cytoskeletal protein. EMBO J. 1998;17:1635–1641. doi: 10.1093/emboj/17.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrath LL, Elgin SCR. Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes & Dev. 1995;9:1263–1277. doi: 10.1101/gad.9.10.1263. [DOI] [PubMed] [Google Scholar]
- Weber V, Harata M, Hauser H, Wintersberger U. The actin-related protein Act3p of Saccharomyces cerevisiae is located in the nucleus. Mol Biol Cell. 1995;6:1263–1270. doi: 10.1091/mbc.6.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston F, Carlson M. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 1992;8:387–391. doi: 10.1016/0168-9525(92)90300-s. [DOI] [PubMed] [Google Scholar]
- Wintersberger U, Kuehne C, Karwan A. Scp160p, a new yeast protein associated with the nuclear membrane and the endoplasmic reticulum, is necessary for maintenance of exact ploidy. Yeast. 1995;11:929–944. doi: 10.1002/yea.320111004. [DOI] [PubMed] [Google Scholar]
- Worcel A, Strogatz S, Riley D. Structure of chromatin and the linking number of DNA. Proc Natl Acad Sci USA. 1981;78:1461–1465. doi: 10.1073/pnas.78.3.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, Crabtree GR. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 1998;95:625–636. doi: 10.1016/s0092-8674(00)81633-5. [DOI] [PubMed] [Google Scholar]