

Abstract
This paper reports a method that combines self-assembled monolayers with matrix assisted laser desorption-ionization time-of-flight mass spectrometry to perform immunoassays on clinical samples. The immunosensors are prepared by immobilizing his-tagged Protein G (or A) to a monolayer presenting the Ni2+ chelates, followed by immobilization of IgG antibodies with specificity for the intended analyte. The SAMDI mass spectrometry technique confirms the presence of the two proteins on the immunosensor and additionally provides a label-free analysis of antigens that bind to the sensor. This paper reports examples of detecting several proteins from human serum, including multi-analyte assays that resolve each analyte according to their mass-to-charge ratio in the SAMDI spectra. An example is described wherein SAMDI is used to identify a proteolytic fragment of cystatin C in cerebral spinal fluids from patients diagnosed with multiple sclerosis and control patients. The SAMDI-TOF immunoassay, which combines well defined surface chemistries for the selective and reproducible localization of analytes with mass spectrometry for label-free detection of analytes, may offer an alternative methodology to address many of the issues associated with standardized clinical diagnostics.
INTRODUCTION
Immunosensors hold a special importance in both basic research and clinical diagnostics. These assays use capture agents (either antibodies or antigens) that are immobilized on a solid phase to determine the concentration of a corresponding analyte in a biological sample. The generality of immunosensors stems from the availability of high affinity and selective antibodies to a broad range of analytes and to common “label-based” formats (using chromogenic, radioactive, or fluorescent reagents) for the detection of analytes.1–4 Recently, much emphasis has been placed on the development of “label-free” detection technologies which hold much promise for diagnostic immunoassays.5, 6 By eliminating the need for labeled reagents these label-free assays offer prospects of reduced time involved for assay development, simplified protocols, lower cost, and realization of immunosensors that perform multiple assays with a common sample. This paper describes the application of SAMDI-TOF mass spectrometry for the label-free detection of analytes bound to antibodies immobilized on a self-assembled monolayer (SAM). This strategy was used to detect multiple protein antigens from humoral fluids, including the detection of a protein marker in cerebral spinal fluid.
Surface plasmon resonance (SPR) spectroscopy and mass spectrometry (MS) are two approaches that currently hold much promise for label-free detection of biomolecules. Recent examples of SPR-based sensors for detection of antibodies include a report by Miura and coworkers showing a competitive assay that examined the presence (or absence) of insulin in serum (at 1 ng/ml) by monitoring the extent of anti-insulin antibody binding to control insulin molecules immobilized on a PEG-based monolayer.7 Homola and coworkers reported the detection of antibodies against Epstein-Barr virus using a synthetic peptide-BSA conjugate immobilized onto the surface of substrates used in an SPR instrument.8 Direct antigen sensors have been developed by Corn and coworkers using a monolayer presenting carboxylic acids to immobilize antibodies for the detection of β2-microglobulin and cystatin C antigens (at 10 ng/mL).9 Also, Koga and coworkers adsorbed antibodies directly onto a gold surface for parallel analysis of up to 400 antigens out of crude mouse tissue homogenates.6, 10, 11 The SPR technique offers the benefits that it can measure low affinity protein interactions in real-time and it provides a good quantitative capability that stems from the linear dependence of signal with mass of analyte. Also, SPR through kinetic data can determine concentrations of biomolecules in solution.
In contrast to SPR, MS methods do not allow in situ kinetic measurements, but they do bring the significant benefit of providing chemical and structural information on the analyte. SPR and related optical methods, for example, do not discriminate between the true analyte and background species that interact with the sensor,11 while mass spectrometry can provide independent measures of each species that interacts with the sensor. Further, by resolving analytes according to their mass to charge ratio, MS methods have been used to monitor multiple analytes simultaneously and to identify post-translationally modified forms of biomolecule targets.12 One of the first applications of MS in concert with immobilized biomolecules was performed by Nelson and coworkers where immobilized trypsin was used to digest proteins directly on a MALDI probe. Nelson also developed a mass spectrometric immunoassay (MSIA) nearly a decade ago with the most recent investigations designed to screen for protein variants across patient populations and identify protein complexes in biological fluids.13–17 This assay started with the application of antibodies to porous frits contained in a pipette-tip followed by exposure to a sample to promote antigen binding, and spotting the analyte directly onto a MALDI-TOF target plate.18 Other reports of MSIA used various strategies to immobilize antibodies and have been extended to planar arrays and fiber optic probes.19–24 Some of this work combined the advantages of optical and mass spectrometric methods by first performing SPR analysis of protein interactions and then removing the substrate for subsequent analysis by MALDI-TOF MS. Surface enhanced laser desorption ionization (SELDI)-TOF mass spectrometry25 has also been adapted to the analysis of individual analytes by using a solid phase resin having an immobilized antibody. In one example, Proudfoot and coworkers used antibodies bound to a solid phase resin to detect the RANTES chemokine including in vivo metabolic modifications in serum.26 An example by Gorenstein and coworkers used a SELDI chip presenting the receptor protein A/G to immobilize antibodies for the detection of ribonucleoproteins in mouse nuclear extracts.27
These immunosensor examples have relied on either physical adsorption or covalent attachment at random amino acid residues to immobilize the antibodies to the sensor, which can have a corresponding loss of control over the orientation and density of the antibody and lead to non-specific adsorption at the surface interface.24, 28–30 These strategies are often empirical and require optimization for each particular antibody. Linnik and coworkers, for example, showed that the binding of an antiphospholipid antibody to an immobilized β2-glycoprotein I antigen strongly depended on the composition of the substrate.31, 32 Other studies have shown that the orientation of the antibody is important for activity, including a report by Nock and coworkers revealing a 10-fold improvement in analyte binding with specific orientation of whole antibodies (or Fab’ fragments) when compared to random orientations.33 In related work, Clark and coworkers showed that the fractional activity of an immobilized antibody may be reduced due to the steric crowding that accompanies immobilization of antibodies at high density.34 Orlando and coworkers have shown that covalent attachment of antibodies on a simple monolayer format can lead to non-specific binding from protein components from serum (although binding a 500 kDa dextran moiety was able to eliminate this).24
Separately, our group has developed surface chemistries that can be applied to a broad range of protein binding and enzyme activity assays that are compatible with several analytical formats, including SPR, radioactivity, fluorescence and mass spectrometry.35–39 Our approach uses self-assembled monolayers of alkanethiolates on gold that are functionalized with oligo(ethylene glycol) groups and maleimide groups.40 The former are important for reducing the non-specific adsorption of proteins41–45 and the latter can be used to immobilize biologically active motifs or functional groups used for subsequent immobilization.36, 46, 47 In an initial application of these methods to immunoassays (with SPR detection), we described the preparation of fusion proteins containing antibody variable (Fv) domains fused to cutinase, a protein that irreversibly binds a phosphonate ligand.48 When the fusion protein is applied to a monolayer that presents the phosphonate capture ligand, the cutinase domain irreversibly binds the substrate, giving an oriented antibody with a density that is determined by the density of the phosphonate ligand in the monolayer. We found that greater than 90% of the immobilized antibodies were active and that this fractional activity was independent of the density of antibodies for small antigens, but decreased with increasing density of antibody for large antigens (due to steric exclusion).
We have also shown that our monolayers are well-suited for analysis by MALDI-TOF MS. This technique, termed self-assembled monolayers for matrix assisted laser desorption ionization mass spectrometry (SAMDI-MS), provides the masses of substituted alkanethiols—permitting enzyme activity assays on immobilized peptides, carbohydrates, and small molecules—and can observe proteins bound to the monolayer with masses approaching 100 kDa.46, 49–52 This paper reports an application of the SAMDI approach to immunosensors and demonstrates label-free detection of protein markers from humoral fluids.
METHODS
Preparation of Monolayers
Monolayers were prepared as reported elsewhere.40 Briefly, 60 nm of titanium followed by 220 nm of gold were evaporated by electron beam onto a microscope cover glass (cat. 12-543-D) from Fisher Scientific (Pittsburg, PA). Self assembled monolayers were generated by immersion of the metallized slides in an ethanolic solution containing an asymmetric disulfide derived from tri(ethylene glycol)- and maleimide-terminated alkanethiolates and a symmetric disulfide derived from the tri(ethylene glycol)-terminated alkanethiolate for 12 hours (1mM, ratios range from 1:9 to 1:49). The triazacyclononane (aza) or nitrilotriacetic acid (NTA) ligands were immobilized by treating the monolayer with the parent thiol-substituted reagents for one hour (1mM, phosphate buffered saline (PBS), pH 7.2). The monolayers were treated with an aqueous solution of NiSO4 for five minutes to generate the Ni2+ complex (1mM, PBS, pH 7.2).
Monolayers were cut into square pieces measuring 4 mm2 and treated for five minutes with His-tagged protein A (48 kDa cat. 6510-00)53, 54 or protein G (27 kDa, cat. 6500-10)55, 56 (10 uL, ~ 1–10 μM, PBS, pH 7.2) from Biovision Inc. (Mountain View, CA, USA). The resulting monolayers were then treated with the IgG-antibodies for five minutes (10 uL, ~5 uM, PBS, pH 7.2). Antibodies to human albumin (cat. A80-129A), human hemoglobin (cat. A80-134A), and human transferrin (cat. A80-128A) were purchased from Bethyl Laboratories Inc. (Montgomery, TX, USA). Anti-human cystatin C (cat. A0451) was purchased from Dako A/S (Carpinteria, CA, USA). Chips were washed under a stream of water and dried with a stream of nitrogen after each step. Protein A or G surfaces could be stored for several days at 4°C prior to use, although, the monolayers are known to be stable for extended periods even at elevated temperatures (~ 1 month at 37°C).44, 57
Antibody/antigen assays
Serum and CSF samples were diluted 1:1 with PBS (pH 7.2), spotted onto the immunosensor (in a volume of 5 uL) and incubated for five minutes. Serum (cat. D119-00-0050) was obtained from Rockland Immunochemicals (Gilbertsville, PA, USA) and plasma (cat. CCN-10) was obtained from Precision BioLogic (Dartmouth, Nova Scotia, Canada). CSF samples, collected under an IRB approved protocol, were generously donated by Dr. Anthony Reder (University of Chicago, Department of Neurology). Quantitation of endogenous human cystatin C (hcysC, 13.3 kDa) in CSF was performed by comparison of peak areas to an internal control. Cystatin C obtained from human urine (cntcysC, 12.5 kDa, cat. 240896) was purchased from EMD Biosciences (San Diego, CA, USA), diluted with PBS, and spiked into the CSF (at various concentrations) prior to application on the immunosensor. Chips were washed under a stream of water (~ 10 sec) after sample incubation and dried under a stream of nitrogen.
Mass spectrometry
Either sinapinic acid (SA) or α-cyano-4-hydroxycinnamic acid (CHCA) at 5–7.5 mg/ml (in acetone, 0.3 uL manually spotted) was used to facilitate protein desorption and ionization from the monolayer surface. Mass analysis was performed on a Voyager DE-PRO Biospectrometry mass spectrometer from Applied Biosystems (Framingham, MA, USA). The instrument has a 337-nm nitrogen laser and typical experimental parameters include: linear mode of operation, manual acquisition, positive ion polarity, 500 nsec delayed extraction, 25000 VDC acceleration voltage, 97.5% grid voltage and 0.3% guide wire. Spectra obtained are the average of ~ 2000 shots. Data were smoothed with a Gaussian routine. To quantitate hcysC, the spectra were baseline corrected prior to integration of peak areas.
RESULTS
Experimental Design
Our approach for immobilizing antibodies and performing immunoassays with SAMDI-TOF mass spectrometry is illustrated in Figure 1. The method is based on immobilization of hexahistidine tagged proteins and therefore begins with a monolayer that presents the aza/Ni2+ ligand at a density of 0.5–5 % (relative to total alkanethiolate) against a background of tri(ethylene glycol) groups. The aza/Ni2+ complex is an alternative to the commonly used NTA ligands and has been described for immobilization of proteins to SAMs.58 Recombinant protein G (or protein A) containing a hexahistidine tag is immobilized to the monolayer and then used to capture an IgG antibody by way of binding to the heavy chain constant region of the antibody (Fc). The specific interactions that mediate the immobilization enforce a defined orientation of the antibody on the surface while not interfering with the antigen binding region on the antibody and therefore are expected to optimize the activity of the immobilized antibody.28–30, 59 Further, this strategy offers control over the density of antibody, which can be particularly relevant to reducing the steric interactions of large antigens at the surface.60
Figure 1.

Method for the formation and application of the SAMDI-based immunosensor. The strategy begins with a maleimide terminated monolayer that is used to immobilize the aza ligand. To immobilize the antibody, the monolayer is treated with Ni2+, recombinant hexahistidine protein G (or protein A), and an appropriate IgG antibody. The monolayer is then treated with humoral fluids (CSF, serum, plasma) containing an internal standard and analyzed with SAMDI-TOF MS. EG3 = tri(ethylene glycol)
Assembly of Surfaces for Immunoassays
We first used SAMDI-TOF mass spectrometry to characterize the assembly of two model immunosensors where either protein A or protein G receptor proteins were used to immobilize cystatin C (cysC) antibody. A solution of either the 48 kDa hisprotein A or 27 kDa hisprotein G (5 μM in PBS with 30% glycerol) was incubated for 5 minutes at room temperature on the monolayer presenting the aza/Ni2+ group at a density of 1% (relative to the total alkanethiolate). Following the incubation, the monolayers were rinsed with water and then incubated with the antibody (5 μM in PBS) for 5 minutes at room temperature. The surface was then rinsed again with water and incubated with cysC (12.5 kDa, 0.3 μM in PBS) for 5 minutes at room temperature. The monolayers were rinsed with water, dried, treated with matrix and analyzed by SAMDI-TOF mass spectrometry.
The SAMDI-TOF spectra reveal the proteins that bind to the surface in each of the steps described above (Figure 2). The 10,000–90,000 m/z region of the mass spectra in Figures 2A and 2B reveal clear peaks for receptor protein A and protein G at 48 kDa and 27 kDa respectively. Multiple charge states and dimeric species are typically observed for both samples. Control experiments conducted in the presence of 300 mM imidazole—which disrupts interaction of the histidine tag with the aza/Ni2+ complex—resulted in minimal retention of the bound protein (data not shown). The antibodies (~150 kDa) efficiently bound to the protein A (or G) on the monolayers (Figures 2C and 2D). We again observed multiple charge states corresponding to the antibody across the 15,000–200,000 m/z region of the spectrum including intense peaks associated with the singly and doubly charged ions at 150,000 m/z and 75,000 m/z, respectively.
Figure 2.

SAMDI-TOF MS was used to monitor each step in the preparation of the immunosensor. The spectra in panels A and B show the immobilization of the protein A and protein G, respectively, onto a monolayer presenting the aza/Ni2+ ligand. Panels C and D show the immobilization of cysC antibody onto the protein A and protein G derivatized surfaces, respectively. Panels E and F show the capture of the cysC on the resulting immunosensors. (#) Unidentified components derived from the stock antibody.
Antibodies that were immobilized as described above efficiently captured the cysC antigen present in the sample (Figures 2E and 2F). Following binding of the antigen to the immobilized antibody, SAMDI revealed peaks for each of the three proteins on the surface—receptor proteins A/G, antibody and antigen—although, the intensity of the peak representing the receptor protein decreased in the presence of the other proteins. This difference may be due to more efficient desorption and ionization of the antibody and antigen from the protein A/G. Interestingly, peaks corresponding to the cysC in a non-covalent complex with the antibody survived the ionization process although we cannot distinguish whether the antigen is still bound to the variable Fv binding region of the antibody or present as a loosely associated gas phase product (see inset, Figure 2E and 2F). These examples demonstrate that SAMDI-TOF, in addition to its potential in performing diagnostic assays (see below), is useful for verifying the integrity of the immunosensor. Measurement of constituently loaded proteins in parallel offers a significant degree of quality control in the development and application of the immunosensor, especially in the presence of complex proteinaceous mixtures, such as serum.
Model Immunoassays
Although this immunosensor strategy requires multiple steps to prepare the surface for an assay, it has the advantage that it only requires a single primary antibody to detect a designated antigen and therefore can readily be applied to a broad range of assays. We first implemented this strategy to detect several antigens from human serum (Figure 3). We used commercially available antibodies raised against the proteins cystatin C (cysC, 13.3 kDa), the subunits of hemoglobin (hem, 15.1 kDa & 15.9 kDa), human serum albumin (HSA, 66.3 kDa), and transferrin (transferrin, 79.5 kDa) from human serum. In each case, the antibodies were immobilized by way of protein G as described in the previous section. For each assay, human serum was diluted with an equal volume of phosphate buffered saline (PBS), 5 uL of sample is incubated for 5 minutes on the immunosensor, washed with water, and dried under a stream of nitrogen prior to application of the CHCA matrix. The spectra in Figure 3 showed species corresponding to the molecular weights of cysC, hemoglobin, HSA, and transferrin were efficiently and selectively bound to the surface from serum and detected by SAMDI-TOF mass spectrometry. In the case of the hemoglobin immunosensor (Figure 3B), both subunits of hemoglobin A—each comprised of an α (15.1 kDa) and a β (15.9 kDa) chain—were detected in parallel.
Figure 3.
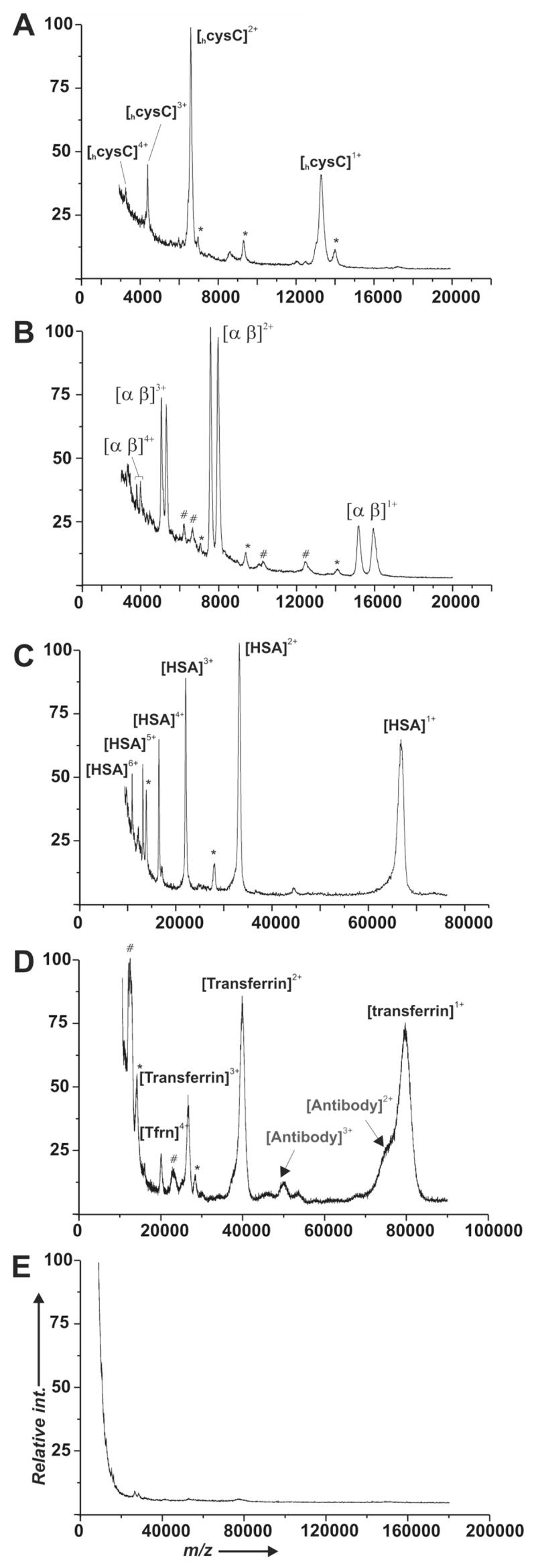
Detection of antigens from human serum using monolayers presenting antibodies for cysC (A), hemoglobin (B), HSA (C), and transferrin (D). Each spectrum reveals prominent peaks for the intended analyte and lower intensity peaks for protein G (*) and unidentified components derived from the stock antibody (#). A control experiment using an identical monolayer that was not treated with Ni2+, protein G or antibody prior to incubation with serum revealed little binding of serum components (E).
The immunoassays presented in Figure 3 were specific for the intended analyte, showing that the surfaces are resistant to non-specific adsorption from other proteins in the serum samples. To further ascertain the role of non-specific binding of serum proteins, we incubated a monolayer presenting only tri(ethylene glycol) groups (in the absence of Ni2+, receptor proteins, or antibodies) with the human serum sample and found a minimal contribution of any single serum component to the base monolayer, with none of these components reproducibly present in the antibody loaded assays (Figure 3E). In several of the assays presented in Figure 3 we did observe low intensity unidentified peaks (typically < 5% peak intensity relative to the base peak) although for the transferrin immunosensor (Figure 3D) there was a dominant cluster of peaks present at ~ 12 kDa. We believe these species derive from protein components that were present during the immobilization of the antibody and appear to have affinity for the protein A/G surface since these same components were observed prior to treatment of the substrate with sample (e.g., Figures 2C & 2D vs. 2E & 2F for cysC).
Multi-Analyte Format
A benefit of mass spectrometry is that the antibodies can be applied as a mixture to the monolayer and need not be arrayed in non-overlapping regions of the substrate to resolve the binding of each antigen—as is the case when fluorescence measurements are used to interrogate the chip. To demonstrate this capability with SAMDI-TOF, we prepared a single monolayer that presented the four antibodies described above and used this substrate to identify analytes from human serum. The four antibodies were mixed in a ratio of 1:2:2:4 for cysC, hem, HSA, and transferrin, respectively, and applied to a monolayer presenting protein G. The substrate was treated with serum for 5 minutes, rinsed, treated with matrix and analyzed by SAMDI. The 5,000–90,000 m/z region of the SAMDI-TOF spectrum shows peaks that correspond to protein G, the antibodies, and each of the antigens in multiple charge states (Figure 4). Nonequimolar amounts of the antibodies were required in order to adequately observe peaks for each of the antigens. The differences in peak intensity presumably reflect a combination of the affinity of the antibody for the antigen and the ionization efficiency of the antigen. Application of SAMDI-TOF for evaluation of multiple proteins offers a significant advantage for clinical diagnostics by significantly reducing the time and sample consumption per assay.
Figure 4.

A monolayer presenting four antibodies was used to simultaneously detect the four antigens cysC, hemoglobin, HSA, and transferrin from serum. The SAMDI-TOF spectrum reveals peaks corresponding to multiple charge states of the antigens and the proteins of the surface.
Quantitation of Antigens
The common strategy for obtaining quantitative data in mass spectrometry experiments relies on the addition of a known standard to a sample, and comparing the intensity (or peak area) of the analyte to that of the standard. We show this strategy for the detection of cysC in human cerebral spinal fluid (CSF) (Figure 5). A truncated form of cysC (trunccysC, 12.5kDa), obtained from human urine, was used as an internal standard. Serial dilutions of the trunccysC were added directly to CSF samples (160–1300 nM, final concentration) and each of the samples was then applied to the immunosensor. SAMDI spectra in the mass range of 11000–15000 m/z show differential binding between the trunccysC and the endogenous cysC (Figure 5A). At higher concentrations of trunccysC, the spectra revealed a shoulder at ~ 11 kDa (in addition to the dominant peak at 12.5 kDa) that we treated as part of the trunccysC peak area.
Figure 5.

Endogenous cysC in CSF was quantitated through the use of a truncated form of cysC (trunccysC) as an internal standard. Several identical samples of CSF were spiked with the internal standard at concentrations ranging from 0.16–1.3 μM. These samples were applied to separate immunosensors to obtain SAMDI spectra in (A). Integration of the peak areas provides a quantitative measure of the ratio of the cysC to the internal standard (B). The assay at 0.49 μM (C) and 0.24 μM internal standard (D) was performed on different days, using monolayers having a range of density of the aza/Ni2+ ligand 0.5% (—), 1% (---), and 4% (…). The comparison of the peak ratios in the SAMDI spectra show the assays were highly reproducible.
Clinical assays based on the enzyme-linked immunosorbent assay (ELISA) provide a linear response across a large concentration range, and provide standards of reproducibility with coefficients of variation (CV) typically less than 10%, and low detection limits (~ 1 pg/ml) for a diverse array of antigens.4 The chart in Figure 5B shows the calculated ratio of the trunccysC peak areas to the total cystatin C area (from Figure 5A) that span the reference range for cystatin C in CSF fluid.61 A linear increase in trunccysC area was observed with a corresponding increase in trunccysC concentration. Using samples of trunccysC at concentrations of 0.24 μM and 0.49 μM we obtained SAMDI spectra on different days, using monolayers having a range of density of the aza/Ni+2 ligand (0.5%, 1.0%, % 4%) (Figures 5C and 5D). The similar peak shapes and ratios observed over the course of these experiments highlights the good reproducibility of this immunoassay. The concentration of endogenous cysC, based on the calculated ratios of the nine peak ratios presented in Figure 5B, was determined to be 0.459 ± 0.030 μM with a CV of 6.5%. In principle, the control antigen should closely mimic the endogenous antigen in its affinity for the antibody and ionization properties. This similarity is apparent for the example presented in Figure 5 since there is a direct competitive relationship between the control and the endogenous antigen. We believe that it will often be possible to identify an appropriate internal standard by selecting a metabolic derivative of the endogenous antigen or by preparing a covalently tagged version of the antigen that does not interfere with the affinity for the antibody.15, 62, 63 The lower limits for detection with this assay will ultimately depend on the affinity of the antibody for the antigen and possibly by the intensity of peaks for background species. For cystatin C we were able to dilute CSF ~ 200-fold and still observe signal above background (data not shown) which translates into a lower limit for detection of ~ 30 ng/ml.
Detection of Post-Translational Modifications in CSF
A recent study has suggested that a proteolytic fragment of cysC may serve as a marker for multiple sclerosis.64 Nath and coworkers used SELDI-TOF to identify a 12.5 kDa form of cysC in the cerebral spinal fluid (CSF) of multiple sclerosis patients. This form of cysC has a mass that is 800 Da lower than that for the parent 13.3 kDa form that is present in CSF from healthy individuals. In an experiment that included 27 diseased patients and 27 control patients, presence of the proteolytically derived fragment of cysC was found to be 100% specific for patients with multiple sclerosis versus other neurological diseases. We evaluated several CSF samples to measure the relative amounts of the 12.5 kDa and 13.3 kDa forms of the cysC protein in clinical samples isolated from patients with and without multiple sclerosis. Seven CSF fluid samples from patients having multiple sclerosis were analyzed and compared to two samples from patients with other neurological diseases, as well as serum and plasma samples from disease-free patients. The 11000–15000 m/z region of each SAMDI-TOF spectrum revealed that the truncated form of the cysC was present in all patients that had multiple sclerosis, while in two cases the normal 13.3 kDa form of the protein was present at ~10% and ~50% of the total cysC captured. In the control samples, the serum, the plasma, and one of the CSF samples showed only the unmodified 13.3 kDa form of cysC, while in one other CSF sample there was a minor amount (~15%) of the observed cysC in the 12.5 kDa form. This species may derive from long-term storage conditions where exposure to temperatures > −20 °C results in a similar truncation as observed in patients with multiple sclerosis.65–67 The effects of storage conditions were not evaluated as part of this study but this example does establish that SAMDI can be applied to the analysis of post-translationally derived markers in actual clinical samples.
DISCUSSION
This paper reports an application of the SAMDI-TOF MS method for performing immunoassays of protein markers in clinical samples. A body of previous work has demonstrated that SAMDI is well suited to the analysis of low molecular weight species, including peptides, carbohydrates and organic molecules. Indeed, SAMDI has been used to perform a range of enzyme activity assays—including kinase, protease, methyltransferase and glycosyltransferase activities—and has been applied to screening small molecule libraries to identify antagonists of enzyme activities.35, 49, 50, 52, 68 The extension of SAMDI from detecting low molecular weight species to large molecular weight proteins, and from analyzing solutions of defined and relatively simple composition to complex samples derived from bodily fluids, has not been previously demonstrated. A first report showed that SAMDI could observe molecular ions corresponding to intact proteins with masses up to approximately 50 kDa.46 That work used surfaces presenting glutathione ligands to immobilize GST-fusion proteins, with the limitation that the rate constant for dissociation of the GST-glutathione interaction limited the stability of the biochip in diagnostic applications. The current work is significant because it provides an additional example showing that SAMDI can analyze large molecular weight protein analytes (up to 150 kDa) and provides the first example that our monolayer surfaces are able to discriminate protein antigen(s) from the other components in complex samples. The previous examples from out group and this current paper highlights a significant benefit of this surface chemistry approach (over SELDI and traditional affinity biochip formats combined with mass spectrometry)69 in that it can be applied to a broad range of bioassays, now including those directed towards antigen detection, protein-protein interactions, and permitting enzyme activity assays on immobilized peptides, carbohydrates, and small molecules.
Several considerations prompted our use of the receptor protein G (or A) to immobilize the antibody to the monolayer. In a previous report that used cutinase-mediated immobilization to give oriented immobilization of antibodies, we showed that control over both density and orientation could be harnessed to optimize the activity of the antibody.60 One limitation for this assay was the necessity for a recombinant cutinase-antibody reagent. The current approach translates the advantages of the cutinase system to the well known use of protein G (or A) to immobilize and orient an IgG antibody through its nonantigenic Fc region.28–30, 59 The protein G (or A)-based assay can be applied to a broad range of immunoassays, provided that an IgG type antibody with reasonable affinity and selectivity for the intended analyte and the receptor protein is available.70 Further, the assay format reported here does not require modification of either the antibody or the analyte. Many immunosensor schemes, for example require chemical or enzymatic modification of the Fc region of the antibody to introduce functionality needed for the immobilization, and additionally require labeling of either antibody or antigen to detect the latter. Because SAMDI can observe the antigen directly, labeling is not required. Unlike sandwich immunoassays, which require two antibodies with high affinity and non-overlapping epitopes on the antigen, the use of SAMDI abrogates the need for the second antibody and substantially streamlines the assay development cycle. Finally, we note that the use of SAMDI as the detection method also serves to verify the integrity of the biosensor, since this technique can verify that each of the intended components is present. This benefit is not of high importance in the development of individual immunoassays—which undergo a stringent assay optimization and validation—but can be very important in array-based experiments where it is not feasible to establish the fidelity of each assay.
Mass spectrometric methods offer a significant benefit over optical methods for label-free detection, including surface plasmon resonance spectroscopy. Because the optical methods measure changes in the refractive index of the medium near the biosensor surface, they do not discriminate between the intended analyte and species that contribute to the background signal. SAMDI, and other methods that use mass spectrometry, provide the masses of the species interacting with the sensor surface and therefore can more efficiently identify, and quantitate, the signal for the intended analyte even when there are significant levels of background species bound to the sensor surface. Further, the mass filtering that is inherent to mass spectrometry methods permits multiple assays to be performed simultaneously. In this work, we showed that four antigens in serum could be analyzed with an immunosensor that presented a mixture of antibodies specific to each analyte. The same experiment would be difficult to perform with fluorescently-labeled reagents because of spectral overlap of the fluorophores, and would be essentially impossible to perform with radiolabeled reagents.
Although mass spectrometry has the clear benefit of differentiating between intended analytes and background, with the current experimental design there is the potential for interference resulting from signal from the receptor protein G (or A) or the antibody. Further, the receptor proteins can immobilize antibodies present in the sample. Proper choice of either protein G (27 kDa) or protein A (48 kDa) can be effective for eliminating spectral overlap between the analyte and the sensor. We also note that the current SAMDI method used ion extraction parameters that were set to provide sufficient signal across a broad m/z range (5000–150,000 m/z) but were not optimized for resolution of each individual analyte. Further optimization of the delayed extraction parameters will give better resolution across narrow mass ranges limiting the chance for interferences.71 Finally, proper selection of matrix can have a profound effect on the resulting charge state distributions from the antigen, protein G/A, and the antibody. In this work we used both sinapinic acid (SA, 5 mg/mL in acetone) and α-cyano-4-hydroxycinnamic acid (CHCA, 7.5 mg/mL in acetone) as the energy absorbing matrices. In each case, approximately 0.3 μL of matrix was spotted by hand with a micropipette and allowed to diffuse across the monolayer. We usually observed signals across the entire surface, but noted a variation of signal intensity and some concentration of the non-covalently bound components at the matrix frontal edge. Typically, SA provided greater S/N for the m/z > 50,000 portion of the spectrum, which offered a more pronounced signal for the antibodies. In these instances, a greater surface area was required for sampling because higher laser intensities were necessary to obtain the spectra, which follow previous observations.22 The CHCA matrix permitted the use of lower laser intensities and therefore provided prolonged sampling across a given area of the surface, while yielding good peak intensities for m/z < 50,000 portion of the spectrum. Signal resulting from the loaded antibodies was sometimes suppressed with CHCA while that of protein A/G molecule and antigen could still be observed (see Figure 3C for HSA vs. Figure 3D for transferrin). This effect may be a response to the high density of antigen loaded across the surface that may interfere with desorption and ionization of the antibody. Alternatively, this observation could result from localized concentration of antigens to regions of the immunosensor that were manually sampled. Control over matrix and matrix deposition will not only have an effect on ion signal intensity and charge state distribution but may help to optimize resolution by providing a more uniform density of analyte across the entire surface. Future efforts should evaluate devices such as aerosol emitting tips or ink jet printers that regulate the deposition of matrix on the surface or harness methods that avoid the use of matrix if they can be adapted to high molecular weight analytes.72–75
Mass spectrometric methods are particularly useful for the characterization of proteins that contain metabolically-derived post-translational modifications or protein variants associated with gene families (or single nucleotide polymorphisms). This capability was highlighted in this paper for the identification of a truncated form of cystatin C in patients with multiple sclerosis and also in the discrimination between the α (15.1 kDa) and β (15.9 kDa) subunits of hemoglobin A. Quantitative comparison of post-translationally derived biomarkers or protein variants could yield valuable information about the stability or expression of the variants or post-translational modifications (PTMs) and prove to be diagnostically useful, as has been highlighted by previous groups when studying proteins present in blood.16, 76, 77 By comparing the ratio of two forms of the same antigen, the need for an internal standard is reduced. For example, α and β thalassemia are inherited diseases of red blood cells that manifests in a deficiency of the respective α and β chains in hemoglobin. The extent of the disease is dependent upon the number of loci that are affected. The SAMDI-TOF immunosensor may offer a simple method to quantitatively compare the ratio of these two subunits (without the use of an internal standard) and thus provide a prognosis for disease.
Despite the clear benefits that protein-based mass spectrometry methods offer in clinical diagnostic assays, they are still not employed in this setting. One reason for this slow acceptance stems from the usual barrier to the introduction of novel methods, but other factors include the need for technicians with specialized training and the need for methods that simplify the preparation, isolation and enrichment of the analytes.78 Standardization of analytical methods has partly been addressed by the commercial SELDI-TOF mass spectrometer. In the traditional SELDI experiment, proteins are partitioned onto a solid support through non-specific interactions with chromatographic resins arrayed onto the surface. In this way, analytes in a complex sample can be enriched through their preferential interaction with a solid phase resin, but cannot be isolated from the sample as is done with immunoassays. This lack of specificity of the plate for an analyte, and the lack of standardized pre-analytical procedures for samples give rise to peak intensity CVs of 10–40%, and limited dynamic range for protein detection in complex mixtures.79 Strategies, such as the SAMDI-TOF immunoassay, which combine well defined surface chemistries for the selective and reproducible localization of analytes with mass spectrometry may offer an alternative methodology to address many of the issues associated with standardized clinical diagnostics. Further improvements to the current assay design could be realized by the covalent attachment of the protein G (or A) to the maleimide surface through a recombinant oligo-cysteine tag.80 Such a design, combined with the known long term stability of the alkanethiolate monolayer,44, 57 could greatly extend the shelf-life of the assay as well as minimize the steps necessary to perform an immunoassay with SAMDI.
CONCLUSIONS
SAMDI-TOF mass spectrometry, which has proven valuable for performing assays of enzyme activities and protein-protein interactions, has now been extended to the label-free detection of proteins found in clinical samples. The design and preparation of self-assembled monolayers that are designed for specific applications with mass spectrometry offers a straightforward method for analyzing protein variants and post-translational modifications in humoral fluids and holds much promise in phenotyping of proteins in diseased samples or across patient populations. Future emphasis in automation of sample handling, matrix deposition, minimizing steps in assay development, and parallel analysis with microarrays will further expand the utility of this approach as a standardized diagnostic tool.
Figure 6.
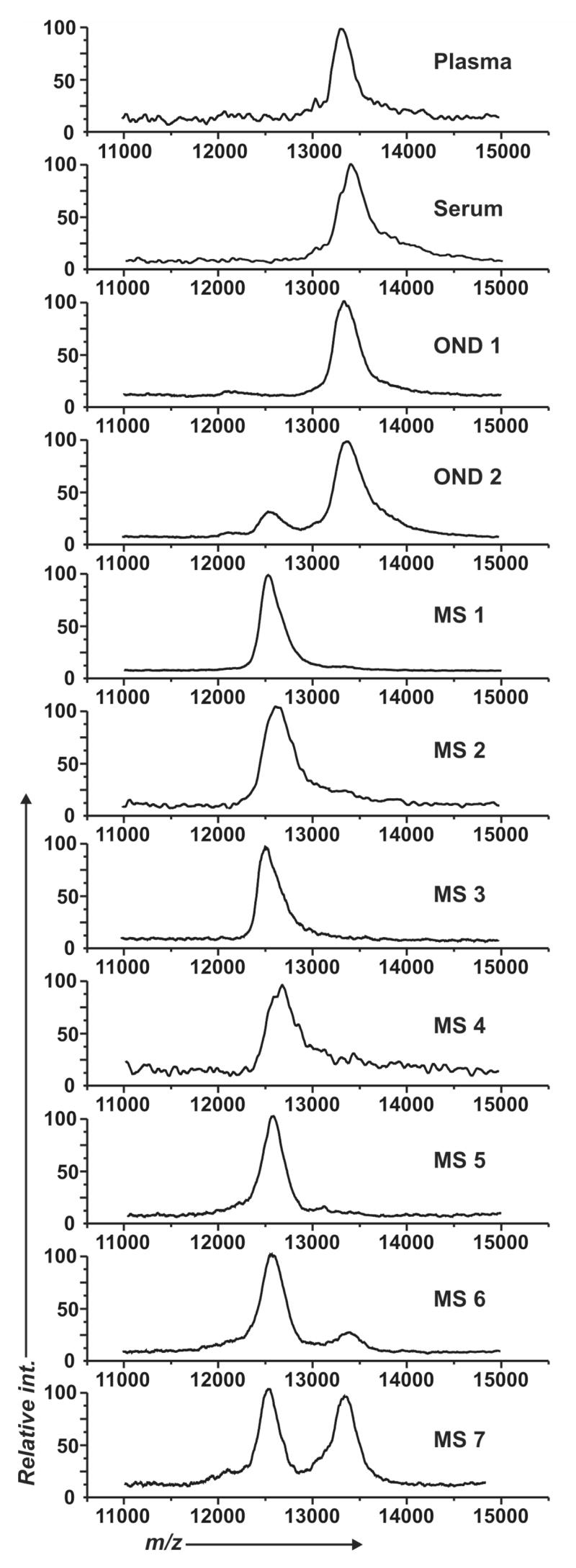
Assessment of in vivo proteolysis of cysC in patients with multiple sclerosis. SAMDI-TOF spectra show the capture of cysC from normal human plasma, normal human serum, two control CSF samples presenting other neurological diseases (OND), and seven CSF samples from patients with multiple sclerosis (MS).
Acknowledgments
The work was supported by the National Institutes of Health, the National Science Foundation, UC Pathology Research Fellowship and the Brain Research Institute.
References
- 1.Haab BB. Proteomics. 2003;3:2116–2122. doi: 10.1002/pmic.200300595. [DOI] [PubMed] [Google Scholar]
- 2.Templin MF, Stoll D, Schrenk M, Traub PC, Vohringer CF, Joos TO. Trends in Biotechnology. 2002;20:160–166. doi: 10.1016/s0167-7799(01)01910-2. [DOI] [PubMed] [Google Scholar]
- 3.Zhu H, Snyder M. Current Opinion in Chemical Biology. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 4.Kingsmore SF. Nat Rev Drug Discov. 2006;5:310–320. doi: 10.1038/nrd2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu X, Xu D, Cheng Q. Proteomics. 2006;6:5493–5503. doi: 10.1002/pmic.200600216. [DOI] [PubMed] [Google Scholar]
- 6.Koga H, Kyo M, Usui-Aoki K, Inamori K. Electrophoresis. 2006;27:3676–3683. doi: 10.1002/elps.200500821. [DOI] [PubMed] [Google Scholar]
- 7.Gobi KV, Iwasaka H, Miura N. Biosens Bioelectron. 2006 doi: 10.1016/j.bios.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Vaisocherova H, Mrkvova K, Piliarik M, Jinoch P, Steinbachova M, Homola J. Biosens Bioelectron. 2006 [Google Scholar]
- 9.Lee HJ, Nedelkov D, Corn RM. Analytical Chemistry. 2006;78:6504–6510. doi: 10.1021/ac060881d. [DOI] [PubMed] [Google Scholar]
- 10.Usui-Aoki K, Shimada K, Nagano M, Kawai M, Koga H. Proteomics. 2005;5:2396–2401. doi: 10.1002/pmic.200401171. [DOI] [PubMed] [Google Scholar]
- 11.Kyo M, Usui-Aoki K, Koga H. Analytical Chemistry. 2005;77:7115–7121. doi: 10.1021/ac050884a. [DOI] [PubMed] [Google Scholar]
- 12.Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 13.Nedelkov D. Expert Rev Proteomics. 2006;3:631–640. doi: 10.1586/14789450.3.6.631. [DOI] [PubMed] [Google Scholar]
- 14.Nedelkov D, Nelson RW. Biosens Bioelectron. 2001;16:1071–1078. doi: 10.1016/s0956-5663(01)00229-9. [DOI] [PubMed] [Google Scholar]
- 15.Nelson RW, Krone JR, Bieber AL, Williams P. Anal Chem. 1995;67:1153–1158. doi: 10.1021/ac00103a003. [DOI] [PubMed] [Google Scholar]
- 16.Nedelkov D, Kiernan UA, Niederkofler EE, Tubbs KA, Nelson RW. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10852–10857. doi: 10.1073/pnas.0500426102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nedelkov D, Kiernan UA, Niederkofler EE, Tubbs KA, Nelson RW. Molecular & Cellular Proteomics. 2006;5:1811–1818. doi: 10.1074/mcp.R600006-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Karas M, Hillenkamp F. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 19.Nelson RW, Krone JR, Jansson O. Anal Chem. 1997;69:4363–4368. doi: 10.1021/ac970538w. [DOI] [PubMed] [Google Scholar]
- 20.Nelson RW, Krone JR, Jansson O. Analytical Chemistry. 1997;69:4369–4374. doi: 10.1021/ac9705374. [DOI] [PubMed] [Google Scholar]
- 21.Nedelkov D, Nelson RW. Trends Biotechnol. 2003;21:301–305. doi: 10.1016/S0167-7799(03)00141-0. [DOI] [PubMed] [Google Scholar]
- 22.Nedelkov D, Tubbs KA, Nelson RW. Electrophoresis. 2006;27:3671–3675. doi: 10.1002/elps.200600065. [DOI] [PubMed] [Google Scholar]
- 23.Brockman AH, Orlando R. Anal Chem. 1995;67:4581–4585. doi: 10.1021/ac00120a024. [DOI] [PubMed] [Google Scholar]
- 24.Brockman AH, Orlando R. Rapid Commun Mass Spectrom. 1996;10:1688–1692. doi: 10.1002/(SICI)1097-0231(199610)10:13<1688::AID-RCM717>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Engwegen JY, Gast MC, Schellens JH, Beijnen JH. Trends Pharmacol Sci. 2006;27:251–259. doi: 10.1016/j.tips.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Favre-Kontula L, Johnson Z, Steinhoff T, Frauenschuh A, Vilbois F, Proudfoot AE. J Immunol Methods. 2006;317:152–162. doi: 10.1016/j.jim.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Yang X, Bowick GC, Herzog NK, Luxon BA, Lomas LO, Gorenstein DG. Biochem Biophys Res Commun. 2006;347:586–593. doi: 10.1016/j.bbrc.2006.06.132. [DOI] [PubMed] [Google Scholar]
- 28.Rao SV, Anderson KW, Bachas LG. Mikrochimica Acta. 1998;128:127–143. [Google Scholar]
- 29.Schramm W, Paek S, Voss G. Immunomethods. 1993;3:93–103. [Google Scholar]
- 30.Lu B, Smyth MR, O’Kennedy R. Analyst. 1996;121:29R–32R. doi: 10.1039/an996210029r. [DOI] [PubMed] [Google Scholar]
- 31.Cockerill KA, Linnik MD, Iverson GM. Clin Immunol. 2004;112:129–135. doi: 10.1016/j.clim.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Iverson GM, Matsuura E, Victoria EJ, Cockerill KA, Linnik MD. J Autoimmun. 2002;18:289–297. doi: 10.1006/jaut.2002.0590. [DOI] [PubMed] [Google Scholar]
- 33.Peluso P, Wilson DS, Do D, Tran H, Venkatasubbaiah M, Quincy D, Heidecker B, Poindexter K, Tolani N, Phelan M, Witte K, Jung LS, Wagner P, Nock S. Analytical Biochemistry. 2003;312:113–124. doi: 10.1016/s0003-2697(02)00442-6. [DOI] [PubMed] [Google Scholar]
- 34.Spitznagel TM, Clark DS. Biotechnology (N Y) 1993;11:825–829. doi: 10.1038/nbt0793-825. [DOI] [PubMed] [Google Scholar]
- 35.Su J, Mrksich M. Angewandte Chemie-International Edition. 2002;41:4715–4718. doi: 10.1002/anie.200290026. [DOI] [PubMed] [Google Scholar]
- 36.Hodneland CD, Lee YS, Min DH, Mrksich M. Proc Natl Acad Sci U S A. 2002;99:5048–5052. doi: 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houseman BT, Huh JH, Kron SJ, Mrksich M. Nature Biotechnology. 2002;20:270–274. doi: 10.1038/nbt0302-270. [DOI] [PubMed] [Google Scholar]
- 38.Yousaf MN, Houseman BT, Mrksich M. Proc Natl Acad Sci U S A. 2001;98:5992–5996. doi: 10.1073/pnas.101112898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houseman BT, Mrksich M. Chem Biol. 2002;9:443–454. doi: 10.1016/s1074-5521(02)00124-2. [DOI] [PubMed] [Google Scholar]
- 40.Houseman BT, Gawalt ES, Mrksich M. Langmuir. 2003;19:1522–1531. [Google Scholar]
- 41.Prime KL, Whitesides GM. Science. 1991;252:1164–1167. doi: 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- 42.Prime KL, Whitesides GM. Journal of the American Chemical Society. 1993;115:10714–10721. [Google Scholar]
- 43.Siegel RR, Harder P, Dahint R, Grunze M, Josse F, Mrksich M, Whitesides GM. Anal Chem. 1997;69:3321–3328. doi: 10.1021/ac970047b. [DOI] [PubMed] [Google Scholar]
- 44.Luk YY, Kato M, Mrksich M. Langmuir. 2000;16:9604–9608. [Google Scholar]
- 45.Mrksich M, Whitesides GM. Poly(Ethylene Glycol) 1997;680:361–373. [Google Scholar]
- 46.Yeo WS, Min DH, Hsieh RW, Greene GL, Mrksich M. Angew Chem Int Ed Engl. 2005;44:5480–5483. doi: 10.1002/anie.200501363. [DOI] [PubMed] [Google Scholar]
- 47.Marin VL, Min DH, Yeo WS, Kay BK, Mrksich M. 2007 unpublished work. [Google Scholar]
- 48.Kwon Y, Han Z, Karatan E, Mrksich M, Kay BK. Anal Chem. 2004;76:5713–5720. doi: 10.1021/ac049731y. [DOI] [PubMed] [Google Scholar]
- 49.Min DH, Tang WJ, Mrksich M. Nature Biotechnology. 2004;22:717–723. doi: 10.1038/nbt973. [DOI] [PubMed] [Google Scholar]
- 50.Min DH, Yeo WS, Mrksich M. Anal Chem. 2004;76:3923–3929. doi: 10.1021/ac049816z. [DOI] [PubMed] [Google Scholar]
- 51.Su J, Bringer MR, Ismagilov RF, Mrksich M. Journal of the American Chemical Society. 2005;127:7280–7281. doi: 10.1021/ja051371o. [DOI] [PubMed] [Google Scholar]
- 52.Min DH, Su J, Mrksich M. Angew Chem Int Ed Engl. 2004;43:5973–5977. doi: 10.1002/anie.200461061. [DOI] [PubMed] [Google Scholar]
- 53.Akerstrom B, Bjorck L. J Biol Chem. 1986;261:10240–10247. [PubMed] [Google Scholar]
- 54.Akerstrom B, Brodin T, Reis K, Bjorck L. J Immunol. 1985;135:2589–2592. [PubMed] [Google Scholar]
- 55.Goding JW. J Immunol Methods. 1978;20:241–253. doi: 10.1016/0022-1759(78)90259-4. [DOI] [PubMed] [Google Scholar]
- 56.Kronvall G, Seal US, Finstad J, Williams RC., Jr J Immunol. 1970;104:140–147. [PubMed] [Google Scholar]
- 57.Harder P, Grunze M, Dahint R, Whitesides GM, Laibinis PE. Journal of Physical Chemistry B. 1998;102:426–436. [Google Scholar]
- 58.Johnson DL, Martin LL. Journal of the American Chemical Society. 2005;127:2018–2019. doi: 10.1021/ja045084g. [DOI] [PubMed] [Google Scholar]
- 59.Kanno S, Yanagida Y, Haruyama T, Kobatake E, Aizawa M. J Biotechnol. 2000;76:207–214. doi: 10.1016/s0168-1656(99)00186-8. [DOI] [PubMed] [Google Scholar]
- 60.Kwon Y, Han ZZ, Karatan E, Mrksich M, Kay BK. Analytical Chemistry. 2004;76:5713–5720. doi: 10.1021/ac049731y. [DOI] [PubMed] [Google Scholar]
- 61. In http://www.biovendor.com/pdf/RD191009100.pdf.
- 62.Niederkofler EE, Tubbs KA, Gruber K, Nedelkov D, Kiernan UA, Williams P, Nelson RW. Anal Chem. 2001;73:3294–3299. doi: 10.1021/ac010143j. [DOI] [PubMed] [Google Scholar]
- 63.Tubbs KA, Kiernan UA, Niederkofler EE, Nedelkov D, Bieber AL, Nelson RW. Anal Chem. 2006;78:3271–3276. doi: 10.1021/ac060013g. [DOI] [PubMed] [Google Scholar]
- 64.Irani DN, Anderson C, Gundry R, Cotter R, Moore S, Kerr DA, McArthur JC, Sacktor N, Pardo CA, Jones M, Calabresi PA, Nath A. Ann Neurol. 2006;59:237–247. doi: 10.1002/ana.20786. [DOI] [PubMed] [Google Scholar]
- 65.Del Boccio P, Pieragostino D, Lugaresi A, Di Ioia M, Pavone B, Travaglini D, D’Aguanno S, Bernardini S, Sacchetta P, Federici G, Di Ilio C, Gambi D, Urbani A. Ann Neurol. 2006 doi: 10.1002/ana.20968. [DOI] [PubMed] [Google Scholar]
- 66.Hansson SF, Hviid Simonsen A, Zetterberg H, Andersen O, Haghighi S, Fagerberg I, Andreasson U, Westman-Brinkmalm A, Wallin A, Ruetschi U, Blennow K. Ann Neurol. 2006 doi: 10.1002/ana.20945. [DOI] [PubMed] [Google Scholar]
- 67.Nakashima I, Fujinoki M, Fujihara K, Kawamura T, Nishimura T, Nakamura M, Itoyama Y. Ann Neurol. 2006 [Google Scholar]
- 68.Su J, Rajapaksha TW, Peter ME, Mrksich M. Analytical Chemistry. 2006;78:4945–4951. doi: 10.1021/ac051974i. [DOI] [PubMed] [Google Scholar]
- 69.Hutchens WT, Yip T. US Patent: 5719060 [Google Scholar]
- 70.Anderson GP, Jacoby MA, Ligler FS, King KD. Biosensors & Bioelectronics. 1997;12:329–336. doi: 10.1016/s0956-5663(96)00074-7. [DOI] [PubMed] [Google Scholar]
- 71.Voyager DE-PRO Biospectrometry Mass Spectrometer Operator Manual.
- 72.Nedelkov D, Nelson RW. Journal of Molecular Recognition. 2000;13:140-+. doi: 10.1002/1099-1352(200005/06)13:3<140::AID-JMR496>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 73.Meier MAR, de Gans BJ, van den Berg AMJ, Schubert US. Rapid Communications in Mass Spectrometry. 2003;17:2349–2353. doi: 10.1002/rcm.1195. [DOI] [PubMed] [Google Scholar]
- 74.Thomas JJ, Shen ZX, Crowell JE, Finn MG, Siuzdak G. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4932–4937. doi: 10.1073/pnas.081069298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peterson DS. Mass Spectrom Rev. 2007;26:19–34. doi: 10.1002/mas.20104. [DOI] [PubMed] [Google Scholar]
- 76.Shimizu A, Nakanishi T, Miyazaki A. Mass Spectrometry Reviews. 2006;25:686–712. doi: 10.1002/mas.20086. [DOI] [PubMed] [Google Scholar]
- 77.Griffith WP, Kaltashov IA. Current Organic Chemistry. 2006;10:535–553. [Google Scholar]
- 78.Verrills NM. Clin Biochem Rev. 2006;27:99–116. [PMC free article] [PubMed] [Google Scholar]
- 79.Albrethsen J, Bogebo R, Olsen J, Raskov H, Gammeltoft S. Clin Chem Lab Med. 2006;44:1243–1252. doi: 10.1515/CCLM.2006.228. [DOI] [PubMed] [Google Scholar]
- 80.Ichihara T, Akada JK, Kamei S, Ohshiro S, Sato D, Fujimoto M, Kuramitsu Y, Nakamura K. J Proteome Res. 2006;5:2144–2151. doi: 10.1021/pr0504889. [DOI] [PubMed] [Google Scholar]