

Abstract
Since the advent of matrix-assisted laser desorption/ionization and electrospray ionization, mass spectrometry has played an increasingly important role in protein functional characterization, identification, and structural analysis. Expanding this role, desorption/ionization on silicon (DIOS) is a new approach that allows for the analysis of proteins and related small molecules. Despite the absence of matrix, DIOS-MS yields little or no fragmentation and is relatively tolerant of moderate amounts of contaminants commonly found in biological samples. Here, functional assays were performed on an esterase, a glycosidase, a lipase, as well as exo- and endoproteases by using enzyme-specific substrates. Enzyme activity also was monitored in the presence of inhibitors, successfully demonstrating the ability of DIOS to be used as an inhibitor screen. Because DIOS is a matrix-free desorption technique, it also can be used as a platform for multiple analyses to be performed on the same protein. This unique advantage was demonstrated with acetylcholine esterase for qualitative and quantitative characterization and also by its subsequent identification directly from the DIOS platform.
Mass spectrometry is quickly becoming an essential tool for characterizing protein function, substrate specificity, and protein identity (1–5) as it complements or, in some cases, supersedes the utility of traditional biological methods (6, 7). For some of the most important proteomics applications, the high sensitivity and accuracy provided by modern mass spectrometry allow for unequivocal characterization and quantitative analysis of proteins and their chemical products (5, 8, 9). Ionization methods such as electrospray ionization used in liquid chromatography/MS and matrix-assisted laser desorption/ionization (MALDI) are the core innovations that allow for mass spectrometry to be used in protein characterization as well as in the determination of protein structure/function relationships (10, 11). MALDI mass spectrometry has been particularly effective as a proteomics tool because of its relatively high tolerance of mixtures and biological contaminants (12, 13); however, its matrix requirement represents a limitation in interference in the low-mass region, preparation time, and the potential to perform sample manipulation after mass analysis. In addition, a prevailing obstacle toward protein characterization by using both MALDI and liquid chromatography/MS is the loss of analyte material during protein separations, chromatographic separations, or functional studies that require the transfer of the sample for subsequent identification. One way to overcome these obstacles would be to both identify and functionally characterize proteins on a single surface.
Desorption/ionization on porous silicon (DIOS), a new method for the generation of intact gas phase ions (14), uses UV laser light to desorb intact analytes from the surface without matrix assistance. The procedure for producing DIOS surfaces involves a simple galvanostatic etching procedure (15), which yields an effective platform for desorption/ionization for a range of biomolecules and organic compounds. Here, we demonstrate the use of DIOS-MS for the identification and functional characterization of proteins as well as protein-catalyzed chemical transformations. Enzyme-catalyzed reactions were monitored by incubating the catalyst and substrate directly on the porous silicon chip for a desired period, after which the mixtures were allowed to dry and the residues were analyzed directly by DIOS-MS. Sample manipulation thereby is minimized and the small volumes used conserve the amount of sample needed for analysis, which can expedite enzymatic reaction analysis (16). Because the protein material presents little interference in the low-mass region, it is easy to monitor product formation quantitatively and thereby measure activity. These functional assays were performed on an esterase, a glucosidase, a lipase, and exo- and endoproteases. After functional characterization, the same protein can be digested with proteolytic enzymes, analyzed by DIOS-MS, and identified by using the mass spectral data with computer-database searching.
Methods
DIOS-MS experiments were performed on a PerSeptive Biosystems Voyager STR (Framingham, MA) equipped with a nitrogen laser (337 nm) and a reflectron mirror. Ions were desorbed from the DIOS surfaces with laser energies ranging from ≈2.1 to 7.0 mJ/3-ns pulse as measured with a single-channel Joulemeter (Molectron, Sunnyvale, CA). Laser intensities were set to optimize signal-to-noise and resolution of analyte mass spectrometry signals and were variable upon the physicochemical properties of the analytes and the DIOS surfaces. Desorbed ions were extracted into the flight tube with 20 kV after a 150 ns delay. DIOS chips were placed directly onto commercial MALDI plates, which have been milled specifically to accommodate the thickness of the surfaces.
The etching conditions for DIOS surfaces have been described previously (15). Briefly, low-resistivity silicon wafers (0.005–0.02 Ω cm−1) were electrochemically etched at 5 mA/cm2 for 1 min with ethanol/HF (25% vol/vol) solution under white-light illumination (50 mW/cm2). In a process referred to as “double-etching,” the porous silicon surfaces were oxidized with ozone followed by immersion for 1 min in an ethanol solution of aqueous HF (5% vol/vol) solution. This process changes the morphology of the surfaces in a way that retards oxidation of the surface without introducing any preferential desorption attributes. Because of the surface characteristics, the “double-etched” surfaces are advantageous for analyses involving sample mixtures such as protein digests or for analysis over an extended period. Interestingly, the standard DIOS chips produced with these etching conditions did not yield strong MS signals for oligosaccharides; however, spot oxidation with hydrogen peroxide on DIOS chips before the sample application yielded reproducible spectra of the sugar and the enzymatic product.
Adenovirus penton protein and Flock House Virus proteins were provided by the laboratories of Glen Nemorrow and Jack Johnson, respectively (The Scripps Research Institute, La Jolla, CA). Acetylcholine esterase from electrophorus electricus (EC 3.1.1.7) and phospholipase A2 from hog pancreas (EC 3.1.0.1.4) were purchased from Fluka, whereas mannosidase II from Jack bean (EC 3.2.1.24) and modified trypsin (EC 3.4.21.4) were purchased from Promega and Glyco (Novato, CA), respectively. Enzymatic reactions were performed in 10 mM ammonium bicarbonate (pH ≈ 7.6) or 10 mM ammonium citrate (pH ≈ 5.0) with an enzyme/protein substrate ratio of ≈1:500 (wt/wt). Enzymatic reactions were sustained on the DIOS surfaces in incubation chambers with periodic (ca. 30 min) buffer refurbishment of the liquid sample. Samples (0.5 μl) were deposited onto DIOS chips in solutions containing less than 25% (vol/vol) organic solvents to minimize spreading on the porous silicon surfaces.
Results and Discussion
Protein Functional Characterization.
Characterizing functional activity can be accomplished by monitoring the interaction of an enzyme with potential substrate molecules, such that the formation of enzyme reaction products helps identify the protein's activity. Thus, the reaction between a 4-nM solution of acetylcholine esterase [AChe, (kcat/KM) = 1.5 × 108 M−1 160 s−1] (17) and a 50-fold molar excess of its natural substrate, acetylcholine (ACh), was monitored by DIOS-MS (Fig. 1). As shown by the appearance of choline (m/z 104) and the disappearance of ACh (m/z 146), the reaction reached completion within 15 min (when performed at pH 8 and 37°C). The presence of the 75-kDa enzyme does not interfere with the detection of any of the small molecules of interest.
Figure 1.

Monitoring the enzymatic production of choline (m/z 104) from the ACh substrate (m/z 146) and acetylcholinesterase. A significant amount of choline was generated within 5 min at 37°C.
With the use of internal standards, DIOS-MS peak intensities also can be used for highly accurate quantitation (15). The pseudo-first-order kinetics of the AChe reaction was examined efficiently by using choline-d9 as internal standard, as shown in Fig. 2. The substrate, product, and internal standard were found to exhibit similar desorption efficiencies, negating any possibility of preferential desorption that might lead to a misleading appearance of product abundance. Interestingly, attempted analogous measurements of the product (m/z 104) by using MALDI or electrospray ionization methods were unsuccessful because of the lack of a reproducibly strong product signal. These results also were consistent with analysis of standard choline. Note that the lack of a strong chromophore on choline would make it difficult to employ UV-visible absorption or emission techniques on the unadorned natural substrate.
Figure 2.

Kinetics plot for the conversion of ACh to choline at 25°C with an initial substrate concentration of 200 μM and an enzyme concentration of 40 pM. Choline-d9 was added as an internal standard at a concentration of 40 μM. The data follow the increase in the product (choline) as a function of time. Each time point is the average and SD of four measurements. (Inset) Linear calibration of choline vs. internal standard used to relate observed choline peak intensities to concentration.
The identification of selective inhibitors of enzymes is an area of both fundamental and practical interest. The ability to observe enzymatic reaction products with DIOS also allows for convenient monitoring of enzyme inhibition. For example, several AChe inhibitors were analyzed for their ability to suppress the deacylation of ACh by measuring the rates of product formation in the presence and absence of inhibitor (Fig. 2). AChe inhibitors are of interest for treatment of cognitive deficits experienced by individuals with dementia (18). Fig. 3 demonstrates the inhibition of acetylcholine esterase over a period of 10 min with two known potent inhibitors used at identical concentrations: (−)–huperzine A (Ki = 4.6 nM) and tacrine (Ki = 31 nM) (18, 19). Note that the reaction is completely stopped by huperzine, and a very small level of conversion is shown in the presence of tacrine, consistent with their relative Ki values. DIOS analysis of another potential inhibitor, 2,6-dimethoxyphenyl-N-butylcarbamate, showed it to be less active under the same conditions. By performing these three reactions in triplicate simultaneously in separate spots on the same DIOS chip, these inhibitors were screened independently within 15 min, including sample preparation time.
Figure 3.

Inhibition of AChe at 37°C was evaluated with several different types of small-molecule inhibitors with a substrate concentration of 200 μM and an enzyme concentration of 4 μM.
Although carbohydrates are notoriously difficult to observe by mass spectrometry, they represent one of the largest and most important classes of biological molecules. DIOS-MS also was used to monitor the exoglycosidase activity of 1–3-(1–6)mannosyl-oligosaccharide α-d-mannohydrolase (commonly known as mannosidase II) on an N-linked carbohydrate. The enzyme (molecular mass, 190,000 Da) preferentially removes the 1→3 linked mannose residue from the nonreducing terminus (20). In this case, the standard DIOS porous silicon material does not provide efficient detection of carbohydrates, perhaps due to the hydrophobic character of the surfaces. A more polar, oxidized DIOS wafer (15) containing surface oxide and hydroxide groups gives much better MS signal intensity for this class of analytes, demonstrating that the surface can be tailored to the nature of the analyte. Although this requires the use of higher laser intensity than is standard for desorption/ionization (thereby diminishing the signal-to-noise ratio somewhat), no fragmentation of carbohydrate analytes is observed.
The enzyme-catalyzed reaction of phospholipase with a small molecule substrate also was examined in these studies. Fig. 4 illustrates the specific enzymatic removal of the sn-2 fatty acyl chain from a phosphatidylcholine lipid. After a 30-min incubation directly on the DIOS chip, the lipase produced an abundance of the lysophospholipid at m/z 411 with the expected satellite sodium and potassium adducts (Fig. 4). After the protein of interest has been functionally characterized through substrate specificity or interaction with an inhibitor, further analysis can identify the protein.
Figure 4.

Monitoring an on-plate enzymatic reaction of the selective removal of a fatty acyl chain from a phospholipid, where substrate is the triacylglycerol phospholipid and product is the lysophospholipid species.
Protein Identification.
Proteins are classified both by their function and by their amino acid sequence. Site-specific proteolytic digestion with enzymes, combined with mass analysis, provide a protein-specific peptide mass map. This unique map combined with database searching allows for protein identification as well as characterization of posttranslational modifications. The absence of MALDI matrix material in DIOS-MS analysis allows us to combine the characterization of protein function, demonstrated above, with protein identification by using standard mass-mapping techniques on the same sample. For this application, it was found that treatment of the chemically oxidized porous silicon material described above with an aqueous HF/ethanol mixture provides a DIOS surface that gives improved signal intensities with mixtures and higher concentrations of analytes, which often are found in protein digests. These so-called “double-etched” surfaces are now a standard DIOS-MS support in our laboratories and are described in detail elsewhere (15).
On-plate digestion and DIOS-MS analysis were used to identify the AChe enzyme used in the aforementioned experiments as a specific catalytic subunit precursor. An in situ tryptic digest of the enzyme was sustained on the porous silicon plate for 4 h at 37°C, followed by drying and direct MS analysis of the residue by DIOS. The mass information was obtained primarily from peptides near the C and N termini, presumably because of the limited access of trypsin to the interior sections of AChe, which are blocked by membrane-spanning regions, disulfide bonds, and glycosylation sites. Even though other less specific enzymes (e.g., pepsin, thermolysin, and chymotrypsin) may provide greater access to the protein interior, the observed peptides nonetheless were sufficient to confirm the identification of the 72-kDa protein (with a probability of 0.95) by using known database sequence information (21). Because of the resistance of AChe toward tryptic degradation, digests performed in solution for 24 h and analyzed by MALDI-MS did not provide significantly more information or more accurate identification than on-plate proteolytic digestion combined with DIOS-MS. We also have observed DIOS-MS spectra of proteolytic digests of β-lactoglobulin, BSA, and flock house capsid proteins performed in solution and deposited directly onto the DIOS plate. Because high-mass species (greater than 20,000 Da) are not desorbed from DIOS surfaces, the intact proteolytic enzyme neither contributes peaks to the spectrum nor interferes with MS detection of smaller fragments. Many of the low-mass proteolytic fragments observed would not be visible with MALDI because of matrix interference. Currently, the mass range of DIOS is limited to less than 18,000 Da, with the most useful data commonly observed at less than 3,500 Da. A DIOS analysis of a tryptic digest of a β-lactoglobulin standard (Fig. 5C) shows the molecular ion of the protein (≈m/z 18,000) in addition to proteolytic fragments as small as 500 Da.
Figure 5.

DIOS mass spectra and database identification of an 18-h solution tryptic digest of BSA (A), an on-plate tryptic digest of adenovirus penton protein at 37°C for 75 min (B), and the 18-h solution tryptic digest of β-lactoglobulin (C). In C, the intact protein (18 kDa) also was observed. Identification was obtained from the profound protein search program (http://129.85.19.192/prowl-cgi/ProFound.exe) linked to the Swiss-Prot database, and search parameters included a 0.5-Da tolerance.
The utility of DIOS analysis for protein identification is best illustrated by the proteolytic digest and the resultant identification of an adenovirus penton protein (Fig. 5B). After simple purification with a 10-kDa filter, the protein was incubated with trypsin on the DIOS chip at 37°C in a reaction volume of 1.0 μl. The mass information obtained from the DIOS spectrum was sufficient not only to identify the type of protein, but also to identify the correct serotype (type II) of the virus (Fig. 5B Inset), distinguishing it from a related serotype, which differs by only a few residues. Other suggested protein database choices were of significantly (orders of magnitude) lower probability.
Although DIOS can have a higher tolerance for biological contaminants than MALDI, spectral quality does diminish in the presence of large quantities of interfering compounds. In these situations the addition of ion-sequestering reagents such as ammonium citrate directly to the sample also can enhance significantly DIOS peak intensities, and this practice is now standard in our lab in the analysis of protein digests (see Methods). It should be noted that such additives do not absorb light at the frequency of the UV laser and, therefore, do not act as a matrix. In the case of adenovirus (Fig. 5B), the sample obtained from ion-exchange chromatography containing a high concentration of salts (40 mM Tris/1 M NaCl) was purified simply with a molecular weight cutoff filter before analysis.
Even with these limitations, the analysis of mixtures of biological molecules is more conveniently performed with DIOS-MS than with other MS techniques, which often require more purification by liquid chromatography or gel electrophoresis before mass spectrometry. Particularly important for identifying characterized proteins, despite the time-consuming manipulations required, are in-gel digests of proteins, which have been investigated previously with MALDI and capillary LC mass spectrometry (22–24). After time-consuming gel washing and destaining steps, significant amounts of salts, surfactants, and involatile buffers still can remain in the sample extracts, and DIOS seems to be suitable for such samples. DIOS-MS of the in-gel digest of 23 pmol BSA was performed, and, additionally, an SDS/PAGE separation of human rhinovirus proteins displayed the predicted Coomassie-stained bands and yielded satisfactory DIOS spectra (data not shown).
Protein Structural Information.
In addition to identifying and monitoring the activity of enzymes, we have used DIOS-MS to gain primary structural information. Thus, on-plate carboxypeptidase Y digestion reactions of bradykinin (1,060 Da) followed by DIOS analysis affords primary sequence information from the C termini to the N termini tripeptide with this on-plate digest (data not shown). Obtaining additional sequence information was limited by carboxypeptidase Y inefficiency at cleaving glycine residues as well as tripeptides (25). This enzymatic approach toward obtaining peptide sequence information requires that the reaction be performed on a single, pure peptide under optimal conditions.
Postsource decay (PSD) analysis is another established means to obtain sequence information and also has been used to identify posttranslational modifications without the addition of chemicals or enzymes to the analyte sample (26). Many molecules analyzed by MALDI do not undergo significant PSD fragmentation, and so the use of this technique with MALDI is limited. In contrast, DIOS-MS provides a fortunate combination of low levels of in-source fragmentation but modest levels of postsource decay when small-molecule ions (having fewer vibrational degrees of freedom and, therefore, fewer pathways to dissipate vibrational energy before fragmentation) are selected for PSD analysis.
The DIOS PSD analysis of the modified phosphopeptide
[Trp-Ala-Gly-Gly-Asp-Ser(PO3H2)-Gly-Glu]
in the negative ion mode revealed the phosphate moiety
(m/z 79 = PO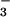 and
m/z 97 =
H2PO
and
m/z 97 =
H2PO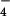 ) as well as several
fragment ions. It has been noted previously from PSD spectra of
phosphopeptides that negative ion spectra in these cases are dominated
by residue losses (b and y ions) from the C termini to the
phosphorylated residue (27). The strong intensity peaks from the loss
of the phosphate moiety and the limited amount of peptide fragmentation
are consistent with the facile loss of the phosphate group.
) as well as several
fragment ions. It has been noted previously from PSD spectra of
phosphopeptides that negative ion spectra in these cases are dominated
by residue losses (b and y ions) from the C termini to the
phosphorylated residue (27). The strong intensity peaks from the loss
of the phosphate moiety and the limited amount of peptide fragmentation
are consistent with the facile loss of the phosphate group.
Conclusions
DIOS is an effective tool for chemical proteomics. These experiments illustrate the utility of DIOS-MS in functional characterization through chemical transformation, monitoring the inhibition of protein-catalyzed reactions, and protein identification. Because DIOS is a matrix-free desorption technique, it has the potential for monitoring sequential reactions on the same surface through multiple mass spectrometric analyses. Without matrix dependence and interference, DIOS-MS extends the observable mass range to small biomolecules. Because small mass ions are readily observed, additional proteomic structural and functional information can be performed. In principle, a variety of substrates could be used to probe important classes of protein function (e.g., kinases, esterases, and peroxidases) either by sequential addition of the protein and substrates as demonstrated in this work or with proteins covalently linked to the porous silicon surface. In the same manner, enzyme inhibitors could be screened rapidly by monitoring the formation of reaction products. Automating these processes will further establish DIOS-MS as an effective tool for characterizing proteins. In addition to facilitating protein characterization, porous silicon also has a demonstrated potential for performing on-chip separation (17), de novo sequencing through postsource decay, and providing quantitative information (without the use of a chromophore). Thus, DIOS offers a platform on which multiple experiments can be performed on the same protein sample and on the same chip.
Abbreviations
- DIOS
desorption/ionization on silicon
- MALDI
matrix-assisted laser desorption/ionization
- ACh
acetylcholine
- AChe
ACh esterase
- PSD
postsource decay
References
- 1.Gevaert K, Vandekerckhove J. Electrophoresis. 2000;21:1145–1154. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1145::AID-ELPS1145>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 2.Bothner B, Chavez R, Wei J, Strupp C, Phung Q, Schneeman A, Siuzdak G. J Biol Chem. 2000;275:13455–13459. doi: 10.1074/jbc.275.18.13455. [DOI] [PubMed] [Google Scholar]
- 3.Haynes P A, Yates J R. Yeast. 2000;17:81–87. doi: 10.1002/1097-0061(20000630)17:2<81::AID-YEA22>3.0.CO;2-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lottspeich F. Angew Chem Int Ed. 1999;38:2476–2492. doi: 10.1002/(sici)1521-3773(19990903)38:17<2476::aid-anie2476>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 5.Pandley A, Mann M. Nature (London) 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 6.Kim S Y, Park K W, Lee Y J, Back S H, Goo J H, Park O K, Jang S K, Park W J. Anal Biochem. 2000;15:42–48. doi: 10.1006/abio.2000.4662. [DOI] [PubMed] [Google Scholar]
- 7.Bantel H, Ruck P, Schulze-Osthoff K. Cell Death Differ. 2000;7:504–505. doi: 10.1038/sj.cdd.4400669. [DOI] [PubMed] [Google Scholar]
- 8.Shevchenko A, Jensen O N, Podtelejnikov A V, Sagliocco F, Wilm M, Vorm O, Mortensen P, Boucherie H, Mann M. Proc Natl Acad Sci USA. 1996;93:14440–14455. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roepstorff P. Curr Opin Biotechnol. 1997;8:6–13. doi: 10.1016/s0958-1669(97)80151-6. [DOI] [PubMed] [Google Scholar]
- 10.Karas M, Hillenkamp F. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita M, Fenn J B. J Phys Chem. 1984;88:4451–4459. [Google Scholar]
- 12.Fenselau C. Anal Chem. 1997;69:661A–665A. doi: 10.1021/ac971831z. [DOI] [PubMed] [Google Scholar]
- 13.Beavis R C, Chait B T. Proc Natl Acad Sci USA. 1990;87:6873–6877. doi: 10.1073/pnas.87.17.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei J, Buriak J, Siuzdak G. Nature (London) 1999;399:243–246. doi: 10.1038/20400. [DOI] [PubMed] [Google Scholar]
- 15.Shen Z, Thomas J J, Averbuj C, Broo K M, Engelhard M, Crowell J E, Finn M G, Siuzdak G. Anal Chem. 2000;73:612–619. doi: 10.1021/ac000746f. [DOI] [PubMed] [Google Scholar]
- 16.Mechref Y, Novotny M V. Anal Chem. 1998;70:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- 17.Voet D, Voet J G. Biochemistry. New York: Wiley; 1995. [Google Scholar]
- 18.Camps P, Cusack B, Mallender W D, Achab R E, Morral J, Torrero D, Rosenberry T L. Mol Pharmacol. 2000;57:409–417. [PubMed] [Google Scholar]
- 19.Szegletes T, Mallender W D, Rosenberry T L. Biochemistry. 1998;37:4206–4216. doi: 10.1021/bi972158a. [DOI] [PubMed] [Google Scholar]
- 20.Yamashita K, Tachibana Y, Nakayama T, Kitamura M, Endo Y, Kobata A. J Biol Chem. 1980;255:5635–5642. [PubMed] [Google Scholar]
- 21.Simon S, Massoulie J. J Biol Chem. 1997;272:33045–33055. doi: 10.1074/jbc.272.52.33045. [DOI] [PubMed] [Google Scholar]
- 22.Patterson S D, Aebersold R. Electrophoresis. 1995;16:1791–1814. doi: 10.1002/elps.11501601299. [DOI] [PubMed] [Google Scholar]
- 23.Jensen O N, Podtelejnikov A, Mann M. Anal Chem. 1997;69:4741–4750. doi: 10.1021/ac970896z. [DOI] [PubMed] [Google Scholar]
- 24.Yates J R, McCormack A L, Eng J. Anal Chem. 1996;68:534A–540A. doi: 10.1021/ac962050l. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi R. In: Methods in Enzymology. Hirs C, Timasheff S, editors. Vol. 47. New York: Academic; 1977. pp. 84–93. [Google Scholar]
- 26.Gorman J J, Purcell A W, Ferguson B L, Lopaticki S, Morrow C J, Mineva E. J Protein Chem. 1998;17:526–527. [PubMed] [Google Scholar]
- 27.Hoffmann R, Metzger S, Spengler B, Otvos L. J Mass Spectrom. 1999;34:1195–1204. doi: 10.1002/(SICI)1096-9888(199911)34:11<1195::AID-JMS881>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]