

Abstract
Ethanolamine and choline are major components of the trypanosome membrane phospholipids, in the form of GPEtn (glycerophosphoethanolamine) and GPCho (glycerophosphocholine). Ethanolamine is also found as an integral component of the GPI (glycosylphosphatidylinositol) anchor that is required for membrane attachment of cell-surface proteins, most notably the variant-surface glycoproteins. The de novo synthesis of GPEtn and GPCho starts with the generation of phosphoethanolamine and phosphocholine by ethanolamine and choline kinases via the Kennedy pathway. Database mining revealed two putative C/EKs (choline/ethanolamine kinases) in the Trypanosoma brucei genome, which were cloned, overexpressed, purified and characterized. TbEK1 (T. brucei ethanolamine kinase 1) was shown to be catalytically active as an ethanolamine-specific kinase, i.e. it had no choline kinase activity. The Km values for ethanolamine and ATP were found to be 18.4±0.9 and 219±29 μM respectively. TbC/EK2 (T. brucei choline/ethanolamine kinase 2), on the other hand, was found to be able to phosphorylate both ethanolamine and choline, even though choline was the preferred substrate, with a Km 80 times lower than that of ethanolamine. The Km values for choline, ethanolamine and ATP were 31.4±2.6 μM, 2.56±0.31 mM and 20.6±1.96 μM respectively. Further substrate specificity analysis revealed that both TbEK1 and TbC/EK2 were able to tolerate various modifications at the amino group, with the exception of a quaternary amine for TbEK1 (choline) and a primary amine for TbC/EK2 (ethanolamine). Both enzymes recognized analogues with substituents on C-2, but substitutions on C-1 and elongations of the carbon chain were not well tolerated.
Keywords: choline kinase, ethanolamine kinase, Kennedy pathway, Trypanosoma brucei
Abbreviations: C/EK, choline/ethanolamine kinase; EK, ethanolamine kinase; GPCho, glycerophosphocholine; GPEtn, glycerophosphoethanolamine; GPI, glycosylphosphatidylinositol; GPSer, glycerophosphoserine; HPTLC, high-performance TLC; LB, Luria–Bertani; MALDI, matrix-assisted laser-desorption ionization; ORF, open reading frame; PtdCho, phosphotidylcholine; PtdEtn, phosphatidylethanolamine; RT, reverse transcription; Tb, Trypanosome brucei; TEV, tobacco etch virus; TOF, time-of-flight; UTR, untranslated region; VSG, variant-surface glycoprotein
INTRODUCTION
The zwitterionic glycerophospholipids GPCho (glycerophosphocholine) and GPEtn (glycerophosphoethanolamine) are the two most abundant phospholipid species in eukaryotic cells and many bacteria [1]. In the parasitic protozoan Trypanosoma brucei, the agent of African sleeping sickness, GPCho accounts for approximately half of all phospholipids in both life-cycle stages, whereas GPEtn forms between 16 and 21% [2]. These phospholipids contribute an important structural role to the membrane and, in addition, determine membrane fluidity and cell-surface charge. Their biosynthesis and utilization is, not surprisingly, implicated in a variety of cellular processes [1]. In mammals, GPCho is an important source of signalling molecules, and the biosynthetic intermediate PtdCho (phosphatidylcholine) has been reported to be a mitogen required for DNA synthesis induced by growth factors [3]. Owing to its physicochemical properties, GPEtn has a tendency to form non-bilayer structures, and its incorporation into membranes affects the processes of membrane trafficking and the folding, stabilization and incorporation of certain proteins into membranes [4]. Moreover, GPEtn is utilized in the biosynthesis of GPI (glycosylphosphatidylinositol), a complex glycolipid that anchors proteins to the exterior leaflet of eukaryotic plasma membranes [5–7]. This is particularly important for the bloodstream form of T. brucei, which expresses a dense cell-surface coat (107 molecules per parasite) of a GPI-anchored VSG (variant-surface glycoprotein). This coat prevents attack by components of the innate immune system and, by undergoing antigenic variation, allows the parasite to evade a specific immune response by the host.
The biosynthesis of the GPI anchor has been genetically [8–10] and chemically [11] validated as a drug target in the bloodstream form of T. brucei. Thus disruption of the biosynthesis of GPEtn could prevent the formation of mature GPI-anchored VSG and lead to clearance of the parasite by the host's immune system.
Owing to the major structural and functional roles of GPCho and GPEtn in T. brucei, disruption of their biosynthetic pathways is likely to interfere with the parasite biology in multiple ways and thus the enzymes involved become of interest as possible novel targets for chemotherapy. Indeed, it has been proposed that the mode of action of lysophospholipid analogues (e.g. miltefosine, edelfosine or ilmofosine) used currently against other trypanosomatid parasites, i.e. Leishmania major, are involved in the inhibition of GPCho biosynthesis [12].
GPEtn and GPCho are synthesized de novo via two branches of the same metabolic pathway, the Kennedy pathway [13] (Figure 1). Ethanolamine and choline are readily phosphorylated to generate PtdEtn (phosphatidylethanolamine) and PtdCho, which are subsequently activated to high-energy donors, CDP-ethanolamine and CDP-choline respectively, before they are coupled to diacylglycerol to form GPEtn and GPCho. For this reason, the two branches of the Kennedy pathway are often referred to as the CDP-ethanolamine or CDP-choline pathways. A second major pathway for the formation of GPEtn is via GPSer (glycerophosphoserine) decarboxylation: GPSer, which is formed through the action of a phosphatidylserine synthase, is decarboxylated by a phosphatidylserine decarboxylase to form GPEtn. In most eukaryotic cells, GPCho can also be generated from GPEtn by three methylation steps. However, according to the tritryp genomes (http://www.genedb.org), these methyltransferase genes are not present in either T. brucei or Trypanosoma cruzi, but interestingly are present in L. major; this has been confirmed experimentally (T.K. Smith, unpublished work).
Figure 1. Pathways for the biosynthesis of the aminoglycerophospholipids GPCho and GPEtn.

The two branches of the Kennedy pathway are (a) the CDP-choline pathway and (b) the CDP-ethanolamine pathway. The relevant steps are discussed in the text. CK, choline kinase (Tb11.18.0017/Tb927.5.1140); CCT, CTP:phosphocholine cytidyltransferase (Tb10.389.0730); CPT, diacylglycerol:CDP-choline cholinephosphotransferase (Tb10.6k15.1570); EK, ethanolamine kinase (Tb11.18.0017/Tb927.5.1140); ECT, CTP:phosphoethanolamine cytidyltransferase (Tb11.01.5730); EPT, diacylglycerol:CDP-ethanolamine ethanolaminephosphotransferase (Tb10.6k15.1570); PSD, phosphatidylserine decarboxylase (Tb09.211.1610); PEMT, phosphatidylethanolamine methyltransferases; DAG, diacylglycerol. The dashed box indicates methyltransferase enzymes which are not present in either T. brucei or T. cruzi, but are present in L. major.
Although the presence of the Kennedy pathway was demonstrated in T. brucei [14], the constituent enzymes have never been characterized. In the present paper, we report investigations into the first steps of both branches of the Kennedy pathway, involving molecular cloning, recombinant expression, purification and detailed kinetic characterization of both ethanolamine and choline kinases (EK and CK respectively) from T. brucei.
EXPERIMENTAL
Organisms and reagents
T. brucei strain 427 was used as a source of genomic DNA. Escherichia coli strains DH5α, XL-1 blue and TOP10 (Invitrogen) competent cells were used for routine manipulation, and strain BL21-Gold(DE3) (Novagen) for protein overexpression. All chemicals were purchased from either Sigma–Aldrich or Fluka. Restriction endonucleases and DNA-modifying enzymes were from New England Biolabs or Promega. [2-3H]Ethanolamine (50.0 Ci/mmol) and [methyl-3H]choline (82.0 Ci/mmol) were purchased from GE Healthcare.
RNA isolation and cDNA synthesis
Total RNA was isolated from bloodstream-form T. brucei using the RNeasy mini kit (Qiagen). A TbC/EK2 (T. brucei choline/ethanolamine kinase 2)-specific cDNA was generated and amplified using a mini-exon-specific forward primer, 5′-CGCGGATCCGAACGCTATTATTAGAACAGTTTCTGTAC-3′, in combination with an ORF (open reading frame)-specific reverse primer, 5′-CTTCTGTTCCACGGGTATAGTC -3′, specific for the sequence of TbC/EK2 using the SuperScript III One step RT (reverse transcription)–PCR kit with Platinum Taq (Invitrogen). The amplified cDNAs were purified (QIAquick PCR purification kit, Qiagen), ligated into pCR®-Blunt II TOPO® (Invitrogen) and sequenced.
Cloning of the TbC/EK1 gene and construction of the pET15b-TbC/EK1 expression vectors
Using the Saccharomyces cerevisiae EK gene sequence as a query, two putative C/EK (choline/ethanolamine kinase) genes were identified in the T. brucei genome database (http://www.genedb.org) using tBlastN. The two putative ORFs (Tb11.18.0017 for TbC/EK1, and Tb927.5.1140 for TbC/EK2) together with ∼350 bp of their 5′- and 3′-UTRs (untranslated regions), were amplified from T. brucei strain 427 genomic DNA using Pfu DNA polymerase and the forward and reverse primers 5′-ATAAGTAAGCGGCCGCCCGCCTAAGTTAGAAGTTGCGCT-3′ and 5′-ATAAGTAAGCGGCCGCTCCAATAGCTCCAGGGAAGGAAAGGGACG-3′ for TbC/EK1; and 5′-ATAAGTAAGCGGCCGCAAGTGCGTTGTGCAGGTCGGCGACGGT-3′ and 5′-ATAAGTAAGCGGCCGCCGTTGGAAGGAGGAAAACGGCCGAGG-3′ for TbC/EK2. The resulting 2.1 kb (TbC/EK1 and UTRs) and 3.7 kb (TbC/EK2 and UTRs) fragments were cloned into pCR®-Blunt-II TOPO®. Clones were sequenced and compared with the annotated GenBank® sequences. A BamHI restriction site, internal to the TbC/EK1 ORF was silenced by site-directed mutagenesis using the forward primer 5′-GTGTTGAGGGGGATAAGCGAATCCATCGCATGGTTCAGC-3′, the reverse primer 5′-GCTGAACCATGCGATGGATTCGCTTATCCCCCTCAACAC-3′ and the QuikChange® site-directed mutagenesis kit (Stratagene).
The TbC/EK1 ORF was PCR-amplified from the TOPO® construct with Pfu polymerase using the forward and reverse primers 5′-GGAATTCCATATGATGGAGGTGGCTGTGGGGCAC-3′ and 5′-CGCGGATCCGCGTTATGAAGATGCACTAAACTC-3′ respectively. The amplicon was purified (QIAquick PCR purification kit; Qiagen), ligated into pCR®-Blunt II TOPO® and sequenced. Using the NdeI and BamHI restriction sites (underlined in primer sequences), the putative TbC/EK1 was ligated into the expression vector pET-15b (Novagen) modified with a TEV (tobacco etch virus) protease cleavage site (in place of a thrombin cleavage site) using the same restriction sites generating the pET-15bTEV-TbC/EK1 construct. The same procedure was employed to ligate the putative TbC/EK2 into the same vector using the forward and reverse primers 5′-GGAATTCCATATGATGGCATTACGACCGTTCCCG-3′ and 5′-CGCGGATCCGCGTCAGGAAAGAAGCCCCTTCTC-3′ respectively.
The D287A and D267A mutants of TbC/EK1 were generated using the QuikChange® site-directed mutagenesis kit with the forward and reverse primers 5′-GGCGCACTGAAGATTATTGcCTTCGAGTATGCAAAACG-3′ and 5′-CGTTTTGCATACTCGAAGgCAATAATCTTCAGTGCGCC-3′ respectively for the D287A mutant and the forward and reverse primers 5′-GAGTACGTGCCACAATGcCCTGCTCAGCGGC-3′ and 5′-GCCGCTGAGCAGGgCATTGTGGCACGTACTC-3′ respectively for the D267A mutant (the lower case letters in the primers highlight the mutated bases). All DNA sequencing was performed by The Sequencing Service (College of Life Sciences, University of Dundee; http://www.dnaseq.co.uk) using Applied Biosystems Big-Dye version 3.1 chemistry on an Applied Biosystems model 3730 automated capillary DNA sequencer.
Expression and purification of TbC/EK1 and TbC/EK2
Overproduction and sample preparation
The pET-15bTEV-TbC/EK1 and the pET-15bTEV-TbC/EK2 constructs were transformed in BL21-Gold(DE3) cells and clones selected on LB (Luria–Bertani) agar plates containing carbenicillin (100 μg/ml). Single colonies were grown at 37 °C in LB medium containing carbenicillin (50 μg/ml) until the D600 was between 0.5 and 0.7, at which point recombinant protein expression was induced with 1 mM IPTG (isopropyl β-D-thiogalactoside) and grown for 16 h at room temperature (25 °C). Cells were harvested by centrifugation at 3500 g for 20 min at 4 °C before being suspended in buffer A (50 mM Tris/HCl, pH 8.0, 300 mM NaCl and 10 mM imidazole). Cells were lysed in the presence of DNase I, either by sonication or using a French press, and the lysate was cleared by centrifugation at 35000 g for 30 min at 4 °C.
Purification and gel filtration
The cleared lysate was applied to a 1 ml HisTrap™ FF crude column or a 5 ml HisTrap™ column (GE Healthcare) pre-loaded with Ni2+. Unbound proteins were removed by washing the column with 15 column vol. of buffer A, whereas TbC/EK1 and TbC/EK2 were eluted with an imidazole gradient in the same buffer.
Fractions containing TbC/EK1 or TbC/EK2 were pooled, dialysed against 50 mM Tris/HCl (pH 8) and 300 mM NaCl and purified further through a Superdex200 16/60 gel-filtration column (GE Healthcare), run in the same buffer at a rate of 1 ml/min. Low and High Molecular Weight Calibration Kits (GE Healthcare) were used to determine the molecular mass and the oligomeric state of the proteins. Purity of protein samples was assessed by SDS/PAGE (4–12 and 10% gels) and MALDI (matrix-assisted laser-desorption ionization)–TOF (time-of-flight)-MS (Proteomics Facility, College of Life Sciences, University of Dundee).
Assay of TbC/EK activity
EK or CK activity was measured by a spectrophotometric coupled assay as described previously [15]. The assay contained 50 mM Mops (pH 7.8), 150 mM KCl, 6 mM MgCl2, 0.5 mg/ml BSA, 1 mM phosphoenolpyruvate, 0.5 mM NADH, 7 units/ml pyruvate kinase, 32 units/ml lactate dehydrogenase and 2 μg/ml TbC/EK1 or 1 μg/ml TbC/EK2. Concentrations of ATP and ethanolamine (or choline) were varied, and the rate of the reaction was monitored by the decrease in absorbance at 340 nm (a consequence of NADH oxidation) in a Shimadzu UV-1601 spectrophotometer. The Enzyme Kinetics module of SigmaPlot (SPSS) was used to analyse and fit the kinetic data.
The pH optimum was assessed by measuring TbC/EK1 and TbC/EK2 activities at various pHs: sodium acetate (pH 5.0), Bis-Tris/propane or Mops (pH 6.5), Tris/HCl (pH 7.0, 8.0 and 8.5), Mops (pH 6.0, 7.0, 7.4 and 7.8) and glycine (pH 9.0). Various ethanolamine and choline analogues were tested as substrates at least in triplicate on 96-well plates using a SpectraMAX 340PC plate reader (Molecular Devices), and those deemed as non-substrates were tested as inhibitors by pre-incubation at 1 mM for 5 min before the addition of ethanolamine or choline (0.1 mM).
Direct EK and CK activity assays were performed by assessing the production of PtdEtn or PtdCho, using a modified method of Kim et al. [16]. Briefly, 2 μg of purified protein was incubated with a reaction mixture (total volume of 50 μl) of 100 mM Mops (pH 7.8), 6 mM MgCl2 and 5 mM ATP, with either 2 mM ethanolamine and 0.2 μCi of [3H]ethanolamine or 2 mM choline and 0.2 μCi of [3H]choline at 37 °C for 20 min and quenched by boiling at 100 °C for 5 min. Substrates and products were separated by HPTLC (high-performance TLC) using silica 60 plates with methanol/0.6% NaCl/ammonium hydroxide (10:10:1, by vol.) as solvent. Radiolabelled species were detected by fluorography at −70 °C after spraying with En3Hance and using Kodak XAR-5 film with an intensifying screen. Unlabelled ethanolamine and PtdEtn standards were run in parallel and visualized by spraying with ninhydrin (0.2% in water-saturated butan-1-ol). Unlabelled PtdCho was run in parallel and visualized by iodine staining.
RESULTS AND DISCUSSION
Genomic identification and cloning of TbC/EK1 and TbC/EK2
Two putative C/EKs were identified in the T. brucei genome database (http://www.genedb.org); these putative ORFs were PCR-amplified from genomic DNA, cloned and sequenced. Two potential initiating methionine residues were detected in the TbC/EK2 ORF; this ambiguity was resolved by amplification and sequencing of the first ∼350 bp of the RT–PCR product using total RNA, a forward primer against the mini-exon and a reverse primer specific for the TbC/EK2 ORF. From the cDNA sequence (Supplementary Figure S1 at http://www.BiochemJ.org/bj/415/bj4150135add.htm), the second methionine residue was identified as being the initiating residue. Interestingly, there seems to be an extra ‘stuffer’ sequence (65 bp) between this and the mini-exon (Supplementary Figure S1), which may represent some form of regulatory element, a concept we are currently investigating.
The predicted molecular masses of the deduced amino acid sequences are 49.1 kDa (431 amino acids) for TbC/EK1 and 70.1 kDa (628 amino acids) for TbC/EK2. An alignment of the translated sequences of TbC/EK1 and TbC/EK2 with previously characterized or putative C/EKs from other organisms (Figure 2) shows significant similarities. Even though the N-terminal region is not generally well conserved among C/EKs, the C-terminal portion contains two highly conserved regions. Brenner's motif (Figure 2) has been identified in many enzymes catalysing phosphotransfer reactions, such as protein kinases and atypical kinases (the superfamily that includes the C/EK family) [17]. The second conserved cluster (Figure 2) is found specifically in members of the C/EK family and is referred to as the choline kinase motif. Previous site-directed mutagenesis studies and structural studies in Caenorhabditis elegans CKα2 [18,19] and the analysis of the crystal structures of human CKα [20] have highlighted the importance of several of the amino acids in substrate binding. Of particular importance are the two aspartate residues marked by an asterisk in the alignment (Figure 2). These residues are involved in the binding of ATP and the stabilization of the transition state of the reaction, and their mutation leads to complete enzyme inactivation [18,20].
Figure 2. A Tcoffee alignment of the predicted amino acid sequences of TbEK1 (formerly TbC/EK1) and TbC/EK2 with CKs and EKs of other eukaryotic organisms: T. cruzi (Swiss-Prot accession number Q4E3A9), L. major (Q4FYH6 and Q4FWX2) and Homo sapiens (Q9HBU6).

Residues are shaded according to percentage similarity using the Jalview 2.08.1 software. The boxes highlight the choline kinase motif and Brenner's phosphotransferase motif. The two aspartate residues marked by an asterisk were mutated in TbEK1 to generate the catalytically inactive mutants D267A and D286A.
Not surprisingly, TbC/EK1 and TbC/EK2 are most closely related to C/EKs from other kinetoplastids. In the region of the alignment shown in Figure 2, TbC/EK1 is 63% identical with a putative C/EK from T. cruzi and TbC/EK2 is 40% identical with a putative C/EK from L. major. The similarity towards previously characterized C/EKs from human, yeast, C. elegans and Plasmodium falciparum is much lower (results not shown), with the exception of Brenner's and the choline kinase motifs.
Recombinant expression and purification of TbC/EK1 and TbC/EK2
In order to confirm that the putative TbE/CK1 and TbE/CK2 are catalytically active, the two ORFs were cloned into the vector pET-15bTEV. This vector encodes an N-terminal hexahistidine epitope tag followed by a TEV protease site, which allowed purification of the recombinant proteins via Ni2+ ion-affinity chromatography (Figures 3A and 3B). MALDI–TOF-MS of the recombinant TbC/EK1 and TbC/EK2 were very close to their theoretical masses of 51655 and 72614 Da respectively. The proteins TbC/EK1 and TbC/EK2 were purified further by gel filtration (Figure 3C). Elution from the gel-filtration column showed that both TbC/EK1 (black arrow) and TbC/EK2 (white arrow) exhibited a dimeric state in solution (Figure 3D). This is similar to most of the C/EKs from other sources [21–23], with the notable exception of the monomeric P. falciparum CK [15]. Typical yields were 10 and 15 mg/l of bacterial culture for TbC/EK1 and TbC/EK2 respectively. TbC/EK1 was found to lose activity over time, being lost completely after a couple of days. It is also prone to precipitation upon freeze–thawing, and therefore assays were preformed on freshly prepared batches. TbC/EK2, on the other hand, was stable, and freeze–thawing did not lead to loss of activity or precipitation.
Figure 3. Expression and purification of recombinant TbEK1 (formerly TbC/EK1) and TbC/EK2 in E. coli.

(A) TbEK1 protein samples from each purification step were separated by SDS/PAGE (4–12% gel) and stained with Coomassie Brilliant Blue. Lane 1, total E. coli cellular protein after induction; lane 2, cleared cell lysate; lane 3, Ni2+ column wash at 20 mM imidazole; lane 4, Ni2+ column wash at 50 mM imidazole; lanes 5–8, subsequent elutions from the Ni2+ column at 250 mM imidazole. MW, molecular mass (in kDa). (B) TbC/EK2 protein samples from each purification step were separated by SDS/PAGE (10% gel) and stained with Coomassie Brilliant Blue. Lane 1, cleared E. coli cell lysate; lanes 2–4, washes; lanes 5–8, peak of TbC/EK2 elution at 220 mM imidazole. MW, molecular mass (in kDa). (C) Protein samples after gel filtration separated by SDS/PAGE. Lane 1, TbEK1 (4–12% gel); lane 2, TbC/EK2 (10% gel). MW, molecular mass (in kDa). (D) Determination of native molecular masses of TbEK1 and TbC/EK2. The elution from the gel-filtration column indicates that both TbEK1 (black arrow) and TbC/EK2 (white arrow) are present as dimers. The following proteins were used as standards: (a) ferritin (440 kDa), (b) conalbumin (75 kDa), (c) carbonic anhydrase (29 kDa), and (d) RNase A (13.7 kDa).
Kinetic analysis of TbC/EK1 and TbC/EK2
Previously characterized C/EKs from various sources [15,21–25] have exhibited different specificity towards the two substrates, choline and ethanolamine. It was therefore necessary to test the activity of the purified proteins in order to ascertain whether they were able to catalyse the phosphorylation of both choline and ethanolamine or if they showed selectivity to each branch of the Kennedy pathway.
TbC/EK1 was found to be able to catalyse the formation of PtdEtn from ethanolamine, in an ATP- and Mg2+-dependent manner (Figures 4A and 4B). However, the enzyme was unable to catalyse the formation of PtdCho from choline, in the presence of ATP and magnesium (Tables 1 and 2). This restricts the specificity of this kinase to the CDP-ethanolamine branch of the Kennedy pathway and, for this reason, TbC/EK1 was renamed TbEK1. To our knowledge, this is the first ethanolamine-specific kinase to be recombinantly expressed, purified to homogeneity and extensively characterized.
Figure 4. Analysis of PtdEtn formation by TbEK1 (formerly TbC/EK1).

(A) Reaction catalysed by TbEK1. EtN, ethanolamine. (B) The substrate ethanolamine (EtN) and the product PtdEtn of the reaction were separated by HPTLC, as described in the Experimental section. Lanes 1–6, autoradiography detection. Lane 1, [3H]ethanolamine standard; lane 3, standard reaction with TbEK1; lanes 2 and 4, no enzyme or no Mg2+ added to the reaction mixture respectively (negative controls); lanes 5 and 6, standard reaction with TbEK1 mutants D267A and D286A respectively; lane 7, ethanolamine standard; lane 8, PtdEtn standard detected by ninhydrin. (C) Enzymatic activities of wild-type (WT) TbEK1 and mutants D267A and D286A assessed by spectrophotometric coupled assay as described in the Experimental section. Results are means±S.D. (n≥3). The inset shows an SDS/PAGE gel of the purified proteins (wild-type and mutants). MW, molecular mass (in kDa).
Table 1. Kinetic values for recombinantly expressed and purified TbEK1 (formerly TbC/EK1) and TbC/EK2.
Results are means±S.D. (n≥3).
| Enzyme | Vmax (μmol/min per mg of protein) | Km choline (μM) | Km ethanolamine (μM) | Km ATP (μM) | kcat (s−1) | kcat/Km choline (s−1·mM−1) | kcat/Km ethanolamine (s−1·mM−1) | kcat/Km ATP (s−1·mM−1) |
|---|---|---|---|---|---|---|---|---|
| TbEK1 | 7.62±0.07 | 0.0±0.0 | 18.4±0.9 | 219.2±29 | 6.56 | – | 356.53 | 29.93 |
| TbC/EK2 | 10.67±0.18 | 31.4±2.6 | 2560±310 | 20.6±1.96 | 12.91 | 411.25 | 5.04 | 626.86 |
Table 2. Substrate specificity of TbEK1 (formerly TbC/EK1) and TbC/EK2 to utilize analogues modified at the amino group of the acceptor substrate.
Results are means±S.D. (n≥3).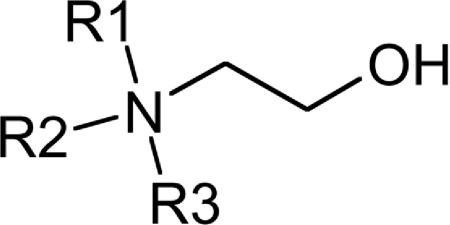
| R1 | R2 | R3 | Compound | TbEK1 relative rate* | TbC/EK2 relative rate† |
|---|---|---|---|---|---|
| -CH3 | -CH3 | -CH3 | Choline | 0.03±1.59 | 100.00±7.97 |
| -H | -H | -H | Ethanolamine | 100.00±1.65 | 33.34±3.34 |
| -H | -H | -CH3 | N-Methylethanolamine | 106.58±9.18 | 135.90±3.64 |
| -H | -CH3 | -CH3 | N,N-Dimethylethanolamine | 76.54±4.86 | 153.33±2.77 |
| -H | -H | -CH2CH3 | N-Ethylethanolamine | 97.38±1.59 | 150.57±8.40 |
| -H | -CH2CH3 | -CH2CH3 | N,N-Diethylethanolamine | 47.75±3.98 | 155.92±4.32 |
| -H | -H | -CH2CH2OH | Diethanolamine | 88.26±0.78 | 138.86±6.30 |
*Analogues are compared with ethanolamine at the constant concentration of 1 mM and a saturating concentration of ATP to give a relative rate (ethanolamine=100).
†Analogues are compared with choline at the constant concentration of 1 mM and a saturating concentration of ATP to give a relative rate (choline=100).
Kinetic analysis showed that TbEK1 displayed a pH optimum at pH 8.0 (Figure 5A), with a Km of 18.4±0.9 μM for ethanolamine (Figure 5B) and a Km of 219±29 μM for ATP (Figure 5C), which is in reasonable agreement with a previously reported Km value of 2.75 μM for EK activity from crude trypanosome homogenates [14]. The Vmax of TbEK1 is 7.62±0.07 μmol/min per mg of protein, corresponding to a kcat of 6.56 s−1 (Table 1). This turnover rate/specific activity compares favourably with the previously reported activities of 346 nmol/min per mg of protein for the yeast EK [16] and 10.73 μmol/min per mg of protein for a soya bean EK [25]. Two catalytically inactive mutants of TbEK1 were generated by substitution of alanine for Asp267 and Asp286 (Figures 4B and 4C), thus confirming the crucial importance of these two residues in the catalytic mechanism of this kinase.
Figure 5. Kinetic analysis of TbEK1 (formerly TbC/EK1).

(A) EK activity was measured spectrophotometrically as a function of pH as described in the Experimental section. Results are means±S.D. of three measurements. (B, C) Determination of TbEK1 Michaelis–Menten constants for ethanolamine and ATP (insets are Lineweaver–Burk plots). (B) ATP concentration was held constant (5 mM), while ethanolamine concentration was varied. (C) Ethanolamine concentration was held constant (2 mM), while ATP concentration was varied.
TbC/EK2, on the other hand, was found to be able to phosphorylate both ethanolamine (Figure 6B) and choline (Figures 6A and 6C), even though choline was the preferred substrate (Tables 1 and 2), with a Km 80 times lower than that of ethanolamine. TbC/EK2 was also dependent upon ATP and Mg2+ for activity (Figures 6B and 6C) with a pH optimum of 7.8 (Figure 7A). Kinetic analysis showed that TbC/EK2 displayed Km values for choline, ethanolamine and ATP of 31.4±2.6 μM, 2.56±0.31 mM and 20.6±1.96 μM respectively with a Vmax of 10.67±0.18 μmol/min per mg of protein and a kcat of 12.91 s−1 (Figures 7B–7D and Table 1). This value is in the range of activities reported for other eukaryotic C/EKs, and more than ten times higher than the CK from P. falciparum [15].
Figure 6. Analysis of PtdCho formation by TbC/EK2.

(A) Reaction catalysed by TbC/EK2. Cho, choline. (B) The substrate ethanolamine (EtN) and the product PtdEtn of the reaction were separated by HPTLC as described in the Experimental section. Lanes 1–7, ninhydrin detection. Lane 1, ethanolamine standard; lane 2, PtdEtn standard; lane 3, standard reaction with TbC/EK2; lanes 4–6, no Mg2+, no ethanolamine or no ATP added to the reaction mixture respectively (negative controls). (C) The substrate choline (Cho) and the product PtdCho of the reaction were separated by HPTLC as described in the Experimental section. Lanes 1–6, autoradiography detection; lane 7, detection by iodine staining. Lane 1, [3H]choline standard; lane 2, standard reaction with TbC/EK2; lanes 3–6, no enzyme, no Mg2+, no [3H]-choline or no ATP added to the reaction mixture respectively (negative controls); lane 7, PtdCho standard visualized by iodine staining.
Figure 7. Kinetic analysis of TbC/EK2.

(A) CK activity was measured spectrophotometrically as a function of pH as described in the Experimental section. Results are means±S.D. of three measurements. (B–D) Determination of TbC/EK2 Michaelis–Menten constants for choline, ethanolamine and ATP (insets are Lineweaver–Burk plots). ATP concentration was held constant (5 mM), while choline (B) or ethanolamine (EtN) (C) concentrations were varied. (D) Choline concentration was held constant (2 mM), while ATP concentration was varied.
Since the TbC/EK2 Km value for ethanolamine is several orders of magnitude higher than that of TbEK1 and the fact that levels of ethanolamine in human plasma are in the micromolar range [26], one can reasonably assume that TbEK1 accounts for the vast majority of the ethanolamine-phosphate formation in the trypanosome. There were no differences in the kinetics between the hexahistidine-tagged and untagged (TEV-cleaved) versions of either TbEK1 or TbC/EK2, thus the tagged versions were used as this decreases preparation time and increases stability, which is especially important for TbEK1.
Acceptor specificity of TbEK1 and TbC/EK2
The ability of the two enzymes to utilize chemically modified substrates for the phosphotransfer reaction was investigated using a variety of ethanolamine and choline analogues, with modifications either at the amino or the hydroxy moieties, or with additional substituents on the two-carbon backbone. Each analogue was compared with ethanolamine (for TbEK1) or choline (for TbC/EK2) at a constant substrate concentration (1 mM) and a fixed saturating concentration of ATP to give a relative rate (ethanolamine/choline=100) (Tables 2–4).
TbEK1 was able to tolerate various modifications at the amino group, including secondary and tertiary amino modifications, despite having been shown earlier not to process choline (Table 2). As shown in Table 3, single methyl and ethyl substituents on the carbon backbone at C-2 [R(−)2-aminobutan-1-ol, S(+)2-aminobutan-1-ol, R(−)2-aminopropan-1-ol and S(+)2-aminopropan-1-ol] are well tolerated, but the introduction of a second methyl group at C-2 (2-amino-2-methylpropan-1-ol) or a negatively charged carboxy group (L-serine) resulted in greatly reduced or non-detectable activity respectively. Methyl substitutions on C-1 (R(−)1-aminopropan-2-ol and S(+)1-aminopropan-2-ol) diminished the ability of the compound to act as a substrate, but to varying degrees in a stereospecific fashion. The increase of the carbon backbone length by one methylene unit (3-aminopropan-1-ol) led to a pronounced decrease in activity, whereas the increase by an ethylene unit (4-aminobutan-1-ol), eliminated substrate activity completely.
Table 3. Substrate specificity of TbEK1 (formerly TbC/EK1) and TbC/EK2 to utilize analogues modified by extra substitutents on the carbon backbone of the acceptor substrate.
Results are means±S.D. (n≥3).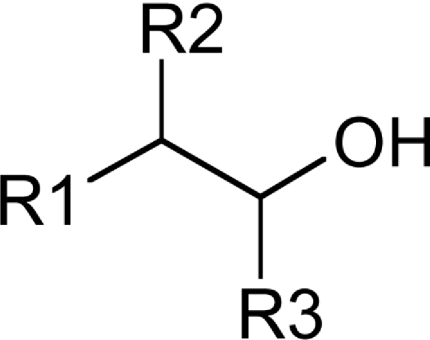
| R1 | R2 | R3 | Compound | TbEK1 relative rate* | TbC/EK2 relative rate† |
|---|---|---|---|---|---|
| -N+(CH3) 3 | -H | -H | Choline | 0.03±1.59 | 100.00±7.97 |
| -NH2 | -H | -H | Ethanolamine | 100.00±1.65 | 33.34±3.34 |
| -NH2 | -CH3 | -H | R(−)-2-Aminopropan-1-ol | 107.91±2.77 | 109.94±15.42 |
| -NH2 | -CH3 | -H | S(+)-2-Aminopropan-1-ol | 93.75±4.02 | 22.38±1.87 |
| -NH2 | -(CH3)2 | -H | 2-Amino-2-methylpropan-1-ol | 25.58±3.98 | 46.41±6.57 |
| -NH2 | -CH2CH3 | -H | S(+)-2-Aminobutan-1-ol | 114.07±5.84 | 40.72±4.93 |
| -NH2 | -CH2CH3 | -H | R(−)-2-Aminobutan-1-ol | 101.74±5.76 | 130.40±9.10 |
| -NH2 | -COOH | -H | L-Serine | 0.94±2.25 | 0.00±0.00 |
| -NH2 | -H | -CH3 | R(−)-1-Aminopropan-2-ol | 52.41±4.50 | 7.16±0.60 |
| -NH2 | -H | -CH3 | S(+)-1-Aminopropan-2-ol | 6.07±2.13 | 1.37±0.15 |
| -CH2NH2 | -H | -H | 3-Aminopropan-1-ol | 37.52±2.96 | 44.52±4.33 |
| (CH2)2NH2 | -H | -H | 4-Aminobutan-1-ol | 0.05±1.93 | 59.95±6.00 |
*Analogues are compared with ethanolamine at the constant concentration of 1 mM and a saturating concentration of ATP to give a relative rate (ethanolamine=100).
†Analogues are compared with choline at the constant concentration of 1 mM and a saturating concentration of ATP to give a relative rate (choline=100).
TbC/EK2 was able to accept analogues modified at the amino group as substrates of the reaction; in fact, secondary and tertiary amino modifications were better than choline. Notably, of the compounds tested, ethanolamine was the poorest acceptor substrate (Table 2); these findings are in contrast with those with CK from S. cerevisiae [24], where N-methylethanolamine and N,N-dimethylethanolamine were as poor substrates as ethanolamine. Compounds with methyl and ethyl alterations on C-2 are good substrates only in the (S) configuration; R(−)2-aminobutan-1-ol and R(−)2-aminopropan-1-ol are much poorer substrates, as well as 2-amino-2-methylpropan-1-ol, which bears two methyl groups at C-2 (Table 3). A reduction of activity is seen with the elongation of the carbon chain, but it is not abolished even when increased by an ethylene unit (4-aminobutan-1-ol). The introduction of a negative charge (L-serine), as well as the introduction of substituents at C-1 [R(−)1-aminopropan-2-ol and S(+)1-aminopropan-2-ol] are not tolerated (Table 3). That the enzyme is relatively more tolerant of substituents on C-2 is in agreement with the previous studies on the yeast orthologue [24]. As expected, modifications at the hydroxy group (Table 4) resulted in no detectable reactivity for either TbEK1 or TbC/EK2.
Table 4. Substrate specificity of TbEK1 (formerly TbC/EK1) and TbC/EK2 to utilize analogues modified at the hydroxy group of the acceptor substrate.
Results are means±S.D. (n≥3).
| R | Compound | TbEK1 relative rate* | TbC/EK2 relative rate† |
|---|---|---|---|
| -OH | Choline | 0.03±1.59 | 100.00±7.97 |
| -OH | Ethanolamine | 100.00±4.84 | 33.34±3.34 |
| -SH | 2-Aminoethanethiol | 0.01±0.51 | 0.96±0.30 |
| -(OCH3)2 | Aminoacetaldehyde dimethyl acetal | 0.73±0.63 | 0.57±0.12 |
| -SO3H | Taurine | 0.00±0.13 | 0.02±0.00 |
| -H | Ethylamine | 0.01±0.26 | 0.00±0.00 |
| -OSO3H | 2-Aminoethyl hydrogen sulfate | 0.54±2.31 | 0.00±0.00 |
| -OOH | Glycine | 0.35±0.72 | 0.85±0.11 |
| -OPO3H2 | Phosphoethanolamine | 0.25±3.32 | 1.35±0.29 |
| -OCH3 | 2-Methoxyethylamine | 0.00±0.02 | 1.49±0.28 |
| -PO3H2 | 2-Aminoethyl phosphonic acid | 0.08±0.66 | 0.80±0.18 |
*Analogues are compared with ethanolamine at the constant concentration of 1 mM and a saturating concentration of ATP to give a relative rate (ethanolamine=100).
†Analogues are compared with choline at the constant concentration of 1 mM and a saturating concentration of ATP to give a relative rate (choline=100).
Analogues deemed as non-substrates were tested as potential inhibitors by pre-incubation at 1 mM for 5 min before the addition of ethanolamine or choline (0.1 mM) to TbEK1 and TbC/EK2 respectively. However, none of the potential inhibitory analogues showed any inhibitory effect (<5%) on either TbEK1 or TbC/EK2 activity.
Previous studies in T. brucei [14] demonstrated that ethanolamine analogues, such as N-methylethanolamine, N-ethylethanolamine, N,N-dimethylethanolamine, S(+)-2-aminobutan-1-ol or R(−)-2-aminobutan-1-ol, can be recognized by the ethanolamine transport system, whereas choline is not. There was ambiguity as to whether these compounds were acting as inhibitors of the transporter or merely competing with ethanolamine to be transported into the cell. If the latter case were true, our results suggest these analogues could be phosphorylated by TbEK1 and/or TbC/EK2, and possibly proceed further in the Kennedy pathway, leading to the formation of unusual PtdEtn analogues, a concept we are currently pursuing.
Conclusions
African sleeping sickness is a neglected disease, which urgently requires new drug therapies. Characterizing fundamental differences in the biochemical pathways and enzymes therein of the parasite and its mammalian host, is an important first step towards this goal. Thus the present study represents the first identification and detailed molecular characterization of the two putative C/EKs in T. brucei. TbEK1 is committed to only the CDP-ethanolamine branch of the Kennedy pathway since it is unable to phosphorylate choline; TbC/EK2, on the other hand, was able to phosphorylate choline much more efficiently than ethanolamine. The substrate specificities for each kinase were determined, providing crucial information on the structural requirements for ligand recognition.
Analysis of the roles of TbEK1 and TbC/EK2 in parasite survival will be crucial to investigate their potential as drug targets for the development of new chemotherapeutic agents against African sleeping sickness and perhaps other protozoan parasites. The similarities in ligand recognition displayed by TbEK1 and TbC/EK2 may guide the design of inhibitors able to target both enzymes at once. Further work will characterize and assess the importance of all steps of the two branches of the Kennedy pathway, as well as their functional role(s) in the biology of the trypanosome.
Online data
Acknowledgments
This work was supported by a Wellcome Trust Senior Research Fellowship 067441 (to T. K. S.), Wellcome Trust grants 082596 and 083481 (to W. N. H.) and a Wellcome Trust Ph.D. studentship (to F. G.).
References
- 1.Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 1997;66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- 2.Patnaik P. K., Field M. C., Menon A. K., Cross G. A. M., Yee M. C., Bütikofer P. Molecular species analysis of phospholipids from Trypanosoma brucei bloodstream and procyclic forms. Mol. Biochem. Parasitol. 1993;58:97–105. doi: 10.1016/0166-6851(93)90094-e. [DOI] [PubMed] [Google Scholar]
- 3.Cuadrado A., Carnero A., Dolfi F., Jiménez B., Lacal J. C. Phosphorylcholine: a novel second messenger essential for mitogenic activity of growth factors. Oncogene. 1993;8:2959–2968. [PubMed] [Google Scholar]
- 4.Bürgemaister M., Birner-Grünberger R., Heyn M., Daum G. Contribution of different biosynthetic pathways to species selectivity of aminoglycerophospholipids assembled into mitochondrial membranes of the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2004;1686:148–160. doi: 10.1016/j.bbalip.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Imhof I., Canivenc-Gansel E., Meyer U., Conzelmann A. Phosphatidylethanolamine is the donor of the phosphorylethanolamine linked to the α-1,4-linked mannose of yeast GPI structure. Glycobiology. 2000;10:1271–1275. doi: 10.1093/glycob/10.12.1271. [DOI] [PubMed] [Google Scholar]
- 6.Menon A. K., Eppingerl M., Mayor S., Schwarz R. Phosphatidylethanolamine is the donor of the phosphoethanolamine group in trypanosome glycosyl-phosphatidylinositol. EMBO J. 1993;12:1907–1914. doi: 10.1002/j.1460-2075.1993.tb05839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menon A. K., Stevens V. L. Phosphatidylethanolamine is the donor of the ethanolamine residue linking a glycosylphosphatidylinositol anchor to protein J. Biol. Chem. 1992;267:15277–15280. [PubMed] [Google Scholar]
- 8.Nagamune K., Nozaki T., Maeda Y., Ohishi K., Fukuma T., Hara T., Schwarz R. T., Sutterlin C., Brun R., Riezman H., Kinoshita T. Critical roles of glycosylphosphatidylinositol for Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10336–10341. doi: 10.1073/pnas.180230697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang T., Milne K. G., Guther M. L. S., Smith T. K., Ferguson M. A. J. Cloning of the Trypanosoma brucei and Leishmania major genes encoding the GlcNAc-phosphatidylinositol de-N-acetylase of biosynthesis that is essential to the African sleeping sickness parasite. J. Biol. Chem. 2002;277:50176–50182. doi: 10.1074/jbc.M208374200. [DOI] [PubMed] [Google Scholar]
- 10.Ferguson M. A. J. Glycosylphosphatidylinositol biosynthesis validated as a drug target for African sleeping sickness. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10673–10675. doi: 10.1073/pnas.97.20.10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith T. K., Crossman A., Brimacombe J. S., Ferguson M. A. J. Chemical validation of GPI biosynthesis as a drug target against African sleeping sickness. EMBO J. 2004;23:4701–4708. doi: 10.1038/sj.emboj.7600456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbina A. J. Mechanisms of action of lysophospholipid analogues against trypanosomatid parasites. Trans. R. Soc. Trop. Med. Hyg. 2006;100:S9–S16. doi: 10.1016/j.trstmh.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy E. P., Weiss S. B. The function of cytidine coenzymes in the biosynthesis of phospholipids. J. Biol. Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- 14.Rifkin M. R., Strobos C. A. M., Fairlamb A. H. Specificity of ethanolamine transport and its further metabolism in Trypanosoma brucei. J. Biol. Chem. 1995;270:16160–16166. doi: 10.1074/jbc.270.27.16160. [DOI] [PubMed] [Google Scholar]
- 15.Choubey V., Guha M., Maity P., Kumar S., Raghunandan R., Maulik P. R., Mitra K., Halder U. C., Bandyopadhyay U. Molecular characterization and localization of Plasmodium falciparum choline kinase. Biochim. Biophys. Acta. 2006;1760:1027–1038. doi: 10.1016/j.bbagen.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Kim K., Kim K. H., Storey M. K., Voelker D. R., Carman G. M. Isolation and characterization of the Saccharomyces cerevisiae EKI1 gene encoding ethanolamine kinase. J. Biol. Chem. 1999;274:14857–14866. doi: 10.1074/jbc.274.21.14857. [DOI] [PubMed] [Google Scholar]
- 17.Scheef E. D., Bourne P. E. Structural evolution of the protein kinase-like superfamily. PLoS Comput. Biol. 2005;1:e49. doi: 10.1371/journal.pcbi.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan C., Kent C. Identification of critical residues of choline kinase A2 from Caenorhabditis elegans. J. Biol. Chem. 2004;276:17801–17809. doi: 10.1074/jbc.M401382200. [DOI] [PubMed] [Google Scholar]
- 19.Peisach D., Gee P., Kent C., Zhaohui X. The crystal structure of choline kinase reveals a eukaryotic protein kinase fold. Structure. 2003;11:703–713. doi: 10.1016/s0969-2126(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 20.Malito E., Sekulic N., Cun W., Too S., Konrad M., Lavie A. Elucidation of human choline kinase crystal structures in complex with the products ADP or phosphocholine. J. Mol. Biol. 2006;364:136–151. doi: 10.1016/j.jmb.2006.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gee P., Kent C. Multiple isoforms of choline kinase from Caenorhabditis elegans: cloning, expression, purification, and characterization. Biochim. Biophys. Acta. 2003;1648:33–42. doi: 10.1016/s1570-9639(03)00106-7. [DOI] [PubMed] [Google Scholar]
- 22.Ayoama C., Liao H., Ishidate K. Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res. 2004;43:266–281. doi: 10.1016/j.plipres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Kim K., Voelker D. R., Flocco M. T., Carman G. M. Expression, purification, and characterization of choline kinase, product of the CKI gene from Saccharomyces cerevisiae. J. Biol. Chem. 1998;273:6844–6852. doi: 10.1074/jbc.273.12.6844. [DOI] [PubMed] [Google Scholar]
- 24.Clary G. L., Tsai C. F., Guynn R. W. Substrate specificity of choline kinase. Arch. Biochem. Biophys. 1986;254:214–221. doi: 10.1016/0003-9861(87)90097-x. [DOI] [PubMed] [Google Scholar]
- 25.Wharfe J., Harwood J. L. Lipid metabolism in germinating seeds: purification of ethanolamine kinase from soya bean. Biochim. Biophys. Acta. 1976;575:102–111. doi: 10.1016/0005-2760(79)90135-8. [DOI] [PubMed] [Google Scholar]
- 26.Baba S., Watanabe Y., Geiyo F., Arakawa M. High-performance liquid chromatographic determination of serum aliphatic amines in chronic renal failure. Clin. Chim. Acta. 1984;136:49–56. doi: 10.1016/0009-8981(84)90246-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.