

Abstract
Spectroscopic responses of the potentiometric probe 2-(4-(dimethylamino)styryl)-1-methylpyridinium iodide (DASPMI) were investigated in living cells by means of a time- and space-correlated single photon counting technique. Spatially resolved fluorescence decays from single mitochondria or only a very few organelles of XTH2 cells exhibited three-exponential decay kinetics. Based on DASPMI photophysics in a variety of solvents, these lifetimes were attributed to the fluorescence from the locally excited state, intramolecular charge transfer state, and twisted intramolecular charge transfer state. A considerable variation in lifetimes among mitochondria of different morphologies and within single cells was evident, corresponding to high physiological variations within single cells. Considerable shortening of the short lifetime component (τ1) under a high-membrane-potential condition, such as in the presence of ATP and/or substrate, was similar to quenching and a dramatic decrease of lifetime in polar solvents. Under these conditions τ2 and τ3 increased with decreasing contribution. Inhibiting respiration by cyanide resulted in a notable increase in the mean lifetime and a decrease in mitochondrial fluorescence. Increased DASPMI fluorescence under conditions that elevate the mitochondrial membrane potential has been attributed to uptake according to Nernst distributions, delocalization of π-electrons, quenching processes of the methyl pyridinium moiety, and restricted torsional dynamics at the mitochondrial inner membrane. Accordingly, determination of anisotropy in DASPMI-stained mitochondria in living cells revealed a dependence of anisotropy on the membrane potential. The direct influence of the local electric field on the transition dipole moment of the probe and its torsional dynamics monitor changes in mitochondrial energy status within living cells.
INTRODUCTION
Cellular energy metabolism is best represented by mitochondrial membrane potential in the range of 150–180 mV, resulting from the proton gradient across the inner membrane (1,2). Stress conditions, such as exposure to reactive oxygen species, uncoupling agents, or ageing (3–5), strongly influence this potential. Thus, visualization of mitochondrial energy status is of significant physiological interest. Potentiometric probes such as cationic and zwitterionic styryl dyes, anionic and cationic cyanines, cationic rhodamines, and anionic and hybrid oxonols (6), are potential tools for determining the membrane potential of mitochondria in living cells.
The styryl dye 2-[4-(dimethylamino)styryl]-1-methylpyridinium iodide (DASPMI) is a low-toxicity (<5 × 10−7 M) specific vital stain of mitochondria in living cells (7–9). Fluorescence intensity of DASPMI is a dynamic measure for the membrane potential of mitochondria. DASPMI's very fast response to transmembrane potential, high sensitivity, and well-definable potentiometric response make it a valuable probe for precise monitoring of the dynamic changes in membrane potential across different mitochondria in a single living cell. The uptake of DASPMI in mitochondria follows Nernst relations (9) and allows for straightforward determination of the mitochondrial energization state (7,8). The mechanism of voltage-sensitive molecular fluorescence and its behavior under conditions that influence the mitochondria energy state in living cells, however, are not yet completely understood. It is difficult to interpret intensity measurements on the single- or sub-mitochondrial level because of thickness variations; however, this problem can be overcome by intensity-independent methods.
Previously, the photophysical properties of DASPMI were investigated by obtaining spectrally resolved fluorescence decays in solvents (10). A three-state model of fluorescence was derived to describe the fluorescence of DASPMI, wherein an initially excited molecule undergoes twist and further solvation. The fluorescence characteristics of DASPMI are influenced by both the polarity and viscosity of the environment, which in general are not discernible. The polarity-based influences manifest themselves as a red shift in the emission spectra, a blue shift in the absorption spectrum, and a decrease in quantum efficiency and lifetime in the case of increasing polarity of the environment. An increase in viscosity results in reduced torsional dynamics, increased lifetimes, and enhancement of fluorescence at the red edge of the emission spectrum. Although such differences exist, it is difficult to identify the individual influences of polarity and viscosity, due to the dependence of torsional dynamics of the dye on the intramolecular charge transfer (ICT) state.
In this article, we embark on a systematic approach to understand the behavior of DASPMI in living cells. From fluorescence decay determinations down to the level of single mitochondria, quantum efficiency and different speeds of mass accumulation of DASPMI have been identified as determinant factors in its behavior, revealing mitochondrial membrane potential. Extraction of emission fingerprints from different mitochondria within a single cell revealed variations in the contribution of different excited states.
Fluorescence anisotropy is a dimensionless quantity that is independent of absolute intensity and environmental influences such as static quenching. Anisotropy is a measure of the mobility of the dye molecules in relation to the excited-state lifetime. Emission anisotropy of DASPMI in mitochondria under high-membrane-potential conditions was high, suggesting restricted torsional flexibility under high-membrane-potential conditions. This observation was further supported by the acquisition of spatially resolved fluorescence decays under conditions that altered the mitochondrial membrane potential. A three-exponential-decay model of successive formation of the locally excited (LE) state, ICT state, and twisted ICT (TICT) state (10) was applied to investigate DASPMI fluorescence properties at different regions inside a single living XTH2 cell and even within single mitochondria. From the observed decays, partial insertion of DASPMI into the membrane wherein it undergoes electrochromism was deduced.
MATERIALS AND METHODS
Chemicals
DASPMI was obtained as a chromatographically pure preparation from the Bayer Research Laboratory (Leverkusen, Germany). Nigericin, oligomycin, carbonyl cyanide m-chlorophenylhydrazone (CCCP), and valinomycin were purchased from Sigma-Aldrich Chemie (Steinheim, Germany). Potassium cyanide (KCN) was purchased from Merck (Darmstadt, Germany). ATP was purchased from Serva Electrophoresis (Heidelberg, Germany). Saponin, glutamate, and malate were purchased from Sigma-Aldrich Chemie.
Cell culture and DASPMI staining
XTH2 cells, an endothelial cell line derived from Xenopus laevis tadpole hearts (11), were grown in Dulbecco's modified minimal essential medium (Gibco, Cergy-Pontoise, France) supplemented with 5% fetal calf serum. Cells seeded on 24-mm-diameter cover glasses at preconfluent density were used for experiments 48 h after trypsinization. These cells were then incubated with 3 μM DASPMI (diluted from aqueous stock solution) for 40 min at 28°C. For microscopic observations, these coverslips were mounted onto a custom-made chamber and rinsed with Hank's balanced salt solution (HBSS) diluted with 20% water to adjust the tonicity to values adequate for amphibian tissues) to remove extracellular DASPMI. For lifetime experiments, cells were kept in HBSS to avoid background fluorescence. XTH2 cells tolerated room temperature (∼25°C) well during the experimentation time; they remained well spread and allowed the observation of single mitochondria.
For permeabilization, cells were incubated for 15 min with 0.01% saponin and 0.5 mM ATP in culture medium (12,13). Cells rinsed with culture medium were then incubated with DASPMI alone or with substrates (malate and glutamate).
For anisotropy measurements, primary chick embryo fibroblast (CEF) cells were grown on Iscove's modified Dulbecco's medium supplemented with 10% fetal calf serum. Cells were maintained in a humidified air, 5% CO2 atmosphere at 41°C. Cells from passages 6 and 28 were seeded on glass coverslips, and for DASPMI staining they were incubated with 3 μM DASPMI for 40 min. Before imaging, the cells were rinsed with culture medium to remove extracellular DASPMI.
Steady-state imaging
Steady-state fluorescence images were obtained using an IX70 inverted microscope (Olympus, Hamburg, Germany). A mercury arc lamp (50 W) was used for excitation, in combination with an FT510 dichroic mirror, 450–490 nm band-pass excitation filter, and emission filters (570DF20, 560DF40, BP515–565 nm, BA590, or BP530–550 nm; Omega Optical, Brattleboro, VT). Imaging was done using an Olympus 100× oil apochromate objective lens and 12 bit Sensi-Cam (Imago, Till Photonics, Herrsching, Germany). Neutral density filters were used at the excitation side to image at a low light level to avoid photobleaching.
Anisotropy measurements were done using a Dual-View microimager (MSMI-DV-FC, Optical Insights, Tucson, AZ) with a polarization/anisotropy filter cube mounted in front of the 12 bit Sensi-Cam. Polarization of the excitation light from the mercury arc lamp was controlled by introducing appropriate polarization filters (AHF Analysentechnik AG, Tübingen, Germany). The resulting Dual-View images on Sensi-Cam were two spatially identical images of the sample in focus that differed only in the polarization used for imaging. The resulting spatially identical, vertically and horizontally polarized images were modified using image-processing functions in MATLAB (The MathWorks, Natick, MA) environment to achieve precisely aligned images. Anisotropy was then calculated using the formula
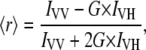 |
where G is the grating factor that has been included to correct for the wavelength response to polarization of the emission optics and detector, and IVV and IVH are the emission intensities measured parallel and perpendicular to the vertically polarized excitation, respectively.
Space- and time-resolved fluorescence decay microscopy
The fluorescence decay microscope used to acquire the spatially resolved fluorescence decays has been described previously (14). In brief, a femtosecond titanium-sapphire laser (model 3980, Spectra-Physics, Darmstadt, Germany) was tuned to 900 nm to obtain an excitation wavelength of 450 nm and repetition rate of 4 MHz after frequency doubling and pulse-picking. The laser was optically aligned to an Olympus IX70 inverted microscope (epiillumination, Olympus IX70, Hamburg, Germany) with a spatial filter system, which also acts as a beam expander. Spatially resolved fluorescence decays were obtained from the projection of the magnified two-dimensional area of the sample in focus onto a time- and space-correlated single-photon counting detector (quadrant-anode (QA) detector, EuroPhoton GmbH, Berlin, Germany). The online acquisition program records a two-dimensional image (256 × 256 pixels) with temporal information associated with each pixel; hence fluorescence decays from specific regions of interest (ROIs) could be constructed for different acquisition times. The reduction in thermal noise of the detector was important and was achieved by water-cooling of the detector by 4°C. The full width half maximum of the instrument response function with the QA detector was ∼200 ps. The spatial resolution was determined by placing a micrometer scale in the sample area of the microscope.
All measurements were performed at room temperature (∼25°C), using an apochromate 100× oil objective (Olympus, Hamburg, Germany), 1.3 NA, to obtain a uniform illumination area of 80 μm diameter. An FT510 dichroic mirror and emission filters (570DF20, BP530–550 nm, and BA590; Omega Optical) were used for imaging.
Fluorescence lifetimes were obtained by analyzing the fluorescence decays by iterative reconvolution with the instrument response function in MATLAB (The MathWorks, Natick, MA). The χ2 goodness-of-fit parameter was reduced by using the Marquardt nonlinear least-square algorithm. The instrumentation accuracy and reliability of analysis have been previously demonstrated (10,14). The fluorescence decays obtained from different regions of DASPMI-stained XTH2 cells were best fitted for a three-exponential fit (Supplementary Material, Fig. S1 in Data S1). The ROIs in some examples were as small as a single mitochondrion. For calculations, a minimum number of 10,000 photon counts were required to achieve a reliable fit of χ2 in the range of 0.9–1.1. The 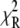 surfaces were examined to determine the range of parameters consistent with the fluorescence decay. Confidence intervals obtained from the surface support analysis were in the range of 10–150 ps for all the data analyzed (Fig. S2 in Data S1). The mean lifetime proportional to steady-state intensity was calculated as 〈τ〉 =
surfaces were examined to determine the range of parameters consistent with the fluorescence decay. Confidence intervals obtained from the surface support analysis were in the range of 10–150 ps for all the data analyzed (Fig. S2 in Data S1). The mean lifetime proportional to steady-state intensity was calculated as 〈τ〉 =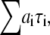 where
where 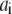 is the normalized relative contribution (
is the normalized relative contribution (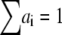 ) of the i fluorescent species characterized by its fluorescent lifetime
) of the i fluorescent species characterized by its fluorescent lifetime 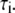 The fractional contributions of each decay time to the steady-state intensity were calculated as
The fractional contributions of each decay time to the steady-state intensity were calculated as 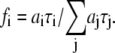
RESULTS
Spatially resolved fluorescence lifetime imaging
Untreated cells in culture
Spatially resolved fluorescence lifetime imaging is the method of choice to relate the complex photophysics of DASPMI to its interaction with mitochondria at different physiological states, i.e., to elucidate how DASPMI fluorescence monitors mitochondrial membrane potential. Using a QA detector, fluorescence decay values can be derived even for single mitochondria within living cells (Fig. 1 and Table 1). In Fig. 1, fluorescence intensities have been color-coded and fluorescence decay has been calculated for ROIs containing one mitochondrion or only a very few organelles. Fluorescence intensity is highest in mitochondria in the perinuclear regions and decreases toward the cell periphery, a behavior often observed in larger cells. A whole cell is shown in Fig. 1; the ROIs with the higher numbers lie in the cell periphery. In the extreme periphery, single mitochondria can no longer be identified because of motion-related blur (this QA picture was taken while the cells were alive and mitochondria were moving within the cytoplasm during the photon collection time).
FIGURE 1.

Pseudo color time-integrated QA image and corresponding decay kinetics (Table 1) in various ROIs of a DASPMI-stained XTH2 cell. The variable mitochondrial membrane potential can be inferred from its intensity distribution. Fluorescence lifetime analysis was performed over various ROIs, including single mitochondria or several organelles. Fluorescence from ROI 3 is primarily from the nucleus, with a minor contribution from mitochondria only. Bar, 5 μm.
TABLE 1.
Fluorescence lifetime analysis of various ROIs in Fig. 1
| ROI | a1 | τ1 (ns) | a2 | τ2 (ns) | a3 | τ3 (ns) | 〈τ〉 (ns) | f1 (%) | f2 (%) | f3 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.65 | 0.091 | 0.31 | 0.544 | 0.04 | 2.343 | 0.322 | 19 | 52 | 29 |
| 2 | 0.74 | 0.085 | 0.23 | 0.528 | 0.03 | 2.162 | 0.249 | 25 | 49 | 26 |
| 3 | 0.70 | 0.118 | 0.24 | 0.739 | 0.06 | 2.711 | 0.423 | 20 | 42 | 38 |
| 4 | 0.71 | 0.101 | 0.26 | 0.591 | 0.03 | 2.286 | 0.294 | 25 | 52 | 23 |
| 5 | 0.74 | 0.221 | 0.22 | 0.747 | 0.04 | 2.357 | 0.422 | 39 | 39 | 22 |
| 6 | 0.75 | 0.216 | 0.22 | 0.780 | 0.03 | 2.503 | 0.409 | 40 | 42 | 18 |
Depending on the intracellular location and physiological state, notable variations in all the three lifetime components and relatively small changes in their contributions are evident. τ1 considerably increases with decreasing fluorescence intensity (Table 1, Fig. 1), and its contribution to overall fluorescence increases. τ2 also increases, but with decreasing fraction. The longest lifetime, τ3, changes considerably and its contribution lies between 3% and 6%. The average lifetime (proportional to the area under the decay curve) decreases at higher fluorescence intensities in the cell shown in Fig. 1. The fractional contribution (f1) of short decay time (τ1) to the steady-state intensity is weaker for regions of higher intensity; it is about half the fractional intensity of the ICT state (f2) for regions of higher membrane potential.
At 3 μmol/L, DASPMI staining no longer is restricted to mitochondria; rather, this weak cation can also be accumulated in the nucleus, very prominently in the nucleolus, and to a minor extent it may remain in the cytoplasm (cytosol; Table 2). In the cytoplasm, nucleus, and nucleolus, the long-lived component τ3 is longer lasting than in mitochondria and f3 contributes more to the overall fluorescence. Also the other two decays have longer duration in the hydrophilic but highly charged environment of the nucleolus, and to a minor extent also in the nucleus and cytosol.
TABLE 2.
Fluorescence lifetime analysis of DASPMI-stained living XTH2 cells
| Condition | a1 | τ1 (ns) | a2 | τ2 (ns) | a3 | τ3 (ns) | 〈τ〉 (ns) | f1(%) | f2(%) | f3(%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Punctated | 0.82 ± 0.09 | 0.065 ± 0.023 | 0.16 ± 0.08 | 0.626 ± 0.082 | 0.02 ± 0.01 | 2.802 ± 0.410 | 0.216 ± 0.065 | 24 ± 3 | 44 ± 5 | 32 ± 4 |
| Elongated | 0.72 ± 0.04 | 0.183 ± 0.041 | 0.24 ± 0.03 | 0.739 ± 0.114 | 0.03 ± 0.01 | 2.610 ± 0.241 | 0.396 ± 0.068 | 34 ± 5 | 45 ± 3 | 21 ± 4 |
| Nucleus | 0.67 ± 0.06 | 0.190 ± 0.071 | 0.25 ± 0.03 | 0.963 ± 0.282 | 0.08 ± 0.03 | 3.020 ± 0.500 | 0.603 ± 0.170 | 21 ± 3 | 39 ± 5 | 40 ± 8 |
| Nucleoli | 0.64 ± 0.07 | 0.225 ± 0.079 | 0.26 ± 0.05 | 1.257 ± 0.243 | 0.10 ± 0.02 | 3.576 ± 0.352 | 0.816 ± 0.084 | 18 ± 6 | 39 ± 1 | 43 ± 5 |
| Cytosol | 0.76 ± 0.16 | 0.222 ± 0.134 | 0.19 ± 0.12 | 1.073 ± 0.718 | 0.06 ± 0.04 | 3.258 ± 0.745 | 0.500 ± 0.066 | 30 ± 23 | 36 ± 3 | 34 ± 19 |
High SDs result from biological variations occurring from measurements on a single-cell or single-mitochondrion level, and not from instrumental inaccuracy.
The different decays found in punctate and elongated mitochondria can be interpreted on the basis of the findings of DASPMI emission in mitochondria exposed to membrane-potential manipulating conditions (Table 3) and in the light of the intracellular differences described. Accordingly, punctate ones exhibit fluorescence decays typical of mitochondria with high membrane potential. In comparison with elongated mitochondria, the fractional intensity of punctated mitochondria pertaining to the LE state (f1) has decreased by 10% and the fractional intensity (f3) of the TICT state has increased by 11%. The decay parameters for elongated and punctated mitochondria were calculated from the fluorescence decays obtained from 20 and 4 different cells, respectively.
TABLE 3.
Influence of lifetime parameters of DASPMI in living XTH2 cells under conditions manipulating membrane potential
| Condition | a1 | τ1 (ns) | a2 | τ2 (ns) | a3 | τ3 (ns) | 〈τ〉 (ns) | f1 (%) | f2 (%) | f3 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | 0.74 ± 0.05 | 0.159 ± 0.051 | 0.23 ± 0.04 | 0.701 ± 0.115 | 0.03 ± 0.01 | 2.540 ± 0.227 | 0.357 ± 0.078 | 33 ± 6 | 45 ± 4 | 22 ± 5 |
| Nigericin | 0.91 ± 0.06 | 0.038 ± 0.028 | 0.07 ± 0.05 | 0.699 ± 0.063 | 0.01 ± 0.01 | 2.868 ± 0.259 | 0.118 ± 0.064 | 27 ± 9 | 41 ± 3 | 32 ± 8 |
| ATP | 0.86 ± 0.06 | 0.093 ± 0.058 | 0.13 ± 0.05 | 0.762 ± 0.104 | 0.02 ± 0.01 | 3.160 ± 0.134 | 0.229 ± 0.113 | 31 ± 7 | 45 ± 4 | 24 ± 4 |
| Glutamate and malate | 0.90 ± 0.06 | 0.080 ± 0.070 | 0.09 ± 0.05 | 0.812 ± 0.141 | 0.01 ± 0.01 | 3.343 ± 0.430 | 0.185 ± 0.132 | 32 ± 9 | 43 ± 1 | 25 ± 8 |
| KCN | 0.73 ± 0.05 | 0.210 ± 0.027 | 0.21 ± 0.04 | 0.767 ± 0.110 | 0.05 ± 0.01 | 2.465 ± 0.308 | 0.445 ± 0.030 | 35 ± 5 | 36 ± 6 | 29 ± 4 |
| CCCP | 0.73 ± 0.03 | 0.171 ± 0.038 | 0.20 ± 0.03 | 0.849 ± 0.205 | 0.08 ± 0.03 | 2.969 ± 0.213 | 0.512 ± 0.110 | 24 ± 3 | 33 ± 6 | 43 ± 7 |
Analysis was performed on several mitochondrial ROIs from 20 control cells and four cells under the influence of a drug. The high SDs result from biological variations occurring from measurements on a single-cell or single-mitochondrion level.
Cells in different physiological conditions
Table 3 shows the mean values of the fluorescence decay kinetics of several DASPMI-stained mitochondrial regions in 20 living XTH2 cells and in four cells exposed to poisons known to manipulate the energy status of a cell (KCN, which inhibits respiration by blocking complex IV function; nigericin, which increases the inner mitochondrial membrane potential by electroneutral K+/H+ exchange; and CCCP, a powerful uncoupler of respiratory chain activity from oxidative phosphorylation due to its protonophore character). In addition, the average lifetime parameters of DASPMI over several mitochondrial regions under conditions that increase the membrane potential, i.e., after treatment with ATP and/or substrate (glutamate and malate) from four permeabilized XTH2 cells are shown.
Control values are in the range of those shown in Table 1. In KCN-treated cells, τ1 is in the range of control mitochondria with locally lower membrane potential (e.g., cell periphery), but a3 is higher than in control conditions. In the presence of nigericin, mitochondria membrane potential can be considered to be close to its maximum. This is represented by almost exclusive emission at very short τ1 and almost no contribution of τ3, indicative of a polar environment. In addition, the τ2 contribution to decay is only one-third that in the control condition. Under the influence of ATP and/or substrate (glutamate and malate), in addition to similar shortening of τ1 as in the case of nigericin, increases in τ2 and τ3 with increasing membrane potential are evident. When treated with CCCP, DASPMI was promptly released in cytosol with a diffuse background. The lifetime values for CCCP-treated cells predominantly arise from diffuse cytosol fluorescence, also evident from the very high contribution (8%) of τ3.
Control cells differ from nigericin-treated cells by 6% higher f1 and 10% less f3. In the presence of KCN or CCCP, f3 is higher than in control conditions. The f1 and f2 values of KCN treated cells are nearly equal, as is the case for peripheral mitochondria shown in Fig. 1.
The f1 values of controls and cells in the presence of ATP and/or substrate are about the same. This is due to the increase of inner membrane rigidity in the very few cases of low-membrane-potential peripheral mitochondria of ATP-treated cells. This behavior in the presence of ATP was in addition to the increase of membrane rigidity with increasing potential. The increase of membrane rigidity with increasing membrane potential is apparent from the increase of τ3 for high-membrane-potential punctate mitochondria or in the presence of nigericin. In isolated mitochondria and vesicles, it was found that lipid fluidity changes and membrane potential were not kinetically coupled (15).
Interpretation of fluorescence decay data
The decrease in lifetime with increasing intensity shown in Table 1 is in accordance with our previous studies of solvents, in which quenching of lifetime with increasing solvent polarity was shown. Although the increase in lifetime can be due to either an increase in viscosity or a decrease in polarity, the reduction in lifetime and quantum efficiency is primarily due to the polarity of the medium. To further understand its viscosity-dependent behavior in living cells, the lifetimes of DASPMI in nucleus, cytosol, and nucleoli were compared with mitochondria of different morphologies (Table 2). An increased contribution of longer lifetimes in the nucleus and nucleoli was evident, with the longest lifetimes in nucleoli. This is consistent with the increased red fluorescence found in the emission fingerprints of nucleus and nucleoli typical for dye immobilization by high viscosity of its environment due to the highly packed nucleoproteins in nucleoli (Fig. S3 A in Data S1). Previously, groove binding of DASPMI with the nucleic acids (16) was reported, which also would result in increased lifetime.
In comparison with elongated mitochondria, the shortening of τ1 in punctate mitochondria (Table 2) signifies the high local mitochondrial membrane potential. The low contribution of f1 in spherical mitochondria, as in the cases of ROIs 1, 2, and 4 (Table 1), wherein the mitochondria have higher membrane potential, is notable. This is in accordance with solvent photophysics, where the charge-transfer state becomes the low-energy state in solvents of higher polarity, and consequently the emission from the LE state diminishes. In comparison to elongated mitochondria, the higher transmembrane potential in punctate mitochondria is accompanied by an increase in compactness of the mitochondrial inner membrane, as suggested by the increase of f3 by 10%.
Further substantiation of the above interpretation came from the influence of ionophores and uncouplers on mitochondrial membrane potential (Table 3 and Fig. S4 in Data S1). Conditions that enhance the mitochondrial membrane potential cause considerable shortening of τ1 (159–38 ps), with a simultaneous increase in its contribution (Fig. S4 in Data S1). DASPMI in energized mitochondria exhibits a behavior similar to that of monoexponential decay (τ = 6 ps) in polar solvent water (10), but still with much longer decay and thus quantum yield. In comparison to control conditions, they are also associated with decreased contributions (a2 and a3) and no considerable changes in lifetimes (τ2 and τ3; Fig. S4 in Data S1). In contrast, the decrease of membrane potential by the addition of KCN changes τ1 from 159 ps to 210 ps (in XTH2 cells, KCN does not fully abolish membrane potential). This shows the direct influence of membrane potential on lifetime (τ1) associated with the LE state and not significantly on charge transfer states (τ2 and τ3), which can be interpreted by dye molecules orienting themselves with the positive pyridinium in the proximity of proton rich intermembrane space and the aniline moiety being located in the hydrophobic regions of the inner membrane.
The increased lifetimes in comparison to solvents are due to the absence of solvation dynamics. Lifetime analysis of red-edge fluorescence (using LP 590 nm) from DASPMI-stained XTH2 cells exhibited a higher contribution of τ3 up to 18%, with a consequently decreased contribution of the shorter lifetime (τ1). This is typical of formation of consecutive excited states from the initial excited molecule, such as LE→ICT→TICT.
Steady-state fluorescence anisotropy
Fluorescence polarization measurements provide useful information on the flexibility of molecules. A high anisotropy means that the probe does not change its molecular configuration and orientation while it is in the excited state. Hence, emission anisotropy of DASPMI-stained mitochondria in living cells of various types was determined using an inverted epifluorescence microscope with a polarizer and Dual-View system attached. Considering the range of fluorescence decay times of several tens of picoseconds to a few nanoseconds (Tables 1–3), anisotropy of DASPMI will reveal intramolecular kinetics (i.e., twist) rather than molecule motions within the membrane.
Representative images of the distribution of polarization-resolved fluorescence of DASPMI in mitochondria of living XTH2 cells are shown in Fig. 2. Apart from regions of higher intensity near the nucleus, submitochondrial zones of higher membrane potential revealed from higher DASPMI intensity were found to have higher anisotropy as well (arrowheads in inserts in Fig. 2). The presence of submitochondrial zones with higher membrane potential has been observed previously using DASPMI and JC1 dyes (17,18).
FIGURE 2.

Distribution of polarization-resolved fluorescence intensities and corresponding anisotropy values of DASPMI-stained XTH2 cells. The very distinct high anisotropy at high-intensity regions (inserts) inside several individual mitochondria is apparent. Such submitochondrial zones of higher membrane potential are known to exist from previous studies in XTH2 cells. The plot of mean anisotropy values and corresponding pixel fluorescence intensities (± 50 units) were fitted to an exponential function of the form 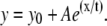
Anisotropy values of DASPMI varied from 0.05 to 0.4 (Fig. 2), indicating a wide range of torsional motions about the flexible single bonds adjoining the olefinic double bond. The twists about the single bonds may well be modulated by the local electric field and membrane fluidity. These two effects are indiscernible by steady-state anisotropy measurements. The increase of anisotropy with membrane potential has been demonstrated by plotting the mean values of anisotropy and standard deviations (SDs) at different pixel intensities (± 50 au) corresponding to total steady-state fluorescence (Fig. 2). Anisotropy values were found to increase exponentially with increasing fluorescence intensity (Fig. 2).
A previous study on the distribution of DASPMI in isolated mitochondria showed a log-normal dependence of DASPMI concentration on mitochondria and membrane potential-mediated changes caused by valinomycin-mediated potassium uptake (9). Accordingly, the exponential correlation between DASPMI fluorescence and its emission anisotropy (Fig. 2) implies a linear dependence of DASPMI emission anisotropy on mitochondrial membrane potential.
Because of the severe loss of dye molecules from the mitochondria in the presence of uncouplers, another physiological condition has been used to demonstrate differences of mitochondrial membrane potential by anisotropy. Decreased mitochondrial membrane potential is a widely found property in senescent cells. Therefore, DASPMI fluorescence anisotropy has been compared in young and old CEFs. The anisotropy in mitochondria of young cells (passage 6) was found to be uniform and higher, in comparison to the old (passage 28) CEF cells. This correlates to the reduced uptake of DASPMI in old CEF cells. Mean values of anisotropy and SDs at different total pixel fluorescence intensities (± 50 units) in young and old cells are shown in Fig. 3. The initial, nearly linear increase of anisotropy in response to mitochondrial membrane potential (as visualized by increased fluorescence intensity) in old cells subsequently coincides at 5000 au intensity with young cells. The higher membrane potential in young CEFs, as apparent from its higher intensity values, exhibits a sharp increase in anisotropy thereafter. The mean anisotropy values and corresponding fluorescence intensities from young and old CEF cells were exponentially correlated (Fig. 3). Hence, membrane-potential-dependent anisotropy in DASPMI has the advantage of giving comparable measures of membrane potential from different cell types stained with different concentrations due to their independence from absolute intensity.
FIGURE 3.

Mean values of anisotropy and SDs at different intensities (± 50 units) in senescent CEFs. Mean pixel fluorescence intensities (± 50 units) from the summation of horizontally and vertically polarized steady-state intensity images of senescent CEFs have been plotted against the mean anisotropy. The plot was fitted to an exponential function of the form 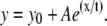 The consistent increase of anisotropy with fluorescence intensity and the loss of membrane potential apparent from low values of anisotropy in senescent CEFs are evident.
The consistent increase of anisotropy with fluorescence intensity and the loss of membrane potential apparent from low values of anisotropy in senescent CEFs are evident.
DISCUSSION
The styryl dye DASPMI has a delocalized π-electron system with hydrophobic and hydrophilic regions. Because of its positively charged methylpyridinium moiety and hydrophobic aniline moiety, the dye is able to diffuse easily between the various compartments of the cell. The electrophoretic force generated by the interaction of DASPMI with the membrane potential across the plasma membrane (−70 mV) and mitochondrial inner membrane (−150 mV) guides the dye into mitochondria. When CCCP was added to the stained XTH2 cells, DASPMI was promptly released from the mitochondria into the cytosol, followed by an accumulation in the nucleoli.
The three-exponential decay kinetics of DASPMI in living cells (as found in glycerol) with lifetimes of 159 ± 51 ps (74% ± 5%), 701 ± 115 ps (23% ± 4%), and 2540 ± 227 ps (3% ± 1%) advocate for a viscous microenvironment. The three-exponential decay kinetics in living cells, representative of the LE, ICT, and TICT states, are also obvious from the emission fingerprint (Fig. S3 B in Data S1). Previously, investigation of time-resolved emission spectra of DASPMI in chloroform revealed a shoulder at shorter wavelengths pertaining to the LE state. The absence of a green shoulder in the emission spectra of nucleoli is in accord with the more polar environment in ribonucleoprotein assemblies (Fig. S3 A in Data S1). The high sensitivity of DASPMI to the mitochondrial membrane potential is evident from its prompt release from mitochondria on the addition of CCCP. Further, the presence of hydrophilic and lipophilic domains, the green shoulder in mitochondrial emission fingerprints, and an absorption maximum between 460 and 470 nm in mitochondria substantiate the localization of DASPMI in the inner mitochondrial membrane.
The direct influence of mitochondrial membrane potential on only the shortest lifetime (τ1) of DASPMI, and its remarkable shortening in energized mitochondria indicate the partial insertion of DASPMI in the outer leaflet of the inner mitochondrial membrane (Fig. 4). This proposition is confirmed by the occurrence of disparate changes between lifetimes pertaining to the LE state (τ1) and charge-shift states of DASPMI-stained energized mitochondria. Furthermore, the >30-fold difference between τ1 and τ3 (in energized mitochondria), excitation maximum of 470 nm in XTH cells (between DMSO and chloroform) and emission maximum of 565 nm (same as chloroform) support our deduction. A blue shift in both excitation and emission spectra is expected for molecules that are only partially inserted into the lipid environment (19–23). The partial insertion of DASPMI in the outer leaflet of the inner mitochondrial membrane ensures a favorable polar environment for the positively charged methylpyridinium moiety and hydrophobic aniline moiety along the nonpolar hydrocarbon chains of lipids. Under this scenario, photoabsorption that is largely confined to the positively charged methylpyridinium moiety can be expected to have an absorption maximum of 470 nm, and emissions predominantly confined to the aniline moiety to have an emission maximum of 565 nm as observed in living cells. This differential solvation has been previously been described for other styryl dyes and has also been observed by us in phospholipids vesicles wherein the absorption maximum was 430 nm and emission maximum of 545 nm (data not shown). In comparison to phospholipid vesicles, the red shift of absorption maxima in mitochondria could be due to partial delocalization of π-electrons under the influence of a local electric field. This assumption of only a partial delocalization of π-electrons, rather than a complete one, is confirmed by the decrease of steady-state anisotropy at longer emission wavelengths (Fig. 5). A complete shift of positive charge to the dimethyl amino group would shift the absorption maxima and anisotropy to higher values, as has been shown to be the case in chloroform (10).
FIGURE 4.

Location and orientation of DASPMI in the mitochondrial inner membrane. DASPMI preferentially locates its positively charged methyl pyridinium ring in the aqueous interface facing the outer mitochondrial membrane, and its hydrophobic moiety along the hydrocarbon chain. As for orientation, it aligns itself along the local electric field for a maximum electrochromic response. Under high-membrane-potential conditions, the delocalization of π-electrons leads to a less polar structure, imposing steric constraints on the single bonds neighboring the olefinic double bond.
FIGURE 5.

Emission wavelength-dependent anisotropy of DASPMI. In response to high-membrane-potential conditions, electrons are withdrawn from the electron-rich aniline moiety, leading to a less polar DASPMI structure. Consequently, an increase in rigidity of the single bonds (i.e., double bond-like character) neighboring the olefinic double bond leads to an increase in anisotropy. The dependence of anisotropy on emission wavelength indicates the gradual change in molecular configuration and hence the transition in dipole moment from the initially excited Franck-Condon level.
The excited-state dynamics of DASPMI depend on the influence exerted by the local electric field on DASPMI charge distribution and its local environment. τ1 attributed to fluorescence from the initially excited state is largely localized in the positively charged methyl pyridinium moiety. τ2 and τ3 are the excited-state lifetimes due to deexcitation from the ICT and TICT states pertaining to the aniline moiety in the lipid environment. The shortening of τ1 in energized mitochondria is due to the increase in concentration of protons in the intracristal space and hence quenching of fluorescence from the methyl pyridinium moiety. Previous electron micrograph studies of DASPMI-stained mitochondria in XTH2 cells supported the proposition that selective accumulation in cristae would render photodamage of inner mitochondrial membrane (17). Quenching of DASPMI fluorescence by energy transfer to cytochromes can almost be excluded because of the prolonged decay times in the presence of cyanide when all the cytochromes should be in the reduced state with strong absorption bands.
The accumulation of DASPMI in mitochondria is a membrane-potential-driven process (8,9) accompanied by a strong change in quantum efficiency while the environment changes from an aqueous solution to the mitochondrial membrane. Although it is much higher than in saline, quantum efficiency in the mitochondrial inner membrane is largely determined by the quenching of fluorescence from the LE state due to higher proton concentrations. Upon addition of CCCP, equal numbers of protons exist across the inner mitochondrial membrane, destroying the membrane potential. Under such conditions, the charge of positively charged methyl pyridinium moiety increases, leading to a more polar structure of DASPMI at the inner membrane, and less quenching of τ1, but now the molecules are able to diffuse freely to other compartments of the cell. When higher concentrations of DASPMI are used to measure mitochondrial membrane potential, other possible localizations and less specific staining will decrease its sensitivity to mitochondrial membrane potential.
The influence of membrane potential on the torsional motions of the probe molecule is expected when considering the partial insertion of DASPMI in the inner mitochondrial membrane, partial delocalization of the π-electron system, and lipid fluidity changes evident from variation of τ3 by as much as 20%. The fluorescence from the LE state corresponds essentially to an electronic configuration found immediately upon excitation. Any changes in inner-membrane fluidity should have a negligible effect on the fluorescence characteristics from the LE state. Under the high-membrane-potential condition, a decreased fractional fluorescence contribution from the LE state leads to an increase in fluorescence from charge transfer states. Depolarization of the fluorescence emission of DASPMI is essentially due to an increase in the fraction from the TICT state, which is associated with fluorescence from a twisted conformation of DASPMI. Consequently, the changes in anisotropy are due to modulation of TICT-state fluorescence by changes in inner membrane fluidity or by changes in the rigidity of single bonds neighboring the olefinic double bond. Since the local electric field changes and membrane fluidity are not kinetically coupled, the fluorescence lifetime distribution of DASPMI is obscured. However, as the membrane rigidity increases with increasing membrane potential, the mean fluorescence anisotropy of DASPMI (within the SD; Figs. 2 and 3) may still correspond linearly to changes in the average mitochondrial membrane potential.
Using the very complex response of DASPMI in living mitochondria to photon absorption, one can derive information regarding energy status as well membrane fluidity. For the first time, to our knowledge, lower membrane fluidity in spherical mitochondria than in elongated ones has been shown by analysis of fluorescence decay parameters. This is an important parameter for inner mitochondrial protein exchange after mitochondrial fusion, an event that is supposed to represent a rescue mechanism in case of protein impairment.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Valentina Strecker for providing young and old CEFs, and Prof. Jacob Piehler for providing the Dual-View system.
This work was supported by the International Max-Planck Research School-Structure and Function of Biological Membranes (to R.R.), the Deutsche Forschungsgemeinschaft (Be 423/22), and the European Commission (project 35150407, MIMAGE).
Editor: Thomas Schmidt.
References
- 1.Chen, L. B. 1988. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4:155–181. [DOI] [PubMed] [Google Scholar]
- 2.Bereiter-Hahn, J. 1990. Behavior of mitochondria in the living cell. Int. Rev. Cytol. 122:1–63. [DOI] [PubMed] [Google Scholar]
- 3.Balaban, R. S., S. Nemoto, and T. Finkel. 2005. Mitochondria, oxidants, and aging. Cell. 120:483–495. [DOI] [PubMed] [Google Scholar]
- 4.Lambert, A. J., and M. D. Brand. 2007. Research on mitochondria and ageing. Aging Cell. 6:417–420. [DOI] [PubMed] [Google Scholar]
- 5.Orrenius, S. 2007. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 39:443–455. [DOI] [PubMed] [Google Scholar]
- 6.Haugland, R. P. 2002. Handbook of Fluorescent Probes and Research Products. Molecular Probes, Eugene, OR. 74–76, 86–96, 830–837, 867.
- 7.Bereiter-Hahn, J. 1976. Dimethylaminostyrylmethylpyridiniumiodine (DASPMI) as a fluorescent probe for mitochondria in situ. Biochim. Biophys. Acta. 423:1–14. [DOI] [PubMed] [Google Scholar]
- 8.Bereiter-Hahn, J., K. H. Seipel, M. Vöth, and J. S. Ploem. 1983. Fluorometry of mitochondria in cells vitally stained with DASPMI or rhodamine 6 GO. Cell Biochem. Funct. 1:147–155. [DOI] [PubMed] [Google Scholar]
- 9.Mewes, W. H., and J. Rafael. 1981. The 2-(dimethylaminostyryl)-1-methylpyridinium cation as indicator of the mitochondrial membrane potential. FEBS Lett. 131:7–10. [DOI] [PubMed] [Google Scholar]
- 10.Ramadass, R., and J. Bereiter-Hahn. 2007. Photophysical properties of DASPMI as revealed by spectrally resolved fluorescence decays. J. Phys. Chem. B. 111:7681–7690. [DOI] [PubMed] [Google Scholar]
- 11.Schlage, W., C. Kuhn, and J. Bereiter-Hahn. 1981. Established Xenopus tadpole heart endothelium (XTH) cells exhibiting selected properties of primary cells. Eur. J. Cell Biol. 24:342. (Abstr.) [Google Scholar]
- 12.Jacob, M. C., M. Favre, and J. C. Bensa. 1991. Membrane cell permeabilization with saponin and multiparametric analysis by flow cytometry. Cytometry. 12:550–558. [DOI] [PubMed] [Google Scholar]
- 13.Saks, V. A., V. I. Velsler, A. V. Kuznetsov, L. Kay, P. Sikk, T. Tiivel, L. Tranqui, J. Olivares, K. Winkler, F. Wiedemann, and W. S. Kunz. 1998. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol. Cell. Biochem. 184:81–100. [PubMed] [Google Scholar]
- 14.Ramadass, R., D. Becker, M. Jendrach, and J. Bereiter-Hahn. 2007. Spectrally and spatially resolved fluorescence lifetime imaging in living cells: TRPV4-microfilament interactions. Arch. Biochem. Biophys. 463:27–36. [DOI] [PubMed] [Google Scholar]
- 15.O'Shea, S. P., S. Feuerstein-Thelen, and A. Azzi. 1984. Membrane-potential-dependent changes of the lipid microviscosity of mitochondria and phospholipid vesicles. Biochem. J. 220:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar, C. V., R. S. Turner, and E. H. Asuncion. 1993. Groove binding of a styrylcyanine dye to the DNA double helix: the salt effect. J. Photochem. Photobiol. Chem. 74:231–238. [Google Scholar]
- 17.Bereiter-Hahn, J., and M. Vöth. 1998. Do single mitochondria contain zones with different membrane potential? Exp. Biol. Online. 3:1–13. [Google Scholar]
- 18.Smiley, S. T., M. Reers, C. Mottolahartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele, and L. B. Chen. 1991. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA. 88:3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loew, L. M., S. Scully, L. Simpson, and A. S. Waggoner. 1979. Evidence for a charge-shift electrochromic mechanism in a probe of membrane potential. Nature. 281:497–499. [DOI] [PubMed] [Google Scholar]
- 20.Loew, L. M., and L. L. Simpson. 1981. Charge-shift probes of membrane potential: a probable electrochromic mechanism for p-aminostyrylpyridinium probes on a hemispherical lipid bilayer. Biophys. J. 34:353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loew, L. M., G. W. Bonneville, and J. Surow. 1978. Charge shift optical probes of membrane potential. Theory. Biochemistry. 17:4065–4071. [DOI] [PubMed] [Google Scholar]
- 22.Loew, L. M. 1996. Potentiometric dyes: imaging electrical activity of cell membranes. Pure Appl. Chem. 68:1405–1409. [Google Scholar]
- 23.Loew, L. M., L. Simpson, A. Hassner, and V. Alexanian. 1979. An unexpected blue shift caused by differential solvation of a chromophore oriented in a lipid bilayer. J. Am. Chem. Soc. 101:5439–5440. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.