

Abstract
There is strong evidence for the importance of genetic factors in idiopathic autism. The results from independent twin and family studies suggest that the disorder is caused by the action of several genes, possibly acting epistatically. We have used cDNA microarray technology for the identification of constitutional changes in the gene expression profile associated with idiopathic autism. Samples were obtained and analyzed from six affected subjects belonging to multiplex autism families and from six healthy controls. We assessed the expression levels for approximately 7,700 genes by cDNA microarrays using mRNA derived from Epstein Barr virus (EBV)-transformed B-lymphocytes. The microarray data was analyzed in order to identify up- or down-regulation of specific genes. A common pattern with nine down-regulated genes was identified among samples derived from individuals with autism when compared to controls. Four of these nine genes encode proteins involved in biological processes associated with brain function or the immune system, and are consequently considered as candidates for genes associated with autism. Quantitative realtime PCR confirms the down-regulation of the gene encoding SEMA5A, a protein involved in axonal guidance. EBV should be considered as a possible source for altered expression but our consistent results make us suggest SEMA5A a candidate gene in the etiology of idiopathic autism.
Keywords: Adolescent, Autistic Disorder, genetics, metabolism, Child, Child, Preschool, Down-Regulation, physiology, Female, Gene Expression, physiology, Genetic Predisposition to Disease, Humans, Male, Membrane Proteins, genetics, metabolism, Nerve Tissue Proteins, genetics, metabolism, Oligonucleotide Array Sequence Analysis, methods, Reverse Transcriptase Polymerase Chain Reaction, methods
Keywords: Autistic disorder, cDNA microarrays, Gene expression, Chromosome 7q31, SEMA5A
Introduction
Autistic disorder (AD) is a neurodevelopmental disorder characterized by impairments in reciprocal social interactions, such as verbal and non-verbal communication, and flexibility as regards interests and activities. The etiology as well as the clinical expression of the disorder is heterogeneous. In idiopathic autism, the symptoms appear during the first years of life. The prevalence of AD is approximately 0.1%, with a higher frequency among boys [1]. Genetic factors behind AD were suggested already by Kanner in 1943 [2], and this has been confirmed by subsequent studies. The average concordance rate for AD is 70% for monozygotic twins compared to 0%–5% for dizygotic twins according to three epidemiological same-sex twin studies [3–5]. Analysis of recurrence risks in families suggests that between two and ten gene loci, possibly more, may be involved [6]. Several full genome scans for autism have been performed, all of which show linkage to a number of susceptibility loci, supporting that several genes may contribute to the disorder [7–11]. Specific gene regions have also been suggested from the identification of patients with different chromosomal abnormalities [12]. Recently, mutations in the neuroligin genes NLGN3 and NLGN4 were reported in a subset of patients with AD [13], and mutations in NLGN4 has been confirmed in a family with X-linked mental retardation with or without autism [14]. The neuroligin genes encode cell-adhesion proteins, suggesting abnormal synapse formation as a key mechanism in the pathogenesis of AD. Still, the genetic factors behind the majority of patients with idiopathic AD remain unknown.
We hypothesize that AD may be associated with constitutional abnormalities in the expression levels of several genes, and we present here a cDNA microarray analysis of 7,700 genes in samples from individuals with familial AD. Abnormalities in the expression profile in patient-derived lymphoblastoid cells are presented and discussed.
Materials and methods
Samples from six subjects with AD belonging to six sib-pair families included in the genome scan performed by Philippe et al [7], and co-segregating for chromosome 7q31, were selected in order to reduce the genetic heterogeneity and to facilitate the identification of common altered patterns in gene expression. Chromosome 7q31 has previously been reported to contain a susceptibility locus for autism. Blood samples were collected from the patients after written informed consent from the parents, and transformed into lymphoblastoid cell lines (LCLs) by Epstein Barr transfection. Diagnosis of AD/childhood autism was confirmed using the DSM-IV diagnostic criteria and Autism Diagnostic Interview-Revised algorithm [15,16]. Organic conditions associated with autism, such as tuberous sclerosis, epilepsy, fragile X syndrome, or chromosomal abnormalities were excluded in the subjects. Clinical features of the patients are presented in table 1. Six LCLs from healthy individuals (3 males and 3 females), without a family history of autism or other neuropsychiatric disorders, were used as controls. This study was approved by the regional ethical committee in Sweden.
Table 1.
Clinical features of patients
| Case | Sex | Agea | Ethnicity | Diagnosis | Onset Months | Regression | Language | ADI-R criteria | CARS | Cognitive levelb | Seizures |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 7.0 | Caucasian | autism | 30 | Yes (at 30 months) | non verbal | Yes | 36.5 | severe mental retardation | – |
| 2 | m | 10.4 | Caucasian | autism | 24 | No | non verbal | Yes | 44 | severe mental retardation | – |
| 3 | m | 13.6 | Caucasian | autism | 35 | Yes (at 35 months) | non verbal | Yes | 37 | mild mental retardation | – |
| 4 | f | 6.9 | Caucasian | autism | 15 | No | delayed (first phrases 60 months) | Yes | 43 | borderline intellectual functioning | – |
| 5 | m | 10.5 | Caucasian | autism | 15 | No | delayed (first words 25 months, phrases 37 months) | Yes | 34 | borderline intellectual functioning | – |
| 6 | m | 4.0 | Caucasian | autism | 19 | No | non verbal | Yes | 44.5 | mild mental retardation | – |
ADI-R, Autism Diagnostic Interview-Revised [15]; CARS, Childhood Autism Rating Scale [42] (scores between 30 and 36.5 indicate mild to moderate autism while scores greater than 36.5 reflect severe autism).
Age in years at which individual was evaluated for inclusion in the study.
Assessed using age and developmentally appropriate instruments, including the Psychoeducational Profile, the Wechsler Intelligence Scale for Children-Revised, and the Leiter.
The LCLs were cultured in RPMI with HEPES, 10% fetal calf serum, 4 μg/ml glutamine and 1000 E/ml penicillin-streptomycin. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. Total RNA was prepared using TRIzol® (Invitrogen) according to the manufacturer’s protocol. The quality of the total RNA was controlled by capillary electrophoresis using a Bioanalyzer 2100 (Agilent Technologies). Only RNA without any sign of degradation was used. In-house produced cDNA microarrays with approximately 7,700 genes in duplicates were used to assess mRNA levels. The cDNA clones were obtained from the Research Genetics sequence-verified human cDNA collection (Invitrogen), and corresponding PCR-products were printed onto microscopic slides. Hybridizations with dye-swap replicates were made for each individual, where RNA from one of the control LCL was used as common reference in all experiments. Two different labeling and hybridization protocols were used: indirect labeling with Micromax™ TSA™ Labeling and Detection Kit (NEN® Life Science products) was used in 15 experiments and direct Cy-dNTP incorporation, using a modified CyScribe™ First Strand cDNA Labelling Kit (Amersham Biosciences) protocol was used in 9 experiments. Eight μg of total RNA were used for the Micromax™ TSA™ labeling and 25 μg of total RNA for the CyScribe labeling. The microarrays were scanned with a GenePix 4000B (Axon instruments) scanner and the fluorescent intensities were quantified with GenePix Pro 3.0 software (Axon Instruments). The data were normalized using a lowess algorithm, which is implemented in the statistical software package R [17]. The normalization was intensity and print-tip dependent and no background subtraction was used. Intensity signals from spots not detected by the quantification software due to low intensities, or spots with artifacts on the arrays were removed in order to exclude irrelevant data. Ratios deviating more than three times between the two dye swap experiments were also removed. A mean value was calculated for each gene and missing values were imputed with a K-nearest neighbor method in SAM (Significance Analysis of Microarrays) [18]. A maximum of two imputations per group and gene was allowed. Two-class SAM was used for identifying candidate genes through an algorithm based on a modified t-test. A score, obtained from the difference between groups and variation within groups, was calculated for each gene and used for ranking. Q-values were produced from permutation of class labels and were used as significance estimates.
Quantitative real-time PCR (qPCR) analysis was performed for SEMA5A, HSPA1L, CCND2 and SAT on total RNA from the same 12 individuals. cDNA was synthesized using M-MLV enzyme (USB®) and random priming and for SEMA5A, HSPA1L, CCND2 and SAT were amplified and detected using TaqMan® Assays-on-Demand™ Gene Expression products (Applied Biosystems) and the ABI PRISM® 7000 Sequence Detection System (Applied Biosystems). mRNA levels of β-actin (ACTB) were used for normalization. The mRNA levels were detected in triplicates and mean values calculated after exclusion of outliers.
Results
After filtration, 2,101 genes remained for further analysis. The majority of the genes were removed due to low expression levels, with resulting signal intensities that could not be detected during the quantification process. Nine genes were identified with significantly altered levels in patient samples. All nine genes were down-regulated, ranging between −1.38 and −1.67 (Table 2). The q-values (0.095) suggest that one of the nine genes may be a false positive and erroneously identified as down-regulated. The magnitude of the changes is low, but in the same range as those found in the microarray study on autism performed by Purcell et al [19]. None of the genes is positioned in the 7q31 region. Table 3 presents the biological functions of the nine genes identified. Four of these genes, SEMA5A, HSPA1L, CCND2 and SAT, encode proteins involved in biological processes associated with brain function or the immune system, and are consequently candidate genes for idiopathic autism. Quantitative PCR confirms the array results for the SEMA5A gene, but not for the HSPA1L, CCND2 and SAT genes (Fig. 1). One outlier in the patient group, case 2, reduced the statistical significance for SEMA5A, resulting in a group mean ratio of 0.96 compared to 2.82 without the outlier. The deviating measurement was well outside three standard deviations from the group mean and thus excluded from the analysis as an outlier [20]. The reason for the outlier in the qPCR experiments is unclear since it does not correspond to the results of the microarray analysis. In conclusion, the cDNA microarray analysis and the quantitative real-time PCR result are consistent and suggest a down-regulation of SEMA5A in the samples analyzed.
Table 2.
Differentially expressed genes identified by SAM analysis
| Gene | Chr. Location | Score | Fold change | q value |
|---|---|---|---|---|
| CSDA | 12p13 | −2.96 | −1.67 | 0.095 |
| SARCOSIN | 2q31 | −2.53 | −1.37 | 0.095 |
| SEMA5A | 5p15 | −2.43 | −1.45 | 0.095 |
| MGST1 | 12p12 | −2.27 | −1.50 | 0.095 |
| SAT | Xp22 | −2.19 | −1.42 | 0.095 |
| CCND2 | 12p13 | −2.17 | −1.65 | 0.095 |
| HSPA1L | 6p21 | −2.16 | −1.66 | 0.095 |
| RPN2 | 20q12-q13 | −2.12 | −1.40 | 0.095 |
| SNX2 | 5q23 | −2.06 | −1.38 | 0.095 |
Score, fold change and q values are derived from SAM.
Table 3.
Biological mechanisms related to nine down-regulated genes
| Gene symbol | Gene name | Molecular function | Biological process |
|---|---|---|---|
| CSDA | Cold shock domain protein A | Transcription co-repressor activity | Cold response |
| SARCOSIN | Sarcomeric muscle protein | Protein binding activity | Striated muscle contraction |
| SEMA5A | Semaphorin 5A | Receptor activity | Neurogenesis |
| MGST1 | Microsomal glutathione S-transferase 1 | Glutathione transferase activity | Cellular defense (against oxidative stress) |
| SAT | Spermidine/spermine N(1)-acetyltransferase | Acetyltransferase activity | Polyamine catabolism |
| CCND2 | Cyclin D2 | Forms serine/threonine kinase holoenzyme complex | Cell cycle regulation |
| HSPA1L | Heat shock 70kDa protein 1-like | Heat shock protein activity | Heat shock response |
| RPN2 | Ribophorin II | Transferase activity | Protein modification |
| SNX2 | Sorting nexin 2 | Protein transporter activity | Intracellular trafficking |
Fig. 1.
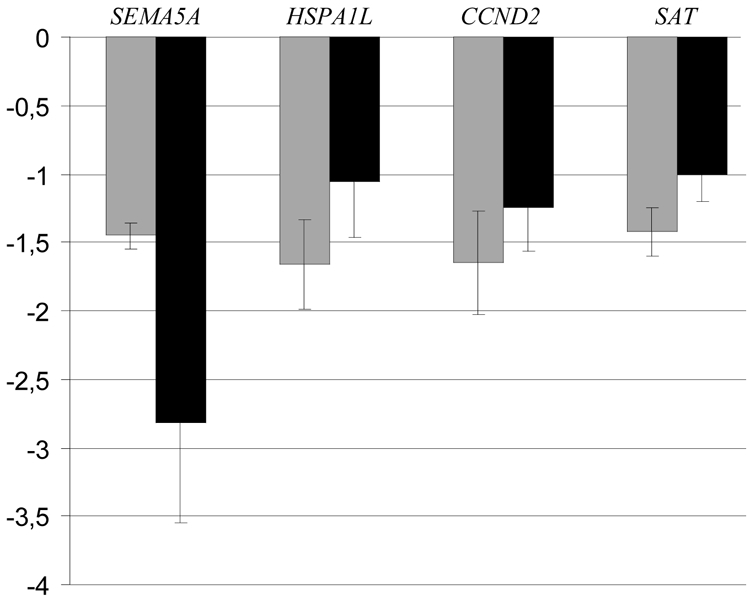
Relative down-regulation of SEMA5A, HSPA1L, CCND2 and SAT mRNA levels in Epstein-Barr virus-transformed cells derived from patients with AD compared to healthy controls. Fold-changes are based on microarray (
 ) and qPCR (■) analysis. Error bars indicate standard deviations.
) and qPCR (■) analysis. Error bars indicate standard deviations.
Discussion
In this report we have used cDNA microarray technology to analyze the expression pattern of 7,700 genes in LCLs from six non-related individuals with idiopathic autism. Each patient was selected from familial forms of AD (sib-pairs), in which at least one chromosome 7q-region segregated with the disorder. The analysis resulted in the identification of nine genes, all of which were down-regulated. None of these genes have been reported previously as associated with autism. Notably, one of the nine genes encodes semaphorin 5A, which is involved in axonal guidance [21]. The SEMA5A gene is positioned at chromosome 5p15, approximately 6 Mb from a marker indicating increased allele sharing in our previous genome-wide scan [7]. Rodent expression of Sema5A has been reported in cerebral cortex, basal ganglia, thalamus and hippocampus in embryonic and postnatal rat brains [22]. Semaphorin 5A is a bifunctional molecule, acting both as attracting and repulsing guidance cues [23,24]. Failure of Sema5A expression leads to abnormal development of the axonal connections in the forebrain of mice [25], which may affect the formation of functional synapses. Furthermore, it has been suggested that haploinsufficiency for SEMA5A is responsible for mental retardation in Cri-du-chat, a syndrome resulting from deletions of chromosome 5p [26]. Synaptic dysfunction of specific neurons is likely to be one etiologic mechanism in AD and this was supported by the identification of mutations in the NLGN3 and NLGN4 genes in a subgroup of patients with autism [13,14]. The NLGN3 and NLGN4 genes encode synaptic cell-adhesion molecules. A down-regulation of SEMASA, with an effect on axonal guidance, may therefore be an analogous candidate mechanism behind AD in some patients.
The HSPA1L, CCND2 and SAT genes were also found to be down-regulated in our microarray study. These genes were of interest due to their involvement in the immune system or brain development [27–34], but differences in mRNA expression levels could not be confirmed by qPCR. This indicates that these genes may be false positives in our microarray analysis.
To our knowledge, no previous findings suggest association between the remaining five genes down-regulated in this study (CSDA, SARCOSIN, MGST1, RPN2 and SNX2) and mechanisms related to brain development or brain function.
Several genes in the 7q31 region, such as RAY1/ST7, WNT2, CORTB2 and RELN have been proposed as candidate genes for autism [35–38]. In addition, linkage and association to the β3 subunit of the GABAA receptor GABRB3 gene on chromosome 15q have been reported by several groups [39–41]. Unfortunately, the WNT2, RELN and GABRB3 genes were not expressed at levels high enough to detect true differences in our LCL samples. The results from a previous microarray analysis of brain specimens suggest that variation in the expression of EAAT1 and GluR1, two members of the glutamate system, are associated with autism [19]. We could not confirm changes in expression of these two candidate genes in our study. The RAY1/ST7, CORTB2, NLGN3 and NLGN4 genes were not present on the microarrays used in this study.
Idiopathic autism is a genetically complex disorder, manifested in the brain. Since idiopathic autism is constitutional, altered transcript levels may be detected also in non-neuronal tissues, such as lymphoblastoid cells, which are used as a model in this study. Consequently, the significance of our findings in the etiology of AD depends on the presence of constitutional changes in mRNA levels from genes expressed in both brain and lymphocytes. The small number of individuals included renders generalized conclusions hazardous, and the risk of separate false positives should be considered. Another limitation is the sensitivity of the microarray system. Small variations in expression levels may be difficult to detect and is further complicated for genes with low constitutional expression levels.
The significance of the down-regulated genes in this study is yet unclear. Further analyses, including a larger number of patient samples and preferably on brain tissue from AD patients, are needed to confirm a possible involvement of these transcripts in the disease process. The microarray technique still provides a useful tool for the investigation of mechanisms behind complex psychiatric disorders, and the method may contribute to clarify both primary and secondary events in these disorders.
Acknowledgments
This work was supported by grants from the Swedish Research Council, the Children’s Cancer Foundation of Sweden, the Swedish Cancer Society, the Swedish Medical Society, the Sävstaholm Society, and the Torsten and Ragnar Söderbergs Fund. The experiments comply with the current laws of Sweden.
References
- 1.Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord. 2003;33:365–82. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- 2.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 3.Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 4.Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–16. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 5.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 6.Pickles A, Bolton P, Macdonald H, Bailey A, Le Couteur A, Sim CH, Rutter M. Latent-class analysis of recurrence risks for complex phenotypes with selection and measurement error: a twin and family history study of autism. Am J Hum Genet. 1995;57:717–26. [PMC free article] [PubMed] [Google Scholar]
- 7.Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, Penet C, Feingold J, Brice A, Leboyer M. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8:805–12. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- 8.International Molecular Genetic Study of Autism Consortium. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet. 1998;7:571–8. doi: 10.1093/hmg/7.3.571. [DOI] [PubMed] [Google Scholar]
- 9.International Molecular Genetic Study of Autism Consortium. A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001;69:570–81. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collaborative Linkage Study of Autism. An autosomal genomic screen for autism. Am J Med Genet. 2001;105:609–15. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 11.Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, Nguyen L, Yang J, Harper C, Thorpe D, Vermeer S, Young H, Hebert J, Lin A, Ferguson J, Chiotti C, Wiese-Slater S, Rogers T, Salmon B, Nicholas P, Myers RM, et al. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillberg C. Chromosomal disorders and autism. J Autism Dev Disord. 1998;28:415–25. doi: 10.1023/a:1026004505764. [DOI] [PubMed] [Google Scholar]
- 13.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthelemy C, Moraine C, Briault S. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–7. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–85. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 16.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington DC: American Psychiatric Press; 1994. [Google Scholar]
- 17.Ihaka R, Gentleman R. R: A language for data analysis and graphics. Journal of Computational & Graphical Statistics. 1996;5:299–315. [Google Scholar]
- 18.Goss Tuscher V, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57:1618–28. doi: 10.1212/wnl.57.9.1618. [DOI] [PubMed] [Google Scholar]
- 20.Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z, Roby P, Glenister P, Thornton C, Thaung C, Stevenson JA, Arkell R, Mburu P, Hardisty R, Kiernan A, Erven A, Steel KP, Voegeling S, Guenet JL, Nickols C, Sadri R, Nasse M, Isaacs A, Davies K, Browne M, Fisher EM, Martin J, Rastan S, Brown SD, Hunter J. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet. 2000;25:440–3. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- 21.Adams RH, Betz H, Puschel AW. A novel class of murine semaphorins with homology to thrombospondin is differentially expressed during early embryogenesis. Mech Dev. 1996;57:33–45. doi: 10.1016/0925-4773(96)00525-4. [DOI] [PubMed] [Google Scholar]
- 22.Skaliora I, Singer W, Betz H, Puschel AW. Differential patterns of semaphorin expression in the developing rat brain. Eur J Neurosci. 1998;10:1215–29. doi: 10.1046/j.1460-9568.1998.00128.x. [DOI] [PubMed] [Google Scholar]
- 23.Kantor DB, Chivatakarn O, Peer KL, Oster SF, Inatani M, Hansen MJ, Flanagan JG, Yamaguchi Y, Sretavan DW, Giger RJ, Kolodkin AL. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron. 2004;44:961–75. doi: 10.1016/j.neuron.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Oster SF, Bodeker MO, He F, Sretavan DW. Invariant Sema5A inhibition serves an ensheathing function during optic nerve development. Development. 2003;130:775–84. doi: 10.1242/dev.00299. [DOI] [PubMed] [Google Scholar]
- 25.Jones L, Lopez-Bendito G, Gruss P, Stoykova A, Molnar Z. Pax6 is required for the normal development of the forebrain axonal connections. Development. 2002;129:5041–52. doi: 10.1242/dev.129.21.5041. [DOI] [PubMed] [Google Scholar]
- 26.Simmons AD, Puschel AW, McPherson JD, Overhauser J, Lovett M. Molecular cloning and mapping of human semaphorin F from the Cri-du-chat candidate interval. Biochem Biophys Res Commun. 1998;242:685–91. doi: 10.1006/bbrc.1997.8027. [DOI] [PubMed] [Google Scholar]
- 27.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 28.Millar DG, Garza KM, Odermatt B, Elford AR, Ono N, Li Z, Ohashi PS. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat Med. 2003;9:1469–76. doi: 10.1038/nm962. [DOI] [PubMed] [Google Scholar]
- 29.Mycko MP, Cwiklinska H, Szymanski J, Szymanska B, Kudla G, Kilianek L, Odyniec A, Brosnan CF, Selmaj KW. Inducible heat shock protein 70 promotes myelin autoantigen presentation by the HLA class II. J Immunol. 2004;172:202–13. doi: 10.4049/jimmunol.172.1.202. [DOI] [PubMed] [Google Scholar]
- 30.Bernal J, Nunez J. Thyroid hormones and brain development. Eur J Endocrinol. 1995;133:390–8. doi: 10.1530/eje.0.1330390. [DOI] [PubMed] [Google Scholar]
- 31.Poguet AL, Legrand C, Feng X, Yen PM, Meltzer P, Samarut J, Flamant F. Microarray analysis of knockout mice identifies cyclin D2 as a possible mediator for the action of thyroid hormone during the postnatal development of the cerebellum. Dev Biol. 2003;254:188–99. doi: 10.1016/s0012-1606(02)00039-8. [DOI] [PubMed] [Google Scholar]
- 32.Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME. Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development. 1999;126:1927–35. doi: 10.1242/dev.126.9.1927. [DOI] [PubMed] [Google Scholar]
- 33.Ingi T, Worley PF, Lanahan AA. Regulation of SSAT expression by synaptic activity. Eur J Neurosci. 2001;13:1459–63. doi: 10.1046/j.0953-816x.2001.01529.x. [DOI] [PubMed] [Google Scholar]
- 34.Kaasinen SK, Oksman M, Alhonen L, Tanila H, Janne J. Spermidine/spermine N1-acetyltransferase overexpression in mice induces hypoactivity and spatial learning impairment. Pharmacol Biochem Behav. 2004;78:35–45. doi: 10.1016/j.pbb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Vincent JB, Herbrick JA, Gurling HM, Bolton PF, Roberts W, Scherer SW. Identification of a novel gene on chromosome 7q31 that is interrupted by a translocation breakpoint in an autistic individual. Am J Hum Genet. 2000;67:510–4. doi: 10.1086/303005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wassink TH, Piven J, Vieland VJ, Huang J, Swiderski RE, Pietila J, Braun T, Beck G, Folstein SE, Haines JL, Sheffield VC. Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet. 2001;105:406–13. doi: 10.1002/ajmg.1401. [DOI] [PubMed] [Google Scholar]
- 37.Cheung J, Petek E, Nakabayashi K, Tsui LC, Vincent JB, Scherer SW. Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics. 2001;78:7–11. doi: 10.1006/geno.2001.6651. [DOI] [PubMed] [Google Scholar]
- 38.Persico AM, D’Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, Wassink TH, Schneider C, Melmed R, Trillo S, Montecchi F, Palermo M, Pascucci T, Puglisi-Allegra S, Reichelt KL, Conciatori M, Marino R, Quattrocchi CC, Baldi A, Zelante L, Gasparini P, Keller F. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry. 2001;6:150–9. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- 39.Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH, Jr, Fang Y, Song CY, Vitale R. Association between a GABRB3 polymorphism and autism. Mol Psychiatry. 2002;7:311–6. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- 40.Cook EH, Jr, Courchesne RY, Cox NJ, Lord C, Gonen D, Outer SJ, Lincoln A, Nix K, Haas R, Leventhal BL, Courchesne E. Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am J Hum Genet. 1998;62:1077–83. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao Y, Cuccaro ML, Hauser ER, Raiford KL, Menold MM, Wolpert CM, Ravan SA, Elston L, Decena K, Donnelly SL, Abramson RK, Wright HH, DeLong GR, Gilbert JR, Pericak-Vance MA. Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am J Hum Genet. 2003;72:539–48. doi: 10.1086/367846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schopler E, Reichler RJ, Renner BR. Irvington. New York: 1986. The Childhood Autism Rating Scale (CARS) for diagnostic screening and classification of autism. [Google Scholar]