

Abstract
The histidine superfamily of nucleotide hydrolases and nucleotide transferases consists of a branch of proteins related to Hint and Aprataxin, a branch of Fhit-related hydrolases, and a branch of GalT-related transferases. While substrates of Fhit and GalT are known and consequences of mutations in Aprataxin, Fhit and GalT are known, good substrates had not been reported for any member of the Hint branch and mutational consequences were unknown for Hint orthologs, which are the most ancient and widespread proteins in the Hint branch and in the histidine triad superfamily. Here we show that rabbit and yeast Hint hydrolyze the natural product adenosine-5′-monophosphoramidate in an active-site dependent manner at second order rates exceeding 1,000,000 M-1 s-1. Yeast strains constructed with specific loss of the Hnt1 active site fail to grow on galactose at elevated temperature. Loss of Hnt1 enzyme activity also leads to hypersensitivity to mutations in Ccl1, Tfb3 and Kin28, which constitute the TFIIK kinase subcomplex of general transcription factor TFIIH, and to mutations in Cak1, which phosphorylates Kin28. The target of Hnt1 regulation in this pathway was shown to be downstream of Cak1 and not to affect stability of Kin28 monomers. Functional complementation of all Hnt1 phenotypes was provided by rabbit Hint, which is only 22% identical to yeast Hnt1 but has very similar adenosine monophosphoramidase activity.
Histidine triad (HIT)1 proteins are a superfamily of nucleotide-binding proteins named for a near C-terminal HisϕHisϕHisϕϕ motif (ϕ, a hydrophobic amino acid) positioned at the α-phosphate of nucleotide substrates (1). The first branch of the superfamily is named for rabbit Hint, which had been purified as an abundant protein from cardiac cytosol by adenosine-affinity chromatography (2) and shown to have homologs in all forms of life (1). Recently, Aprataxin, a gene located at 9p13 that is inactivated in ataxia with oculomotor apraxia, the second most common of the autosomal recessive ataxias, was identified as a member of the Hint branch of the HIT superfamily (3,4). Human Fhit (5), which functions as a tumor suppressor protein in human (6-9) and murine (10,11) epithelial tissues, is the prototypical member of the second branch of the HIT superfamily. Fhit homologs have been found in fungi (12) and animals (13-16) and exhibit diadenosine polyphosphate hydrolase activity. A third branch of the HIT superfamily contains more distantly related nucleotide transferases including galactose-1-phosphate uridylyltransferase (GalT), which is the enzyme deficient in galactosemics (1), budding yeast diadenosine tetraphosphate phosphorylases Apa1 and Apa2 (17), and adenylylsulfate:phosphate adenylyltransferase (18). Ironically, though Hint is the most ancient and widespread of the HIT proteins, reasonable Hint substrates remained unidentified and the consequences of mutations in Hint or Hint orthologs were unknown (19).
Recently, human Hint was identified as a two-hybrid partner of Cdk7 and evidence was presented for a genetic interaction between yeast Hnt1 and a yeast Cdk7 homolog, Kin28 (20). The putative physical interaction between Hint/Hnt1 and Cdk7/Kin28 and the fact that hnt1 deletion lowers the restrictive temperature of a temperature-sensitive mutation in kin28 were interpreted to suggest that Hint/Hnt1 alters Cdk7/Kin28 substrate specificity by physical association (20). Cdk7 is a cyclin-dependent kinase that associates with cyclin H (21) and MAT1 (22,23) to form the mammalian cyclin-dependent kinase activating kinase activity (CAK). The ternary CAK complex is, in turn, part of general transcription factor TFIIH that contains the catalytic activity to phosphorylate C-terminal heptad repeats of RNA polymerase II large subunit (24,25). This activity, termed C-terminal domain kinase (CTDK), is required at post-initiation steps in TFIIH function (26). In yeast, CAK and CTDK activities are separated not only by virtue of different requirements for regulatory subunits but also by virtue of different catalytic subunits. In S. cerevisiae, the CAK for Cdc28 is encoded by monomeric Cak1 (27-29) while the major CTDK of yeast is Kin28, which is TFIIH-associated (30,31) and lacks CAK activity (32). In addition to phosphorylating the activation-loop of Cdc28, Cak1 phosphorylates the activation-loop of Kin28 (33,34). Because the CAK, Cak1, and CTDK, Kin28, could both be considered orthologous to Cdk7 and because Kin28 is a substrate of Cak1, we considered the two-hybrid and yeast genetic data (20) to be equivocal in identifying the direct target of Hnt1 regulation in yeast. Further, we were interested in determining whether an enzymatic or a nonenzymatic property of Hnt1 is responsible for Cak1 or Kin28 regulation. To avoid species-specific differences in terminology, we refer to the kinase subcomplex of TFIIH, i.e., the yeast Kin28-Ccl1-Tfb3 complex and the mammalian Cdk7-cyclin H-MAT1 complex, as TFIIK (30,31).
In this study we discover that loss of hnt1 alone produces a temperature-sensitive phenotype on galactose media and that hnt1 deficiency leads to synthetic loss of viability with hypomorphic alleles of cak1, ccl1 and tfb3. Additionally, hnt1 produces synthetic phenotypes with all temperature-sensitive alleles of Kin28 examined, with nonphosphorylatable Kin28, and with Kin28 overexpression. Despite the report of a physical interaction (20), none of the phenotypes correlate with absence of Hnt1 protein but rather with loss of Hnt1 enzymatic activity. We also demonstrate that Hnt1 and mammalian Hint are enzymes that hydrolyze unusual adenosine nucleotides such as adenosine-5′-monophosphoramidate (AMPNH2) to AMP plus a presumptive ammonia leaving group and that the active site histidine we show biochemically to be required for AMPNH2 hydrolysis is required for biological function and not required for protein stability. Though rabbit Hint is only 22% identical to yeast Hnt1, the enzymatic activity of Hint is substantially the same as that of Hnt1 and expression of the rabbit enzyme fully suppressed all hnt1Δ phenotypes. These data suggested that accumulation of an Hnt1 substrate in hnt1 deletion strains, likely an unusual adenosine mononucleotide and/or a nucleotidylated protein substrate, may inhibit function of Cak1 or Kin28 such that either protein kinase cannot tolerate mutational destabilization in itself or the other molecule. Though Cak1 has an unusual ATP-binding site, we show that strains without Cak1 are strongly Hnt1 and Hnt1 active site-dependent and thus Cak1 is not the target of Hnt1 regulation. Thus, the findings that Hnt1 enzyme activity is limiting for growth of cells with increased Kin28 abundance, reduced Kin28 phosphorylation, and destabilization of Kin28 or the additional TFIIK components Ccl1 and Tfb3, reveal an unanticipated nucleotide-dependent form of Kin28 regulation.
EXPERIMENTAL PROCEDURES
Yeast molecular biology and strain constructions
Standard yeast media, growth conditions and genetic manipulations were used (35). Galactose-containing media (Gal) contained 2% galactose while galactose plus raffinose-containing media (GalRaf) contained, in addition, 1% raffinose. To create diploid yeast strains heterozygous for hnt1 deletion, primers 4748 (primer sequences are provided in Table 2, which is available as a supplement) and 4749 were used to amplify a TRP1 fragment (plasmid pRS414 (36) as template) and a URA3 fragment (plasmid pRS416 (36) as template) with HNT1-homologous ends and transformed into wild-type diploid strain SEY6210.5 (37). Transformants were selected on SDC -trp media and SDC -ura media and screened using diagnostic PCR primers HNT1-5 and HNT1-3. A trp+ isolate heterozygous for hnt1 disruption named BY1 and a ura+ isolate heterozygous for hnt1 disruption named BY8 were allowed to sporulate and tetrads were dissected, generating haploid hnt1Δ progeny including BY1-2a, BY8-3b, BY8-4a, and BY8-5c. Additionally, a hnt1Δ::kanMX4 PCR product generated with primers 4748 and 4749 and plasmid pRS400 as template (36) was used to transform haploid wild-type MATα wild-type strain GF312-17c (28) and MATa cdc28-4 strain GF2412 to geneticin-resistance to produce hnt1Δ:kanMX4 strain BY158 and hnt1Δ::kanMX4 cdc28-4 strain BY169.
Strain BY8-3b was crossed with kin28-ts2 strain JGV117, kin28-ts3 strain JGV105, kin28-ts7 strain JGV111, and kin28-ts8 strain JGV112 (38) to generate four strains heterozygous for hnt1Δ and kin28 alleles named BK2, BK3, BK7 and BK8, respectively, which were dissected to yield hnt1Δ kin28 double mutant haploid progeny including BK2-2a, BK3-8b, BK7-1c and BK8-2a.
To introduce hnt1Δ into the cak1-civ1ts4 background, hnt1Δ strain BY158 was mated with cak1 strain GF2351 (28) and a double mutant strain BY161 was obtained by tetrad dissection. This strain was backcrossed to wild-type strain SEY6210 (37) and strain BY164 of genotype hnt1Δ cak1-civ1ts4 was obtained by tetrad dissection.
To generate strain BY185 of genotype hnt1Δ::kanMX4 cdc28Δ0 cak1Δ0 kin28Δ0 carrying CDC28-4324 and alleles of KIN28 on a plasmid, wild-type diploid strain SEY6210.5 (37) was disrupted with a PCR-generated cdc28Δ::loxP-kanMX-loxP disruption cassette generated by amplification of plasmid pUG6 (39) with primers 5122 and 5123. The cdc28Δ::loxP-kanMX-loxP heterozygous diploid strain BY172 was confirmed by amplification with diagnostic primers 5126 and 5127 and then transformed with plasmid pSH47, which expresses cre recombinase from the GAL1 promoter (39). A geneticin-sensitive derivative of genotype cdc28Δ0 / CDC28 was cured of plasmid pSH47, named strain BY173, and subjected to transplacement of cak1Δ::loxP-kanMX-loxP using primers 5124 and 5125. Heterozygosity for the cak1 disruption in strain BY175 was confirmed with primers 5130 and 5131 and the cak1Δ0 allele was recovered in strain BY176 as above. kin28Δ::loxP-kanMX-loxP replacement of strain BY176 was accomplished with primers 5132 and 5133 and confirmed with primers 5134 and 5135 to generate strain BY177, whose geneticin-sensitive kin28Δ0 derivative was named strain BY178. This strain was transformed with a YCpTRP1 plasmid carrying CDC28-4324 (40) and pB192, which expresses URA3 and KIN28-Thr162Ala, and subjected to tetrad dissection. Segregant BY178-6a scored as trp+ and ura+ and, additionally, 5-fluoroorotic acid sensitive, and contained cdc28Δ0, cak1Δ0 and kin28Δ0 disruption alleles as evidenced by diagnostic PCR. This strain was used to derive the hnt1Δ::kanMX4 disruptant BY185 using primers 4748 and 4749, as confirmed by diagnostic primers HNT1-5 and HNT1-3. LEU2 plasmids pB164, pB165 and pB183, which express KIN28, KIN28-Thr162Ala, and KIN28-Thr162Glu, respectively, were then transformed into strain BY185 with eviction of plasmid pB192.
To generate a ccl1-ts4 hnt1Δ double mutant and a tfb3-rig2ts23 hnt1Δ double mutant, hnt1Δ strain BY8-4a was mated with ccl1-ts4 strain GF2093 (41) and tfb3-rig2ts23 strain GF2217 (42) to generate diheterozygous diploid strains BK10 and BK13, respectively. Diploids were allowed to sporulate and tetrads were dissected, yielding double mutant segregants BK10-5a and BK13-1c, respectively.
To generate strain BY155-8c of genotype hnt1Δ kin28Δ carrying alleles of KIN28 on a plasmid, a kin28Δ::kanMX4 construct was generated with primers 5046 and 5047 and plasmid pRS400 as template (36). Diheterozygous strain BY155, whose genotype was confirmed by diagnostic PCR with primers 5048 and 5049, was then transformed with URA3 KIN28 plasmid pB192. Isolate BY155-8c was obtained by tetrad dissection and, after transformation with plasmids pB164 or pB165 containing LEU2 and KIN28 or KIN28-Thr162Ala, was cured of plasmid pB192. Haploid strain YGL26 (34) of genotype kin28Δ, expressing HA-tagged Kin28 from plasmid pGK13 (32), was used to produce strain BY186 (genotype hnt1Δ kin28Δ, expressing HA-tagged Kin28 from plasmid pGK13) by direct transformation with the hnt1Δ::kanMX4 construct.
Plasmid constructions
E. coli strain XL-1 Blue was used for bacterial cloning and plasmid amplification. Bacterial media and standard molecular biology techniques were as described (43). All plasmid constructions were confirmed by DNA sequencing. To create plasmid pB42 containing HNT1 for expression in yeast with its endogenous promoter, genomic DNA from strain SEY6210 (37) was amplified using primers 4787 and 4795. The amplified product was cleaved with BamHI and KpnI, cloned into BamHI and KpnI-digested pRS423 (44) to generate plasmid pB42. Plasmid pB150 was generated by site-directed mutagenesis (45) of pB4 using primer 5052 to generate the hnt1-His116Ala allele. To create plasmid pB159 containing rabbit HINT under the control of the GAL1 promoter, HINT cDNA was amplified from pSGA02-HINT (2) using primers 5085 and 5086. The product was then digested with BamHI and XhoI, and cloned into BamH1 and XhoI-cleaved plasmid pRS425GAL1 (46). To create plasmid pB216 containing HNT1 with a C-terminal triple FLAG tag (47) for expression in yeast, HNT1 was amplified with primers HNT1-5 and 5190, and the BamHI and XhoI-cleaved product was cloned into BamHI and XhoI-cleaved pRS413 (36). To create plasmid pB217 containing the triple FLAG-tagged hnt1-His116Ala allele, site-directed mutagenesis was performed with primer 5052.
To create plasmid pB24 containing the HNT1 cDNA in plasmid pSGA02 (48) for bacterial expression, primers 4739 and 4740 were used to amplify a product from oligo dT-primed yeast cDNA, which was digested with NdeI and XhoI and cloned into NdeI and XhoI-cleaved pSGA02. To create plasmid pB195 containing HNT1-His116Ala for bacterial expression, plasmid pB24 was subjected to site-directed mutagenesis using primer 4766.
To create plasmid pB176, containing PTC2 under the control of the GAL1 promoter, genomic DNA from wild-type strain SEY6210 (37) was amplified using primers 5108 and 5109. The amplified product was digested with BamHI and EcoRI and cloned into BamHI and EcoRI-cleaved plasmid pRS426GAL1 (46).
To create plasmid pB164, containing KIN28 expressed from its own promoter, genomic DNA from SEY6210 was amplified using primers 5066 and 5067. The product was digested with BamHI and EcoRI, cloned into BamHI plus EcoRI-cleaved plasmid pRS415 (36). Site directed mutagenesis of plasmid pB164 using primer 5075 generated plasmid pB165 containing KIN28-Thr162Ala. Mutagenesis of pB164 using primer 5116 generated pB183 containing KIN28-Thr162Glu. To construct plasmid pB192, the BamHI-XhoI fragment of plasmid pB165 was transferred to BamHI and XhoI-cleaved pRS416 (36). To create plasmid pB153, the KIN28 cDNA was amplified from oligo dT-primed yeast cDNA using primers 5054 and 5055 and inserted into pGEM (Promega). The KIN28 cDNA was excised from plasmid pB153 with SpeI and XhoI and inserted into SpeI plus XhoI-cleaved pRS425GAL1 to construct plasmid pB194.
Purification of recombinant Hint and Hnt1 proteins
Rabbit and yeast Hint proteins were expressed in E. coli from plasmids pSGA02-HINT (2), pB24 (HNT1) and pB194 (hnt1-His116Ala) according to published procedure (2). Frozen and thawed cell pellets (250 mg wet weight) were lysed in 12.5 ml buffer A (20 mM Tris pH 7.5, 150 mM NaCl) with one EDTA-free protease inhibitor cocktail tablet (Roche). Lysates were cleared by centrifugation and further cleared after precipitation with 1 mg protamine sulfate. Ammonium sulfate was added to 40% saturation and the 40% ammonium sulfate pellet was dialyzed against buffer A and clarified by centrifugation. Rabbit Hint and yeast Hnt1-His116Ala were purified by AMP-agarose (Sigma) affinity chromatography using a 4 ml column, washing with 24 column volumes of buffer A, and eluting with 3 column volumes of buffer A with 200 μM adenosine for Hint and 10 mM adenosine for Hnt1-His116Ala. Yeast Hnt1 was purified on a POROS 20 HQ column (PE Biosystems) using 20 mM Tris pH 7.5 , 2% glycerol as a loading and running buffer and NaCl as an eluant. Homogenous enzyme preparations were dialyzed against buffer A, concentrated to 10 mg/ml, and stored at -80 °C.
Enzymatic assays
Compounds tested as potential substrates were from Sigma, except AMP-N-alanine methyl ester and AMP-N-ε-(N α-acetyl lysine methyl ester), which were synthesized according to a modification of the synthesis of AMP-N-ε-lysine (49), and human DNA ligase I adenylylated intermediate, a gift of Alan E. Tomkinson, which was prepared as described (50). Compounds were screened for hydrolysis in 10 μl reactions containing 1 mM substrate, 0.5-2.0 μg enzyme, 0.5 mM MgCl2, 20 mM Na PIPES at pH 7.2 for 5-12 hr at 37 °C. Reactions repeated without MgCl2 were within 10% of the values reported. Reactions were diluted to 100 μl and analyzed by liquid chromatography on a MonoQ HR 5/5 column (Pharmacia) with mobile phases that were suitable mixtures of water and 1.25 M NH4 HCO3, pH 8. Ultraviolet absorbing peaks corresponding to nucleoside monophosphate products were integrated and converted to nmole using standard curves for AMP and ratios of appropriate extinction coefficients to that of AMP. Activity on p-nitrophenyl TMP was assayed colorimetrically at 400 nm as described (51). Human DNA ligase I adenylylated intermediate (50 pmol) was treated with 1 μg of enzyme in 50 μl of ligase buffer (30 mM Tris HCl, 50 mM NaCl, 0.5 mM DTT, 1 mM EDTA, 10% glycerol, pH 7.5) for 1 hr at 37 °C and analyzed by liquid chromatography for release of AMP and for the size and retention time of the ligase peak at 280 nm. Compounds that yielded activity greater than 0.005 nmol min-1 μg-1 were assayed further in 50-200 μl reactions with 0.2 mM substrate, 0.005-1 μg enzyme and 20 mM PIPES, pH 7.2 for 10 min at 37 °C. Reactions were stopped by adding NaOH to adjust the pH to 11, except for ester-containing reactions, which were stopped by freezing on dry ice. For saturation curves, reactions were performed in 200 μl with 0.5-3 ng enzyme. In some cases 10 mM Na BES or 10 mM imidazole HCl at pH 7.2 was used as buffer because of an interfering peak in PIPES. The low Km of the rabbit enzyme made it necessary to calculate initial rates from reactions that exceeded 10% hydrolysis with corrections as described (52). Thus, the reported Km of 68 nM should be considered an upper limit.
Evaluation of Hnt1 and Kin28 abundance in vivo
To examine steady state abundance of FLAG-tagged wild-type and active-site mutant Hnt1 proteins, strain BY8-5c was transformed with empty vector pRS413 and with plasmids pB216 and pB217, which encode the two FLAG-tagged Hnt1 constructs. Transformants were grown at 37 °C in SGalC -his media to an optical density (600 nm) of 0.6. Washed cell pellets were frozen at -20 °C and 0.4 g samples of cells were lysed with 0.8 g glass beads (0.5 mm, Sigma) in 1 ml of cold 150 mM Tris acetate, pH 7.5, 300 mM (NH4)2SO4, 1 mM spermidine, 1 mM dithiothreitol, 10% glycerol, 1x Complete protease inhibitor cocktail (Roche) with 7 one-minute pulses of a Mini-8 Beadbeater (Biospec). Twenty μg of total protein (determined by Biorad assay) from the clarified lysate of each sample was electrophoresed (15% SDS-PAGE) and transferred to an Immobilon-P membrane (Millipore). The transfer was probed with a 1:1000 dilution of anti-FLAG M2 monoclonal antibody (Sigma) and the washed blot was probed with a 1:2000 dilution of sheep anti-mouse IgG-horseradish peroxidase conjugate (Amersham), washed, and developed with ECL reagents (Amersham) as instructed. To detect HA-tagged Kin28 protein, strains YGL26 and BY186 were grown to an optical density (600 nm) of 0.6 in YPGal and YPD medium at 30 °C, 34 °C and 37 °C. Total soluble protein was extracted as above, electrophoresed (12% SDS-PAGE) and transferred as above. The Western blot was produced with a 1:1000 dilution of ascites fluid from monoclonal antibody HA.11 (Covance) and secondary antibody and detection system as above.
RESULTS
hnt1 mutants are gal- at 39 °C
Budding yeast contain one ortholog of HINT, HNT1, one ortholog of FHIT, HNT2 (1,19), and one ortholog of the Hint-related enzyme Aprataxin, HNT3. To explore the consequences of loss of the HINT ortholog, we generated disruption alleles hnt1Δ::TRP1 and hnt1Δ::URA3 and introduced them into diploid strains. Diploids heterozygous for hnt1Δ were allowed to sporulate, were dissected, and resulted in four viable haploid segregants per tetrad, indicating that HNT1 is neither required for spore germination nor for viability. Careful characterization of hnt1Δ mutants in comparison with isogenic HNT1 isolates revealed that hnt1Δ mutants do not grow on rich or synthetic galactose-containing media at 39 °C (Fig. 1A). Transformation of hnt1Δ mutants with a plasmid carrying an intact HNT1 gene restored full growth on galactose at 39 °C, but empty vector did not. As shown in Fig. 1B, hnt1Δ mutants display no temperature-sensitivity on glucose medium.
Fig. 1. Hnt1 enzyme activity is necessary for growth on galactose at 39 °C and complemented by rabbit Hint.

(A) An hnt1Δ strain (BY8-5c) was transformed with plasmids containing either HNT1 (pB42), hnt1-His116Ala (pB150), rabbit HINT cDNA under GAL1 control (pB159), or an empty vector (pRS423), and colonies were grown at 39 °C on SGalRafC media for six days.
(B) Strain BY8-5c transformants containing either HNT1 (pB42), hnt1-His116Ala (pB150), or empty vector (pRS423) were grown at 39 °C on SDC media for four days.
(C) Strain BY8-5c transformants containing either empty vector (pRS413, lane 1), FLAG-tagged HNT1 (pB216, lane 2) or FLAG-tagged hnt1-His119Ala (pB217, lane 3) were grown in SGalC -his at 37 °C and subjected to a Western blot with anti-FLAG antibody. The lane 1 control demonstrates the specificity of the signal and an arrow marks the predicted size of FLAG-tagged Hnt1.
Mammalian and yeast Hint proteins possess adenosine 5′-monophosphoramidase activity
Rabbit Hint was initially purified as an abundant cytosolic protein that binds adenosine agarose (2). Crystal structures of Hint bound to GMP, 8-Br-AMP and adenosine showed that a majority of the residues conserved in the HIT superfamily are located in positions to interact with bound nucleotide and that Hint is structurally related to Fhit and to GalT (1), which are enzymes that utilize a conserved histidine to perform hydrolysis or transfer reactions at the α phosphate of a bound nucleotide (13,53). It was also reported that human Hint has an extremely weak ADPase activity (kcat/Km = 8.5 M-1 s-1) (54). Discovery of a physiological assay for yeast Hnt1 prompted us to explore a range of nucleotide substrates for rabbit and yeast Hint proteins. Rabbit Hint and yeast Hnt1 proteins were expressed in E. coli and purified to homogeneity by AMP-agarose affinity or ion exchange chromatography. As shown in Table 1, under the conditions of our assay, which could detect as little as 30 fmol of nucleoside monophosphate released per minute per microgram of enzyme, ATP was detectable as a weak substrate of rabbit Hint (130 fmol min-1 μg-1) but not yeast Hnt1. Purine nucleotides such as ITP, 8-Br-ATP, GTP and GDP-glucose, pyrimidine mononucleotides such as CTP, CDP, UTP, UDP, UDP-glucose and p-nitrophenyl TMP, deoxy pApA, diadenosine polyphosphates ApppA through AppppppA, and acetylCoA were not detectably cleaved. Purine nucleoside diphosphates IDP, 8-Br-ADP, GDP and ADP and the smallest purine dinucleoside diphosphate-related compounds such as AppA, NAD, NADH and ADP-ribose were moderately better substrates than ATP, with adenosine diphosphate showing better cleavage than the other purine nucleoside diphosphates. Incubation with AMPSO4 and AMPαS indicated that sulfate and sulfur are much better leaving groups than phosphate for Hint as these compounds were cleaved 20 to 50-fold better by rabbit and yeast enzymes than ADP.
In a focused search for adenosine mononucleotides with a leaving group other than phosphate, we found that AMP morpholidate, a nucleotide with 5′ phosphoramidate (P-N) linkage to a six-membered ring, was cleaved 200-fold better than ADP by rabbit Hint and 2000-fold better than ADP by yeast Hnt1, bringing the hydrolysis rates up to the order of 1 nmol min-1 μg-1. Substitution of the morpholine ring with alanine methyl ester or N-α-acetyl lysine methyl ester, which maintained a phosphoramidate linkage to AMP, produced substrates about two-fold better than AMP morpholidate. The smallest possible amine substitution of AMP, AMPNH2, was the best substrate, being consumed at 1.7 and 7.6 nmol min-1 μg-1 by rabbit and yeast enzymes, respectively.
We performed initial rate determinations as a function of the concentration of AMPNH2. As shown in Fig. 2, AMPNH2 is hydrolyzed to AMP plus a presumptive ammonia leaving group with kcat and Km values of 0.20 ± 0.05 s-1 and 68 ± 15 nM (n=3) for the rabbit enzyme and values of 0.87 ± 0.25 s-1 and 870 ± 150 nM (n=3) for the yeast enzyme. To test whether these activities depend on the conserved middle histidine of the HIT motif, which is required for catalytic activity of Fhit (13,55-57), a His116Ala mutant form of yeast Hnt1 was generated, purified from E. coli, and incubated with the same substrates. No AMP was detected (Table 1). As the HPLC assay detects enzyme activity at levels 100,000-fold lower than those observed from enzymatic hydrolysis of AMPNH2, active-site dependence was shown conclusively. Moreover, discovery of AMPNH2 as a substrate allowed the demonstration that Hint and Hnt1 are over 100,000-fold more proficient on this substrate (kcat/Km for rabbit and yeast, 2.9 × 106 M-1 s-1 and 1.0 × 106 M-1 s-1 , respectively) than on ADP (54).
Fig. 2. Hint proteins are adenosine monophosphoramidases.
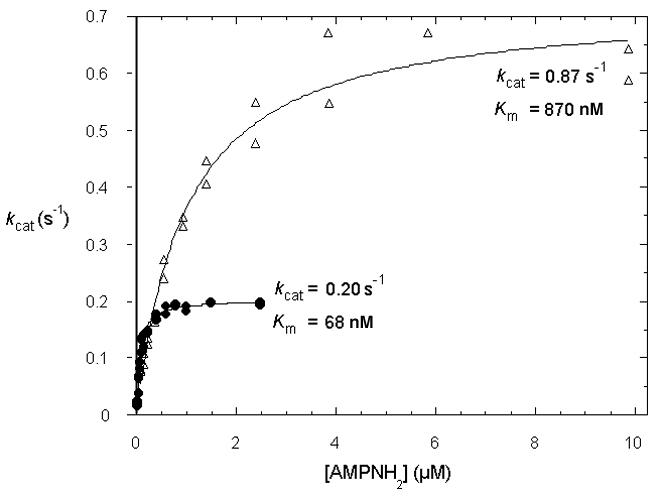
Substrate-concentration dependent rates (per monomer active site) for AMPNH2 hydrolysis to AMP plus the presumptive ammonia leaving group by rabbit Hint (•) and yeast Hnt1(Δ).
Review of the literature shows that AMPNH2 is found in eukaryotic cells (58), is synthesized from AMPSO4 and ammonia by an enzyme that can be found in a wide variety of bacteria and eukarya (59), and is hydrolyzed to AMP plus ammonia by an enzyme from rat liver that, like Hint, is a homodimer of 14 kDa subunits (60). Because that nucleoside monophosphoramidase activity was reported not to require magnesium (60), we repeated all Hint and Hnt1 assays without magnesium and found that all metal-free activities were within 10% of the values reported in Table 1. The rat liver enzyme was reported to be active on AMP-N-aminohexylamine and the Hint/Hnt1 enzymes are active on the similar substrate AMP-N-ε-(N-αacetyl lysine methyl ester) (Table 1). Failure to hydrolyze the Lys ε amino-linked AMP intermediate of DNA ligase I may be due to steric hindrance of AMP in the ligase active site cavity (50,61). Thus, Hint was likely already purified as nucleoside-5′-monophosphoramidase (60). Hint and Hnt1are postulated to function in vivo by hydrolyzing AMPNH2 or a related substrate, here provisionally termed AMP-X, to AMP plus the protonated leaving group.
Hnt1 enzymatic activity is necessary for biological function and can be functionally complemented by rabbit Hint
To test whether Hnt1 enzymatic activity is necessary for complementation of the hnt1Δ gal- phenotype at elevated temperature, the His116Ala allele of hnt1, which has no detectable enzymatic activity in vitro, was constructed for plasmid expression in yeast under the control of the HNT1 promoter. As shown in Fig. 1A, hnt1-His116Ala showed no complementation, demonstrating that the active site histidine residue is necessary for physiological function of the protein. Though the yeast Hnt1-His116Ala polypeptide had been purified from E. coli (above) and did not exhibit reduced steady-state abundance in vivo or instability as a purified protein, we wished to eliminate the possibility that loss of function of hnt1-His116Ala was caused by protein instability rather than loss of enzymatic activity. We therefore constructed C-terminally triple FLAG-tagged (47) HNT1 and hnt1-His116Ala alleles, and expressed them from the HNT1promoter on centromeric plasmids. As expected, FLAG-tagged HNT1 complemented the gal- phenotype of hnt1 at 39 °C while FLAG-tagged hnt1-His116Ala did not (not shown). Having validated the constructs, we probed soluble lysates for presence of the FLAG-tagged antigens at 37 °C, which is the highest temperature at which the FLAG-tagged hnt1-His116Ala strain can be grown. As shown in Fig. 1C, the hnt1-His116Ala allele did not diminish protein expression and thus, Hnt1 enzymatic activity is necessary for physiological function. The rabbit HINT cDNA, encoding a polypeptide with a conserved nucleotide-binding site and, as shown above, conserved enzymatic activity, but retaining only 22% sequence identity to Hnt1 (1), was put under the control of the GAL1 promoter. Rabbit HINT fully suppressed the gal- phenotype of the hnt1Δ strain (Fig. 1A).
Loss of Hnt1 enzymatic activity produces synthetic loss of viability with temperature sensitive alleles of kin28 and cak1
Recently it was reported that human Hint has a strong two-hybrid interaction with Cdk7 and that hnt1 deletion lowers the restrictive temperature of kin28-ts3 on galactose-containing media (20). These workers also showed a weak two-hybrid interaction between Kin28 and Hnt1 and that a small fraction of overexpressed human Hint could be immunoprecipitated with Cdk7. The results were taken together to suggest that Hint physically associates with Cdk7 and functions to increase substrate specificity for CTD phosphorylation (20). We reasoned that evidence for a physical interaction between Hnt1 and Kin28 could be bolstered if there were allele-specificity in the dependence of kin28-ts strains for HNT1. However, deletion of hnt1 in strains carrying seven different temperature-sensitive alleles of kin28 invariably reduced the restrictive temperature on galactose-containing media. Synthetic interactions with four alleles are shown in Fig. 3A. At temperatures permissive for single kin28-ts mutations that are selective for the corresponding kin28-ts hnt1Δ double mutants, single mutants are slightly enlarged with respect to wild-type cells while double mutants are highly elongated and cease cell division. To test whether the heightened requirement for Hnt1 of kin28 mutants reflects an enzymatic or nonenzymatic property of Hnt1, we transformed hnt1Δ kin28-ts8 strain BK8-2a with plasmids expressing wild-type HNT1, hnt1-His116Ala, rabbit HINT, or with an empty vector. As shown in Fig. 3B, yeast Hnt1 and rabbit Hint proteins were fully functional while yeast Hnt1 with an active-site mutation was without function. Thus, Hnt1 enzymatic activity rather than the Hnt1 polypeptide is conditionally essential in supporting hypomorphic forms of Kin28. Additionally, with weak alleles of kin28 such as kin28-ts8, hnt1 deletion reduces the restrictive temperature on glucose as well as galactose media (not shown).
Fig. 3. Synthetic loss of viability between hnt1Δ and temperature sensitive alleles of kin28 and cak1.

(A) Micrographs of strains carrying an hnt1 deletion with the indicated temperature-sensitive alleles of kin28 grown in SGalRafC -leu at 30 to 32 °C, bearing multicopy plasmids containing either wild-type HNT1 (pB42) or no insert (pRS423).
(B) An hnt1Δ kin28-ts8 strain (BK8-2a) was transformed with plasmids containing either HNT1 (pB42), rabbit HINT cDNA under GAL1 control (pB159), hnt1-His116Ala (pB150), or an empty vector (pRS423), and grown at 34 °C on SGalRafC media for six days.
(C) An hnt1Δ cak1-civ1ts4 strain (BY164) was transformed with plasmids containing either HNT1 (pB42), rabbit HINT cDNA under GAL1 control (pB159), hnt1-His116Ala (pB150), or an empty vector (pRS423), and grown at 34 °C on SGalRafC media for six days.
To test whether mutation of Cak1, which is not only the CAK for Cdc28 (27-29) but also the CAK for Kin28 (33,34), also increases the cellular requirement for Hnt1, a strain bearing the cak1-civ1ts4 allele was disrupted for hnt1 to generate strain BY164. As with kin28-ts3, cak1 destabilization is synthetically less viable with Hnt1 deletion or ablation of Hnt1 enzymatic activity and the double mutant phenotype is rescued by expression of rabbit Hint (Fig. 3C). Thus, Hnt1 enzyme activity appears to function as a positive regulator of either Cak1 or Kin28 in yeast and the Hnt1 requirement is most apparent at elevated temperature on galactose media.
We reasoned that a Hint substrate chemically related to AMPNH2, provisionally termed AMP-X, may inhibit one of the protein kinases in this pathway. By Occam’s Razor, we sought to account for all experimental facts with a single target of Hnt1 regulation, either Cak1 or Kin28, and discover the cellular reasons why mutation of the other kinase also enforces a requirement for Hnt1.
Cak1 is not the target of Hnt1 regulation
Because the initial models under consideration propose that AMP-X, an adenosine nucleotide, is a specific inhibitor of a protein kinase, Cak1 as an enzyme with an unusual ATP-binding site (27-29), was a good initial candidate as the direct target of Hnt1 regulation. Specifically, because Cak1 lacks the canonical GlyXGlyXXGly sequence that nearly all protein kinases use to bind ATP and is uniquely resistant to 5′-fluorosulfonylbenzoyladenosine (62), we considered it possible that Cak1 is uniquely sensitive to inhibition by a Hint/Hnt1 nucleotide substrate. Though CAK1 is an essential gene, it is possible to bypass the essential function with the multiply mutated, CAK1-independent allele of CDC28, CDC28-4324 (40). Strains containing a cak1 deletion suppressed by expression of CDC28-4324 as the sole source of Cdc28 were constructed and then subjected to hnt1 deletion. If Hnt1 were to function solely as a positive regulator of Cak1, then a strain without Cak1 would not be expected to show Hnt1-dependence. Nonetheless, as shown in Fig. 4A, deletion of hnt1 produces slow growth in strains without Cak1.
Fig. 4. Cak1 is not the target of Hnt1 regulation and strains with destabilized Kin28-binding proteins are limited By Hnt1 enzyme activity.

(A) Yeast strain BY185 (genotype hnt1Δ::kanMX4 cdc28Δ0 cak1Δ0 kin28Δ0, carrying the cak1-independent CDC28-4324 allele on a plasmid and the indicated alleles of KIN28 on plasmids) was transformed additionally, with pRS423 (indicated by hnt1Δ) or pB42 (indicated by HNT1). Transformants were streaked on SGalC -leu -his media and grown at 30 °C for 5 days.
(B) Yeast strain BY1-2a (genotype hnt1Δ strain) was transformed with pRS423 (indicated by hnt1Δ) or pB42 (indicated by HNT1) and with multicopy GAL1-PTC2 plasmid pB176 or empty vector pRS416. Transformants were grown at 30 °C on SGalRafC -his -ura media for 4 days.
(C) An hnt1Δ ccl1-ts4 strain (BK10-5a) was transformed with plasmids containing either HNT1 (pB42), rabbit Hint cDNA (pB159), hnt1-His116Ala (pB150), or an empty vector (pRS423), and grown at 35°C on SGalRafC for 6 days.
(D) An hnt1Δ tfb3-rig2ts23 strain (BK13-1c) was transformed with plasmids containing either HNT1 (pB42), rabbit Hint cDNA (pB159), hnt1-His116Ala (pB150), or an empty vector (pRS423), and grown at 34 °C on SGalRafC media for 6 days.
Two additional experiments were inconsistent with Cak1 as the target of Hnt1 regulation. If Cak1 were to be inhibited in hnt1 cells, the expected consequences would be reduction in Cdc28 activating phosphorylation (27-29), and reduction in Kin28 phosphorylation (33,34). To determine whether cells with a reduced level of phosphorylated Cdc28 are hyperdependent on Hnt1, we tested whether hnt1Δ produces synthetic loss of viability with overexpression of Ptc2, a Cdc28 activation-loop phosphatase (63), and with temperature-sensitive mutation of cdc28. Overexpression of Ptc2 rendered cells temperature-sensitive on galactose medium, but neither GAL1-driven PTC2 (Fig. 4B) nor the cdc28-4 genotype (data not shown) was sensitized to HNT1 status and thus a mechanism involving reduced Cdc28 activation by Cak1 in hnt1 strains is not experimentally supported.
Hnt1-deficiency causes synthetic loss of viability with mutant Kin28-binding proteins
Given the evidence inconsistent with Hnt1 as a positive regulator of Cak1, we considered the possibility that Hnt1 might be a positive regulator of Kin28. We reasoned that if Hnt1 enzymatic activity is a positive regulator of Kin28, then hnt1 deletion might cause synthetic loss of viability with destabilization of Ccl1 and Tfb3, the two other TFIIK components of general transcription factor TFIIH. Precedents for such a prediction are observations that temperature-sensitive mutations in the cyclin H homolog Ccl1 (41) and the MAT1 homolog Tfb3 (42,64), show synthetic loss of viability with destabilizing mutations in Kin28. Accordingly, the hnt1Δ::URA3 deletion was introduced into strains bearing ccl1-ts4 and tfb3-rig2ts23 mutations and plasmid pB42 that expresses HNT1 was added back to evaluate synthetic loss of viability. As shown in Fig. 4C and 4D, growth of hnt1 ccl1 and hnt1 tfb3 strains is limited by Hnt1 enzyme activity: Both double mutants show synthetic loss of viability that is relieved by yeast or rabbit Hint proteins and not relieved by expression of hnt1-His116Ala. Thus, synthetic interactions with Kin28 and Kin28-interacting proteins, Cak1, Ccl1 and Tfb3, suggest that Kin28 function is limited by Hnt1 enzyme activity.
Hnt1 deficiency is synthetically less viable with nonphosphorylatable Kin28
Given that Kin28 is phosphorylated by Cak1 on Thr162 (33,34), we reasoned that the basis for Cak1-dependence of hnt1 deletion strains might be that underphosphorylated Kin28 is hypersensitive to HNT1 status. The nonphosphorylatable KIN28-Thr162Ala allele is known to be intragenically synthetically less viable with kin28-ts16 and to be synthetically less viable with destabilizing mutations in tfb3 (34). However, prior reports were in apparent disagreement about the phenotype of nonphosphorylatable Kin28 on its own. While one report presented evidence that KIN28-Thr162Ala does not complement kin28-ts3 on galactose (33), another report found no phenotypic consequence of KIN28-Thr162Ala in experiments performed on glucose (34). Though the authors of earlier report discovered existence of two additional mutations that might have been responsible for noncomplementation (65), the apparent conflict might have been explained on the basis that the phenotypic assays differed in carbon source. In fact, while the restrictive temperature for wild-type strains differs in different backgrounds, the KIN28-Thr162Ala mutation reduces that temperature with respect to isogenic wild-type strains. As shown in Fig. 5, nonphosphorylatable Kin28 reduces the restrictive temperature on galactose by ∼1 to 2 °C while hnt1Δ reduces the restrictive temperature by 1 to 2 °C. The effects are additive, such that double mutants cannot grow at temperatures permissive for either single mutant. Thus, synthetic loss of viability on galactose between KIN28-Thr162Ala and hnt1Δ accounts for synthetic loss of viability between cak1 and hnt1. Additionally, it is now possible to reconcile the observations that while cells with a single KIN28-Thr162Ala mutation are aphenotypic on glucose media (34), they are moderately temperature-sensitive on galactose media (33). Further, in the experiment presented in Fig. 4A, we examined whether the amino acid at the Cak1 phosphorylation site of Kin28 affects the phenotype of cak1Δ hnt1Δ or cak1Δ HNT1 cells. While KIN28-Thr162Ala is synthetically less viable with hnt1Δ (Fig. 5), the nonphosphorylatable allele is not synthetically less viable with hnt1Δ in the absence of Cak1 (Fig. 4A). This confirms that what Cak1 does for Kin28 is simply to phosphorylate it at Thr162 and that hnt1Δ sensitizes cells to hypophosphorylated Kin28.
Fig. 5. Nonphosphorylatable Kin28 is limited by Hnt1 for growth on galactose.

Strain BY155-8c (genotype hnt1Δ kin28Δ, carrying KIN28 or KIN28-Thr162Ala on plasmids pB164 and pB165, respectively) was transformed with HNT1 plasmid pB42 or with empty vector pRS423, and transformants were grown at 39 °C and 37 °C on SgalC -his -leu media for 6 days.
Kin28 protein is not conditionally null in hnt1 mutants
Destabilized Tfb3 with nonphosphorylatable Kin28, a genetic combination that is synthetically less viable than either single mutation, leads to Kin28 protein degradation at the nonpermissive temperature (34). As hnt1 deletion also is also synthetically less viable with KIN28-Thr162Ala, we tested whether Kin28 might be degraded in hnt1 mutants on galactose as a function of temperature, and discovered that Kin28 is not destabilized by hnt1 deletion. As shown in Fig. 6A, Kin28 protein has a similar steady-state abundance in cells grown at increasing temperatures and the protein level is not reduced as a function of hnt1 deletion. Even in hnt1Δ cells at 37 °C in galactose media, which are nearing the nonpermissive temperature, Kin28 protein levels are not reduced.
Fig. 6. Kin28 protein is stable in hnt1 mutants and Kin28 overexpression in hnt1 mutants inhibits growth on galactose.
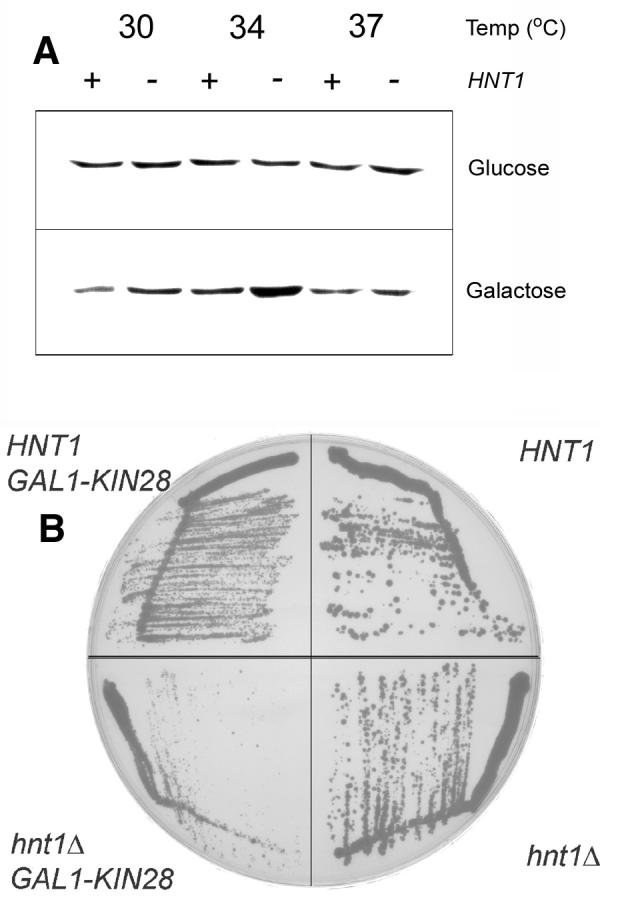
(A) Isogenic strains varying only at the HNT1 locus, expressing HA-tagged Kin28 were grown at the indicated temperatures in YDP or YPGal and subjected to a Western blot with anti-HA antibody.
(B) Strain BY8-5c (genotype hnt1Δ) was transformed with HNT1 plasmid pB42 or empty vector pRS423 and GAL1-KIN28 plasmid pB194 or the empty pRS425GAL1 control and transformants were grown at 37 °C on SGalC -his -leu media for 6 days.
Synthetic loss of viability of hnt1 with temperature-sensitive alleles of kin28, ccl1 and tfb3 suggested Kin28 might be conditionally null in hnt1Δ cells or disadvantaged in complex formation. With the observation that Kin28 is not degraded but stable in hnt1 strains, we tested whether Kin28 overexpression from the GAL1 promoter is deleterious to hnt1 mutants. As shown in Fig. 6B, GAL1-driven KIN28 expression mildly inhibits growth of HNT1 cells and seriously inhibits growth of hnt1 mutant cells. Thus, a straightforward interpretation of the disadvantage that hnt1 mutants have, particularly in combination with mutations in TFIIK components or excessive or nonphosphorylated Kin28, is that the Hint/Hnt1substrate AMP-X inhibits Kin28 complex formation.
DISCUSSION
HIT superfamily proteins are conserved as nucleotide-binding proteins (1). Whereas enzymes in the Fhit and GalT branches are found in subsets of living organisms, Hint orthologs are the ancestral prototypes that contain representatives in archaea, bacteria, and eukarya (19). To this date, no phenotypic consequence of loss of a Hint ortholog alone had been demonstrated in any organism. Loss of Aprataxin, a human Hint-branch protein whose yeast ortholog is Hnt3, is responsible for the progressive neurological disease ataxia with oculomotor apraxia (3,4) though biochemical substrates or a requirement for enzymatic activity have not been examined. Here we show that in yeast, Hnt1 is conditionally essential for growth on galactose media at elevated temperature. As was shown earlier, hnt1 deletion reduces the permissive temperature for kin28-ts3 cells (20), but this effect is not allele-specific for kin28, and synthetic reduction of viability by hnt1 deletion was discovered with temperature-sensitive mutations in cak1, ccl1, and tfb3, with the nonphosphorylatable KIN28-Thr162Ala allele, and with KIN28 overexpression. The single hnt1Δ mutant phenotype and the double mutant phenotypes allowed an evaluation of the requirement of the Hnt1 active site. Complete suppression by rabbit Hint protein, which shares only 22% sequence identity with Hnt1, and lack of complementation by Hnt1-His116Ala in every phenotypic assay suggested that Hint homologs have an enzymatic activity responsible for HNT1-function in vivo. We looked for an enzymatic activity in purified Hint and Hnt1 preparations, beyond the earlier report of an ADPase activity of 8.5 M-1 s-1 (54), and discovered that AMPNH2 is hydrolyzed by these enzymes in an active site-dependent manner with specificity constants of 2.9 × 106 M-1 s-1 and 1.0 × 106 M-1 s-1 , respectively. Though we do not know whether AMPNH2 is the authentic in vivo substrate of Hnt1, which is provisionally termed AMP-X, we note that AMPNH2 is found in eukaryotic cells (58) and that the enzyme that produces it from AMPSO4, a precursor of all organosulfur compounds, and ammonia has been found in a wide variety of organisms of bacterial and eukaryotic origin (59).
Because of the active-site dependence of Hnt1 function in vivo and in vitro, and synthetic loss of viability with kin28 and cak1, we proposed that Hint substrate AMP-X inhibits either Cak1 or Kin28 when hnt1 is inactivated. Genetic analysis showed that a Cak1 function is made more important in hnt1-deficient cells but this is not due to inhibition of Cak1 because cells that were constructed to live without Cak1 are strongly Hnt1-dependent. Additionally, while hnt1 deletion attenuates cells to the stability of known Kin28-binding proteins Ccl1 and Tfb3, implicating Kin28 as the direct target of AMP-X, a synthetic phenotype between hnt1 and KIN28-Thr162Ala revealed that the Cak1-dependence can be explained by an hnt1-inhibited form of Kin28 requiring phosphorylation by Cak1.
Kin28/Cdk7 may not be the only target of Hnt1/Hint regulation. Indeed, in bacteria, failure to cleave AMPNH2 or a related compound is likely to have completely different consequences as bacteria do not have a Cdk7 homolog. Additionally, it has been appreciated for several years that nucleoside-based prodrugs, such as 2′,3′-dideoxy-2′,3′-didehydro-thymidine can be made more potent by conversion to a 5′ phosphoramide. For example, 2′,3′-dideoxy-2′,3′-didehydro-thymidine containing an α-phosphate linked to alanine methylester is readily intracellularly available and is converted to the triphosphate after hydrolysis of the nucleoside monophosphoramide by an unknown intracellular enzymatic activity (66). The apparent identity of Hint to nucleoside monophosphoramidate hydrolase (60) and the fact that Hint and Hnt1 liberate AMP from AMP alanine methyl ester suggest that Hint may be the enzyme responsible for phosphoramide prodrug maturation in vivo.
Accumulation of a Hint/Hnt1 substrate may inhibit Kin28 either by reducing Kin28 function in a way that enforces dependence on Kin28 binding proteins and Cak1 activation or by reducing the ability of Kin28 to form a complex with TFIIK components Ccl1 and Tfb3 at elevated temperatures. The stability of Kin28 in hnt1deletion strains and the synthetic phenotype of Kin28 overexpression provide some support for Hnt1 as a positive regulator of Kin28 complex assembly and potentially indicate that a form of Kin28 accumulates in hnt1 cells that has dominant interfering activity, particularly on galactose media. Finally, while the idea that Hnt1 promotes Kin28 function simply by complex formation (20) appears to be incorrect, the two hybrid interactions reported could be explained if Hnt1 has a protein substrate, namely a nucleotidylated form of Kin28. The possibility that these enzymes have modified proteins as substrates is supported by the observation that Hint and Hnt1 readily hydrolyze a lysyl-AMP derivative. Ongoing work is designed to elucidate the biosynthesis and the identity of AMP-X, other possible targets of Hint and Hnt1 regulation, and whether Aprataxin and Hnt3 function as adenosine monophosphoramidases on either small molecule or macromolecular substrates.
Acknowledgements
We thank Edward Winter and Mark Solomon for helpful discussions and yeast strains, Alan E. Tomkinson for adenylylated human DNA ligase I, and Tia M. Maiolatesi and Edward M. Carlin for help with figures.
Footnotes
This work was supported by research grant CA75954 from the National Cancer Institute to C.B. and MCB9982645 from the National Science Foundation to L.D.B.
- HIT
- histidine triad
- GalT
- galactose-1-phosphate uridylyltransferase
- CAK
- cyclin-dependent kinase activating kinase
- CTDK
- RNA polymerase II carboxy-terminal domain kinase
- AMP-NH2
- adenosine-5′-monophosphoramidate.
REFERENCES
- 1.Brenner C, Garrison P, Gilmour J, Peisach D, Ringe D, Petsko GA, Lowenstein JM. Nat. Struct. Biol. 1997;4:231–238. doi: 10.1038/nsb0397-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilmour J, Liang N, Lowenstein JM. Biochemical Journal. 1997;326:471–7. doi: 10.1042/bj3260471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S. Nat Genet. 2001;29:184–8. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- 4.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M. Nat Genet. 2001;29:189–93. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 5.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 6.Siprashvili Z, Sozzi G, Barnes LD, McCue P, Robinson AK, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L, Schwartz G, Pierotti MA, Croce CM, Huebner K. Proc. Natl. Acad. Sci. USA. 1997;94:13771–13776. doi: 10.1073/pnas.94.25.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji L, Fang B, Yeh N, Fong K, Minna JD, Roth JA. Cancer Res. 1999;59:3333–3339. [PubMed] [Google Scholar]
- 8.Sard L, Accornero P, Tornielli S, Delia D, Bunone G, Campiglio M, Colombo MP, Gramegna M, Croce CM, Pierotti MA, Sozzi G. Proc. Natl. Acad. Sci. USA. 1999;96:8489–8492. doi: 10.1073/pnas.96.15.8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dumon KR, Ishii H, Vecchione A, Trapasso F, Baldassarre G, Chakrani F, Druck T, Rosato EF, Williams NN, Baffa R, During MJ, Huebner K, Croce CM. Cancer Res. 2001;61:4827–4836. [PubMed] [Google Scholar]
- 10.Fong LYY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Dolsky R, Druck T, McCue PA, Croce CM, Huebner K. Proc. Natl. Acad. Sci. USA. 2000;97:4742–4747. doi: 10.1073/pnas.080063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dumon KR, Ishii H, Fong LYY, Zanesi N, Fidanza V, Mancini R, Vecchione A, Baffa R, Trapasso F, During MJ, Huebner K, Croce CM. Proc. Natl. Acad. Sci. USA. 2001;98:3346–3351. doi: 10.1073/pnas.061020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y, Garrison PN, Barnes LD. Biochem. J. 1995;312:925–932. doi: 10.1042/bj3120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes LD, Garrison PN, Siprashvili Z, Guranowski A, Robinson AK, Ingram SW, Croce CM, Ohta M, Huebner K. Biochem. 1996;35:11529–11535. doi: 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- 14.Pekarsky Y, Campiglio M, Siprashvili Z, Druck T, Sedkov Y, Tillib S, Draganescu A, Wermuth P, Rothman JH, Huebner K, Buchberg AM, Mazo A, Brenner C, Croce CM. Proc. Natl. Acad. Sci. USA. 1998;95:8744–8749. doi: 10.1073/pnas.95.15.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pace HC, Hodawadekar SC, Draganescu A, Huang J, Bieganowski P, Pekarsky Y, Croce CM, Brenner C. Curr Biol. 2000;10:907–17. doi: 10.1016/s0960-9822(00)00621-7. [DOI] [PubMed] [Google Scholar]
- 16.Draganescu A, Hodawadekar SC, Gee KR, Brenner C. J. Biol. Chem. 2000;275:4555–4560. doi: 10.1074/jbc.275.7.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huebner K, Garrison PN, Barnes LD, Croce CM. Ann. Rev. Genet. 1998;32:7–31. doi: 10.1146/annurev.genet.32.1.7. [DOI] [PubMed] [Google Scholar]
- 18.Bruser T, Selmer T, Dahl C. Journal of Biological Chemistry. 2000;275(3):1691–8. doi: 10.1074/jbc.275.3.1691. [DOI] [PubMed] [Google Scholar]
- 19.Brenner C, Bieganowski P, Pace HC, Huebner K. J. Cell. Physiol. 1999;181:179–187. doi: 10.1002/(SICI)1097-4652(199911)181:2<179::AID-JCP1>3.0.CO;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korsisaari N, Makela TP. J. Biol. Chem. 2000;275:34837–34840. doi: 10.1074/jbc.C000505200. [DOI] [PubMed] [Google Scholar]
- 21.Fisher RP, Morgan DO. Cell. 1994;78:713–24. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- 22.Fisher RP, Jin P, Chamberlin HM, Morgan DO. Cell. 1995;83:47–57. doi: 10.1016/0092-8674(95)90233-3. [DOI] [PubMed] [Google Scholar]
- 23.Devault A, Martinez AM, Fesquet D, Labbe JC, Morin N, Tassan JP, Nigg EA, Cavadore JC, Doree M. Embo Journal. 1995;14:5027–36. doi: 10.1002/j.1460-2075.1995.tb00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serizawa H, Makela TP, Conaway JW, Conaway RC, Weinberg RA, Young RA. Nature. 1995;374:280–2. doi: 10.1038/374280a0. [DOI] [PubMed] [Google Scholar]
- 25.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Nature. 1995;374:283–7. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 26.Akoulitchev S, Makela TP, Weinberg RA, Reinberg D. Nature. 1995;377:557–60. doi: 10.1038/377557a0. [DOI] [PubMed] [Google Scholar]
- 27.Kaldis P, Sutton A, Solomon MJ. Cell. 1996;86:553–64. doi: 10.1016/s0092-8674(00)80129-4. [DOI] [PubMed] [Google Scholar]
- 28.Thuret JY, Valay JG, Faye G, Mann C. Cell. 1996;86:565–76. doi: 10.1016/s0092-8674(00)80130-0. [DOI] [PubMed] [Google Scholar]
- 29.Espinoza FH, Farrell A, Erdjument_Bromage H, Tempst P, Morgan DO. Science. 1996;273:1714–7. doi: 10.1126/science.273.5282.1714. [DOI] [PubMed] [Google Scholar]
- 30.Feaver WJ, Svejstrup JQ, Henry NL, Kornberg RD. Cell. 1994;79:1103–9. doi: 10.1016/0092-8674(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 31.Valay JG, Simon M, Dubois MF, Bensaude O, Facca C, Faye G. Journal of Molecular Biology. 1995;249:535–44. doi: 10.1006/jmbi.1995.0316. [DOI] [PubMed] [Google Scholar]
- 32.Cismowski MJ, Laff GM, Solomon MJ, Reed SI. Molecular and Cellular Biology. 1995;15:2983–92. doi: 10.1128/mcb.15.6.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espinoza FH, Farrell A, Nourse JL, Chamberlin HM, Gileadi O, Morgan DO. Molecular and Cellular Biology. 1998;18:6365–73. doi: 10.1128/mcb.18.11.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimmelman J, Kaldis P, Hengartner CJ, Laff GM, Koh SS, Young RA, Solomon MJ. Molecular and Cellular Biology. 1999;19:4774–87. doi: 10.1128/mcb.19.7.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaiser C, Michaelis S, Mitchell A. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press; Plainview, NY: 1994. [Google Scholar]
- 36.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Yeast. 1998;14:115–32. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Robinson JS, Klionsky DJ, Banta LM, Emr SD. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valay JG, Simon M, Faye G. Journal of Molecular Biology. 1993;234:307–10. doi: 10.1006/jmbi.1993.1587. [DOI] [PubMed] [Google Scholar]
- 39.Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. Nucleic Acids Research. 1996;24:2519–24. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cross FR, Levine K. Molecular and Cellular Biology. 1998;18:2923–31. doi: 10.1128/mcb.18.5.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valay JG, Dubois MF, Bensaude O, Faye G. Comptes Rendus de L Academie Des Sciences. Serie Iii, Sciences de La Vie. 1996;319:183–9. [PubMed] [Google Scholar]
- 42.Faye G, Simon M, Valay JG, Fesquet D, Facca C. Molecular and General Genetics. 1997;255:460–6. doi: 10.1007/s004380050518. [DOI] [PubMed] [Google Scholar]
- 43.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short Protocols in Molecular Biology. 3rd Wiley; New York: 1995. [Google Scholar]
- 44.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 45.Kunkel TA, Roberts JD, Zakour RA. Methods in Enzymology. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 46.Mumberg D, Muller R, Funk M. Nucleic Acids Research. 1994;22:5767–8. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herren B, Pech M. J Recept Res. 1993;13:725–738. doi: 10.3109/10799899309073689. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh S, Lowenstein JM. Gene. 1997;176:249–255. doi: 10.1016/0378-1119(96)00260-0. [DOI] [PubMed] [Google Scholar]
- 49.Rossomando EF, Hadjimichael J. International Journal of Biochemistry. 1986;18:481–4. doi: 10.1016/0020-711x(86)90193-x. [DOI] [PubMed] [Google Scholar]
- 50.Tomkinson A, Totty N, Ginsburg M, Lindahl T. Proc. Natl. Acad. Sci. USA. 1991;88:400–404. doi: 10.1073/pnas.88.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Razzell WE. Methods in Enzymology. 1963;6:236–258. [Google Scholar]
- 52.Lee H-J, Wilson IB. Biochem. Biophys. Acta. 1971;242:519–522. doi: 10.1016/0005-2744(71)90144-6. [DOI] [PubMed] [Google Scholar]
- 53.Wedekind JE, Frey PA, Rayment I. Biochemistry. 1996;35:11560–11569. doi: 10.1021/bi9612677. [DOI] [PubMed] [Google Scholar]
- 54.Lima CD, Klein MG, Hendrickson WA. Science. 1997;278:286–90. doi: 10.1126/science.278.5336.286. [DOI] [PubMed] [Google Scholar]
- 55.Lima CD, Damico KL, Naday I, Rosenbaum G, Westbrook EM, Hendrickson WA. Structure. 1997;5:763–774. doi: 10.1016/s0969-2126(97)00231-1. [DOI] [PubMed] [Google Scholar]
- 56.Pace HC, Garrison PN, Robinson AK, Barnes LD, Draganescu A, Rosler A, Blackburn GM, Siprashvili Z, Croce CM, Huebner K, Brenner C. Proc. Natl. Acad. Sci. USA. 1998;95:5484–9. doi: 10.1073/pnas.95.10.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abend A, Garrison PN, Barnes LD, Frey PA. Biochem. 1999;38:3668–3676. doi: 10.1021/bi981895j. [DOI] [PubMed] [Google Scholar]
- 58.Fankhauser H, Berkowitz GA, Schiff JA. Biochemical and Biophysical Research Communications. 1981;101:524–32. doi: 10.1016/0006-291x(81)91291-2. [DOI] [PubMed] [Google Scholar]
- 59.Fankhauser H, Schiff JA, Garber LJ. Biochemical Journal. 1981;195:545–60. doi: 10.1042/bj1950545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuba M, Okizaki T, Ohmori H, Kumon A. Int J Biochem. 1994;26:235–45. doi: 10.1016/0020-711x(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 61.Gumport RI, Lehman IR. Proc. Natl. Acad. Sci. USA. 1971;68:2559–63. doi: 10.1073/pnas.68.10.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Enke DA, Kaldis P, Holmes JK, Solomon MJ. Journal of Biological Chemistry. 1999;274:1949–56. doi: 10.1074/jbc.274.4.1949. [DOI] [PubMed] [Google Scholar]
- 63.Cheng A, Ross KE, Kaldis P, Solomon MJ. Genes and Development. 1999;13:2946–57. doi: 10.1101/gad.13.22.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feaver WJ, Henry NL, Wang Z, Wu X, Svejstrup JQ, Bushnell DA, Friedberg EC, Kornberg RD. Journal of Biological Chemistry. 1997;272:19319–27. doi: 10.1074/jbc.272.31.19319. [DOI] [PubMed] [Google Scholar]
- 65.Espinoza FH, Farrell A, Nourse JL, Chamberlin HM, Gileadi O, Morgan DO. Molecular and Cellular Biology. 2000;20:1898. doi: 10.1128/mcb.18.11.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balzarini J, Karlsson A, Aquaro S, Perno C-F, Cahard D, Naesens L, DeClercq E, McGuigan C. Proc Natl Acad Sci USA. 1996;93:7295–7299. doi: 10.1073/pnas.93.14.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]