

Abstract
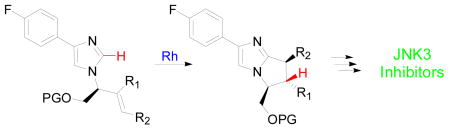
The efficient preparation of the privileged bicyclic bis-arylimidazole kinase inhibitor scaffold was accomplished using rhodium-catalyzed C-H activation and intramolecular alkylation. The key C-H activation/alkylation step represents one of the first evaluations of acyclic stereocontrol in catalyzed C-H activation/olefin alkylation processes. Several inhibitors of JNK3 were prepared using this sequence, with the most potent inhibitor having an IC50 value of 1.6 nM.
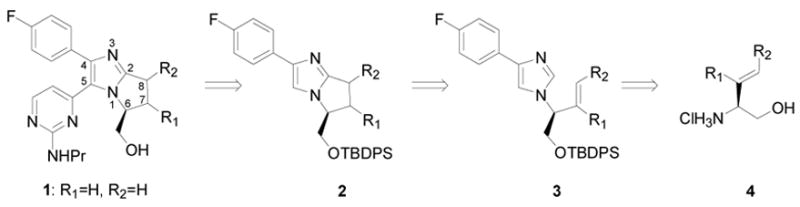
The privileged bis-arylimidazole framework is found in a variety of potent kinase inhibitors, some of which have entered clinical trials. Bicyclic bis-arylimidazole derivatives, such as the c-jun N-terminal kinase 3 (JNK3) inhibitor 1 (Figure 1),1 have recently been developed to enhance affinity and particularly selectivity, which remains a key challenge in kinase inhibitor drug discovery efforts.
Figure 1.
Retrosynthesis of Bicyclic Bis-Arylimidazole Kinase Inhibitors
Bicyclic bis-arylimidazole inhibitors, exemplified by inhibitor 1, represent challenging synthetic targets. Indeed, the synthesis of 1 required 14 linear steps and was accomplished in less than 6% overall yield.1 Herein, we report an efficient asymmetric synthesis of 1 in 11 linear steps and 13% overall yield with the key bicyclic imidazole core generated through catalytic C-H bond functionalization. We also successfully incorporated substituents in the C7 and C8 positions, substitution patterns difficult to access by the previously reported synthetic route, and in doing so observed the first examples of acyclic stereocontrol in metal-catalyzed C-H bond activation. Moreover, evaluation of the compounds synthesized by this route resulted in the identification of a JNK3 inhibitor even more potent than 1.
In our retrosynthetic analysis of the bicyclic bis-arylimidazole framework, we envisioned installing the C5 pyrimidine by a cross-coupling with 2 (Figure 1). Synthesis of the bicyclic imidazole core would be accomplished via rhodium-catalyzed C-H activation/annulation of 3. A van Leusen cycloaddition could be employed to generate 3 from 4, which can be readily prepared from commercially available starting material.
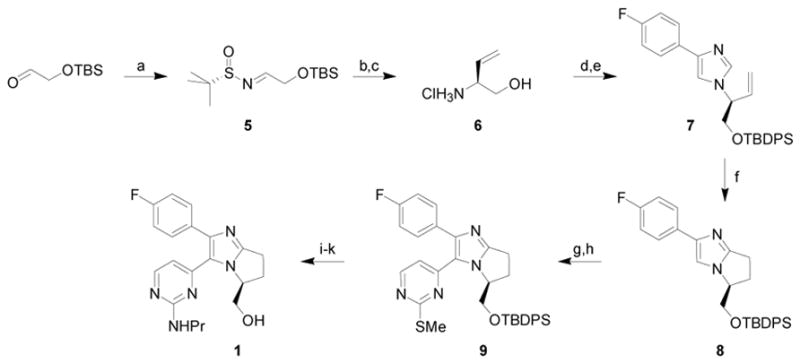
The synthesis of inhibitor 1 commenced with the condensation of (SS)-tert-butanesulfinamide and commercially available tert-butyldimethylsiloxyacetaldehyde to provide 5 in 86% yield (Scheme 1).2 The addition of vinylmagnesium bromide to 5 proceeded with 91:9 dr, and after chromatography, the major diastereomer was obtained in 69% yield. Acidic cleavage of the silyl and tert-butanesulfinyl groups provided 6 in nearly quantitative yield.2 Condensation of 6 with glyoxylic acid followed by treatment with 4-fluorophenyl tosylmethyl isonitrile3 generated the desired enantiomerically pure imidazole in 92% yield.4 Protection of the resulting primary alcohol as a tert-butyl diphenyl silyl (TBDPS) ether provided 7 in 98% yield.
Scheme 1.
Synthesis of Inhibitor 1
Conditions: (a) (SS)-tert-butanesulfinamide, CuSO4, CH2Cl2, 86%; (b) vinylmagnesium bromide, CH2Cl2, 0 °C to rt, 69% (single diastereomer); (c) 4N HCl, CH3OH, 99%; (d) 4-fluorophenyl tosylmethyl isonitrile3, glyoxylic acid, K2CO3, DMF, 92%; (e) TBDPSCl, iPr2EtN, DMAP, CH2Cl2, 98%; (f) [RhCl(coe)2]2, PCy3, MgBr2, toluene, 180 °C, 50%, 92% ee; (g) Br2, CH2Cl2, −78 °C, 94%; (h) 2-methylthio-4-trimethylstannylpyrimidine,6 Pd2(dba)3·CHCl3, PPh3, LiCl, CuI, dioxane, 170 °C, 85%; (i) OXONE®, THF, H2O, 79%; (j) propylamine, 78%; (k) Bu4NF, THF, 100%.
Due to the steric hindrance introduced by the C6 substituent, forcing conditions were required to achieve good conversion in the C-H activation/annulation step. Ultimately, cyclization of 7 was accomplished by using 5% [RhCl(coe)2]2 and 15% PCy3 to generate the active catalyst with 5% MgBr2 as an additive and toluene as solvent at 180 °C to provide 8 in 50% yield and with 92% ee (Scheme 1).5 Olefin isomerization and olefin reduction products were also isolated in 11% and 6% yield, respectively. Competitive olefin isomerization has been shown to occur under these conditions and is likely responsible for the minor erosion of enantiomeric excess observed during the cyclization.5c
Treatment of 8 with Br2 resulted in bromination of the imidazole ring at the C5 position in 94% yield. The resulting bromide was subjected to Stille cross coupling conditions in the presence of 2-methylthio-4-trimethylstannylpyrimidine6 to provide 9 in 85% yield (Scheme 1).7 The requisite amine was generated by oxidation of the thioether to the sulfone (79% yield) followed by addition of propylamine (78% yield). Quantitative Bu4NF cleavage of the silyl ether provided 1 in 13% overall yield.
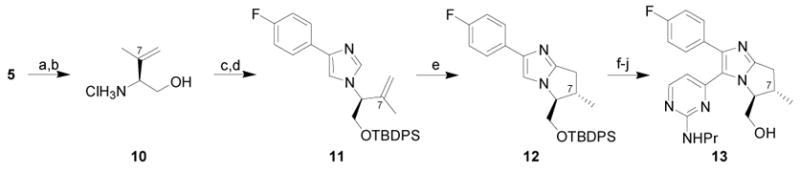
To demonstrate the flexibility of our synthetic approach toward bicyclic bis-arylimidazole systems and to explore acyclic stereocontrol in the C-H activation/annulation step, we generated derivatives of 1 containing methyl substituents at the C7 or C8 positions. By employing isopropenylmagnesium bromide in place of vinylmagnesium bromide in the previously described sequence we were poised to generate a derivative with a C7 methyl substituent (Scheme 2). The sequence proceeded smoothly with excellent yields and selectivity for the generation of enantiomerically pure 11 (78% from 5).
Scheme 2.
Synthesis of C7 Methyl Bicyclo bis-Arylimidazole Derivative
Conditions: (a) isopropenylmagnesium bromide, CH2Cl2, −78 °C to rt, 90% (single diastereomer); (b) 4N HCl, CH3OH, 96%; (c) 4-fluorophenyl tosylmethyl isonitrile, glyoxylic acid,3 K2CO3, DMF; (d) TBDPSCl, iPr2EtN, DMAP, CH2Cl2, 86% (over 2 steps); (e) [RhCl(coe)2]2, PCy3, MgBr2, toluene, 180 °C, 61%, 3:1 dr, 98% ee; (f) Br2, CH2Cl2, −78 °C, 96%; (g) 2-methylthio-4-trimethylstannylpyrimidine,6 Pd2(dba)3·CHCl3, PPh3, LiCl, CuI, dioxane, 170 °C, 66%; (h) OXONE®, THF, H2O, 77% (single diastereomer); (i) propylamine, 93%; (j) TBAF, THF, 87%.
Treatment of 11 with the previously described rhodium-catalyzed C-H activation/annulation conditions resulted in the formation of 12 in 61% yield as a 3:1 mixture of inseparable diastereomers (Scheme 2).5 The tetrasubstituted olefin isomerization and olefin reduction products also were isolated in 24% and 3% yields, respectively. For operational simplicity the diastereomers were separated at a later stage in the synthesis.
Bromination of 12 proceeded in 96% yield, and subsequent Stille cross-coupling installed the requisite arene at the C5 position in 66% yield (Scheme 2).7 Oxidative sulfone formation proceeded in quantitative yield and allowed for separation of the diastereomeric mixture by silica gel chromatography. Sulfone displacement and silyl ether cleavage then provided the C7 methyl derivative 13 in 15% overall yield from the commercially available tert-butyldimethylsiloxy-acetaldehyde.
The C8 methyl derivative was generated in a similar fashion (Scheme 3). The addition of allylmagnesium bromide to imine 5 proceeded with excellent yield and selectivity. Subsequent transformations generated enantiomerically pure 15 in excellent overall yield (67% from 5). Treatment of 15 with [RhCl(coe)2]2 in the presence of PCy3 and MgBr2 at 180 °C provided the bicyclic imidazole core with 86:14 dr and allowed for the isolation of 16 as a single diastereomer in 52% yield with 92% ee (Scheme 3).5 Consistent with previous studies, olefin isomerization to the internal disubstituted position occurs prior to annulation.5c Completion of the synthesis was accomplished by bromination of 16, Stille cross-coupling7 to install the pyrimidine, oxidation and formal nucleophilic displacement of the resulting sulphone, and silyl ether cleavage to provide 17 (61% from 16). The overall yield for the preparation of the C8 methyl derivative 17 from the common aldehyde starting material was 18%.
Scheme 3.
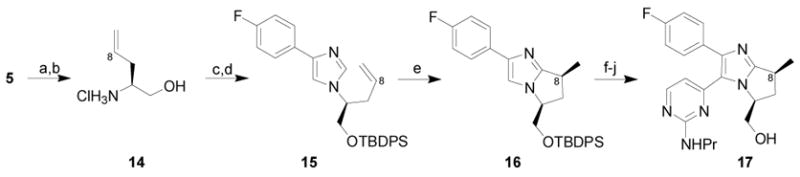
Synthesis of C8 Methyl Bicyclo Bis-Arylimidazole Derivative
Conditions: (a) allylmagnesium bromide, CH2Cl2, −78 °C to rt, 92% (single diastereomer); (b) 4N HCl, CH3OH, 94%; (c) 4-fluorophenyl tosylmethyl isonitrile,3 glyoxylic acid, K2CO3, DMF; (d) TBDPSCl, iPr2NEt, DMAP, CH2Cl2, 78% (over 2 steps); (e) [RhCl(coe)2]2, PCy3, MgBr2, toluene, 180 °C, 52%, 92% ee; (f) Br2, CH2Cl2, −78 °C, 80%; (g) 2-methylthio-4-trimethylstannylpyrimidine,6 Pd2(dba)3·CHCl3, PPh3, LiCl, CuI, dioxane, 170 °C, 84%; (h) OXONE®, THF, H2O; (i) propylamine, 91% (over 2 steps); (j) TBAF, THF, 99%.
The ability of compounds 13 and 17, and their enantiomers ent-13 and ent-178 prepared by employing (RS)-tert-butanesulfinamide in the previously described synthetic sequences, to inhibit JNK3 were determined (Table 1). While potency comparable to 1 was observed for 13, 17 and ent-17, a three to four-fold increase in potency was observed for ent-13.
Table 1.
Inhibition of c-Jun N-Terminal Kinase 3
| Inhibitor | IC50 (nM)a,b | Inhibitor | IC50 (nM)a,b |
|---|---|---|---|
| 1 | 5.38 (±1.40) | ||
| 13 | 5.29 (±1.25) | ent-13 | 1.63 (±0.34) |
| 17 | 4.85 (±0.54) | ent-17 | 8.10 (±1.82) |
Homogeneous time resolved fluorescence assay performed in quadruplicate.
IC50 values confirmed using standard radioactivity assay (see Supporting Information).
In summary, we have developed an efficient and flexible synthetic route for the generation of the potent kinase inhibitor 1 with the key transformation being intramolecular alkylation via rhodium-catalyzed C-H bond activation. By utilizing this intramolecular alkylation, our approach easily allows for the introduction of substituents at the C7 and C8 positions, derivatives that would be much more difficult to access by the previously published route to 1. Determination of the selectivity profiles of the synthesized analogues and preparation of even more potent compounds are in progress.
Supplementary Material
Complete experimental details and spectral data for all compounds described. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the NIH GM069559 to J.A.E., the Director and Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC03-76SF00098 to R.G.B. Support for M.Y. by Sankyo is also gratefully acknowledged.
References
- 1.Graczyk PP, et al. Bioorg & Med Chem Lett. 2005;15:4666–4670. doi: 10.1016/j.bmcl.2005.07.076. [DOI] [PubMed] [Google Scholar]
- 2.Tang TP, Volkman SK, Ellman JA. J Org Chem. 2001;66:8772–8778. doi: 10.1021/jo0156868. [DOI] [PubMed] [Google Scholar]
- 3.Sisko J, Mellinger M, Sheldrake PW, Baine NH. Tetrahedron Lett. 1996;37:8113–8116. [Google Scholar]
- 4.Sisko J, Kassick AJ, Mellinger M, Filan JJ, Allen A, Olsen MA. J Org Chem. 2000;65:1516–1524. doi: 10.1021/jo991782l. [DOI] [PubMed] [Google Scholar]
- 5.(a) Weidemann SH, Lewis JC, Ellman JA, Bergman RG. J Am Chem Soc. 2006;128:2452–2462. doi: 10.1021/ja0576684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tan KL, Bergman RG, Ellman JA. J Am Chem Soc. 2002;124:3203–3205. doi: 10.1021/ja0281129. [DOI] [PubMed] [Google Scholar]; (c) Tan KL, Bergman RG, Ellman JA. J Am Chem Soc. 2001;123:2685–2686. doi: 10.1021/ja0058738. [DOI] [PubMed] [Google Scholar]
- 6.Ahaidar A, Fernandez D, Danilon G, Cuevas C, Manzanares I, Albericio F, Joule JA, Alvarez M. J Org Chem. 2003;68:10020–10029. doi: 10.1021/jo035332b. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez M, Fernandez D, Joule JA. Tetrahedron Lett. 2001;42:315–317. [Google Scholar]
- 8.Researchers at Easai had previously demonstrated that ent-1 had inhibitory activity comparable to that of 1.1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental details and spectral data for all compounds described. This material is available free of charge via the Internet at http://pubs.acs.org.