

Abstract
Liver microsomes are widely used to study xenobiotic metabolism in vitro and covalent binding to microsomal proteins serves as a surrogate marker for toxicity mediated by reactive metabolites. We have applied liquid chromatography-tandem mass spectrometry (LC-MS-MS) to identify protein targets of the biotin-tagged model electrophiles 1-biotinamido-4-(4′-[maleimidoethylcyclohexane]-carboxamido)butane (BMCC) and N-iodoacetyl-N-biotinylhexylenediamine (IAB) in human liver microsomes. The biotin-tagged peptides resulting from in-gel tryptic digestion were enriched by biotin-avidin chromatography and LC-MS-MS was used to identify 376 microsomal cysteine thiol targets of BMCC and IAB in 263 proteins. Protein adduction was selective and reproducible and only 90 specific cysteine sites in 70 proteins (approximately 25% of the total) were adducted by both electrophiles. Differences in adduction selectivity correlated with different biological effects of the compounds, as IAB, but not BMCC induced ER stress in HEK293 cells. Targeted LC-MS-MS analysis of microsomal glutathione-S-transferase cysteine 50, a target of both IAB and BMCC, detected time-dependent adduction by the reactive acetaminophen metabolite N-acetyl-p-benzoquinoneimine during microsomal incubations. The results indicate that electrophiles selectively adduct microsomal proteins, but display differing target selectivities that correlate with differences in toxicity. Analysis of selected microsomal protein adduction reactions thus could provide a more specific indication of potential toxicity than bulk covalent binding of radiolabeled compounds.
Liver microsomes are widely used to study the metabolism of drugs and other xenobiotics in vitro. Microsomes are membrane vesicles that consist largely of ER1, with lesser contributions from plasma membranes, lysosomes, peroxisomes, nuclear membranes and cytoplasm and have high specific contents of CYP, UGT, GST, FMO and other xenobiotic-metabolizing enzymes (1). In vitro microsomal metabolism studies provide a useful indication of primary oxidative pathways for metabolism of drugs and are widely employed in exploratory drug metabolism and safety evaluation (2). Microsomal metabolism studies also are widely used in toxicity screening, as reactive metabolites bind covalently to microsomal proteins (3). This covalent binding often reflects the tendency of a compound to produce covalent binding and toxicity in vivo (4).
Despite the widespread use of in vitro microsomal covalent binding to assess the formation of reactive metabolites, very little is known about the microsomal protein targets of reactive metabolites. In common practice, covalent binding assays detect only bound radioactivity after incubations with radiolabeled substrates. Levels of covalent binding are low (typically in the range of pmol mg−1 microsomal protein) and potential for modification of perhaps hundreds of microsomal proteins has precluded the characterization of covalent binding in any molecular detail (3).
Identification of the protein targets of covalent binding becomes important in three contexts. First, elucidation of electrophile targeting of proteins at the sequence and structure level provides new insights into the factors that govern the susceptibility of proteins to damage. Second, drugs and chemicals whose metabolites bind covalently to proteins nevertheless may differ considerably in toxicity (5, 6). Identification of targets and binding patterns whose modification is associated with toxic outcomes could enable prediction of toxicity with greater specificity. Finally, microsomal covalent binding is also significant because microsomes are formed from ER, which plays a key role in cellular stress responses.
ER stress describes the cellular response to unfolded proteins, perturbations in protein thiol-disulfide redox status and protein glycoslyation abnormalities in the ER (7, 8). The ER stress response is thought to be triggered by binding of the ER resident chaperone Bip/GRP78 to unfolded or misfolded proteins in the ER lumen, thereby dissociating BiP from and activating the proteins PERK, ATF6 and IRE1. BiP is a member of the Hsp70 family of protein chaperones and increases in levels of unfolded proteins are thought to lead to BiP dissociation from these mediators, which then down regulate protein translation and up regulate transcription of ER stress response genes and activate complementary stress pathways (8). The ER stress pathway is induced by reactive electrophiles and thus appears to govern susceptibility and adaptation to chemical toxicity (9, 10). Recent work also indicates that S-nitrosylation of ER proteins contributes to ER stress associated with neurodegenerative disease (11).
Previous work has applied immunoblotting methods or radiolabeling to detect and identify a small number of ER proteins adducted by reactive metabolites of halothane, diclofenac, bromobenzene, acetaminophen and other xenobiotics (12–20). Immunochemical approaches are limited by the availability, sensitivity and specificity of antibodies against adducts, whereas radiolabeling is limited by the specific activity of radiolabel. In both cases, detection of bands or spots on 1D or 2D gels led to the identification of protein(s) that co-migrate with the detected label. These analyses identified protein and peptide sequences, but did not identify specific adducts at the level of amino acid sequence. This reflects not only the limitations of the methods used (e.g., Edman sequencing), but also the difficulty of detecting adducts in the presence of excess unmodified proteins in complex mixtures.
The use of affinity-tagged model electrophiles has recently provided new opportunities to probe the specificity and selectivity of covalent protein binding by model electrophiles. Biotin-tagged electrophiles containing electrophilic functional groups can be used to probe complex proteomes for protein targets that display affinity and high reactivity towards specific chemotypes (21, 22). Application of biotin-tagged model electrophiles together with shotgun proteome analyses by LC-MS-MS has recently enabled sequence-specific identification of over 500 electrophile protein targets in cytoplasmic and nuclear protein fractions from human cells (23).
Here we have applied the same approach to identify protein targets of biotin-tagged model electrophiles BMCC and IAB (Figure 1) in human liver microsomes. The biotin-tagged peptides resulting from in-gel tryptic digestion were enriched by biotin-avidin chromatography and LC-MS-MS was used to identify 376 microsomal cysteine thiol targets of BMCC and IAB in 263 proteins. Protein adduction was selective and reproducible and correlated with different biological effects of the compounds, as IAB, but not BMCC induced ER stress in HEK293 cells. The results indicate that electrophiles selectively adduct microsomal proteins, but display differing target selectivities. Assessment of microsomal protein adduction selectivity thus could provide a more specific indication of potential toxicity than bulk covalent binding of radiolabeled compounds.
Figure 1.

Structures of IAB and BMCC.
EXPERIMENTAL PROCEDURES
Chemicals and reagents
BMCC, IAB and DTT were obtained from Pierce (Rockford, IL). Streptavidin sepharose was from Amersham Bioscience (Piscataway, NJ) and an Alexa Fluor 680-conjugated streptavidin was from Molecular Probes (Eugene, OR). Mass spectrometry grade trypsin (Trypsin Gold) was purchased from Promega (Madison, WI) and other reagents were from Sigma (St. Louis, MO). Antibody to BiP/GRP78 was from BD Transduction Laboratories (San Jose, CA). Antibody to α-actin was from Abcam (Cambridge, MA). Antibody to calreticulin was from Affinity Bioreagents (Golden, CO). Alexa Fluor 680-conjugated anti-mouse IgG secondary antibody was from Molecular Probes (Junction City, OR).
Human liver microsomes and cell culture
Human liver microsomes were generously provided by Drs. F.P. Guengerich and M.V. Martin. Human livers were obtained from organ donors through Tennessee Donor Services (Nashville, TN), stored at −80°C and used to prepare microsomes by differential ultracentrifugation as described previously (24, 25). The final microsomal pellet was washed and resuspended in 10 mM Tris acetate buffer (pH 7.4) containing 1 mM EDTA and 20% glycerol. Protein concentrations were estimated using the bichinchoninic acid method according to the supplier’s recommendations (Pierce Chemical Co., Rockford, IL) using bovine serum albumin as a standard. The isolated microsomes were stored at −80°C. For treatment with electrophiles, 3–5 mg mL−1 of microsomes were incubated with 100 μM BMCC or IAB in 50 mM Tris-HCl, 5 mM EDTA, pH 8.0 at 37°C for 30 min and the reactions were stopped by adding DTT to a final concentration of 10 mM at room temperature. The samples then were suspended in LDS 4X loading buffer (Invitrogen, Carlsbad, CA) for SDS-PAGE.
HEK293 cells were obtained frozen at low passage from Master Cell Bank cultures from GIBCO Invitrogen Cell Culture (Grand Island, NY). Cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 μg/mL penicillin and 100 μg/mL streptomycin. Cells at approximately 90% confluence in 100 mm plates were washed with PBS and treated with up to 100 μM of electrophile or equal volumes of vehicle (DMSO at 0.3% of total volume) delivered in 4 mL DMEM with 5% fetal bovine serum.
SDS-PAGE and western blot analyses
Microsomal proteins were separated on a NuPAGE 10% SDS-PAGE gel (Invitrogen, Carlsbad, CA) using NuPAGE MOPS SDS running buffer. Each gel lane was loaded with 40 μg microsomal protein in 4X LDS buffer. Proteins on the gels were stained with colloidal Coomassie blue (Invitrogen). For detection of electrophile adduction, BMCC- or IAB-treated microsomal proteins separated on gels were transferred to PVDF membranes and biotin-labeled proteins were detected by immunoblotting with Alexa Fluor 680-conjugated streptavidin (Molecular Probes, Junction City, OR). For the analysis of BiP induction by electrophiles, HEK293 cells were treated with IAM, IAB, BMCC or NEM for 0, 3, 8 and 24 h and then lysed in NP-40 buffer (1% NP-40, 50 mM Tris-Cl, pH7.4, 150 mM NaCl, 5 mM EDTA) with protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Proteins (15 μg) were separated on 10% SDS-PAGE gels and transferred to PVDF membranes for western blot. BiP was detected by immunoblot analysis with anti-BiP at a 1:500 dilution. Anti-α-actin (loading control) was used at a 1:3000 dilution. Detection was done with an Alexa Fluor 680-conjugated anti-mouse secondary antibody. Immunoblot images were acquired and processed using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
To detect adduction of BiP and calreticulin by western blotting, microsomes (400 μg protein) treated with BMCC or IAB were incubated with streptavidin sepharose high performance beads (GE Life Sciences, Piscataway, NJ) to capture biotinylated proteins. The supernatant (designated “flow-through”) was collected and the beads then were washed twice with RIPA buffer and once with water. Bound proteins then were eluted with 25% acetonitrile/1M acetic acid for 1 h at room temperature, followed by a second elution overnight. Proteins taken from lysates, from flow-through and eluate fractions were separated on 10% SDS-PAGE gels and transferred to PVDF membranes for western blotting. BiP and calreticulin in these fractions were detected by immunoblot analysis with anti-BiP at a 1:500 dilution or anti-calreticulin at 1:1000 dilution. Detection and image analysis were done as described above.
In-gel digestion and peptide extraction
SDS-PAGE gel lanes containing microsomal proteins were cut into 18 slices comprising the entire molecular weight range and each individual gel slice was further cut into 1 mm pieces, which were washed in 100 mM ammonium bicarbonate and subjected to reduction and alkylation with DTT and iodoacetamide as described (26). Gel pieces were then dehydrated with acetonitrile and residual solvent was evaporated in vacuo in a SpeedVac vacuum centrifuge (Thermo-Fisher Scientific, Waltham, MA). Dried gel pieces were rehydrated on ice in 25 mM ammonium bicarbonate containing 10 μg mL−1 of trypsin for 10 min and then incubated overnight at 37°C. Digested peptides were extracted twice from the gel pieces with 1% aqueous formic acid/60% acetonitrile for 15 min and then evaporated to dryness in a SpeedVac. Samples were resuspended in 100 mM ammonium bicarbonate for affinity capture.
Affinity capture of biotinylated peptides
Biotinylated peptides from each gel slice were subjected to affinity capture using 0.1 mL of a 50/50 (w/v) slurry of streptavidin sepharose high performance beads. Beads first were equilibrated with 100 mM ammonium bicarbonate and then peptide samples were incubated for 30 min with streptavidin beads with gentle rotation. The beads were washed three times with 100 mM ammonium bicarbonate and twice with deionized water, then bound peptides were eluted with 5 % formic acid/70 % acetonitrile for 30 min twice followed by a third elution overnight. The mixtures were centrifuged at 5,000 g for 5 min and the supernatants were collected and evaporated in a SpeedVac and resuspended in 0.1 % aqueous formic acid for LC-MS-MS analysis.
LC-MS-MS analyses and identification of protein adducts
Peptide samples were analyzed on a Thermo LTQ linear ion trap instrument (Thermo Electron, San Jose, CA) equipped with a Thermo Surveyor solvent delivery system, autosampler and a microelectrospray source and equipped with a 6 cm (100 μm inner diameter) fused silica capillary C18 desalt pre-column operated as described previously (27). Peptides were loaded and desalted for 15 min with 2% acetonitrile/0.1% formic acid and then resolved by reverse-phase chromatography on an 11 cm fused silica capillary column (100 μm inner diameter) packed with Jupiter C-18 (5 μm) (Phenomenex Inc., Torrance, CA) with the flow set at 700 nL min−1. The mobile phase consisted of 0.1 % formic acid in either HPLC grade water (A) or acetonitrile (B). Peptides were eluted initially with 98% A for 15 min, then a linear gradient to 75 % A by 50 min, then held at 10% A from 65–74 min and the programmed to 95 % A at 75 min and returned to 100% A from 80–95 min.
MS-MS spectra of the peptides were acquired using data-dependent scanning in which one full MS spectrum (mass range 400–2000 m/z) was followed by three MS-MS spectra. MS-MS spectra were recorded using dynamic exclusion of previously analyzed precursors for 30s with a repeat duration of 2 min. The MS-MS spectra were matched to human database sequences (UniRef100; http://www.pir.uniprot.org/database/download.shtml) with the SEQUEST database search algorithm (28). S-carboxamidomethylation of Cys (+57 amu), oxidation of Met (+16), BMCC adduction at Cys (+533 amu and +549 for oxidized form) and IAB adduction at Cys (+382 and +398 for oxidized form) were specified as dynamic modifications to identify spectra of adducts. Criteria for acceptance of MS-MS spectrum to database sequence matches for adducted peptides have been described previously (23).
Microsomal metabolism of acetaminophen
Microsomal metabolism of acetaminophen to NAPQI was confirmed by trapping this metabolite as its GSH conjugate by modification of previously described methods (29, 30). The reaction mixture contained 500 μM substrate compound, 1 mg mL−1 microsomal protein, 10 mM GSH, 1 mM NADP+, 10 mM glucose-6-phosphate, 1 U mL−1 glucose-6-phosphate dehydrogenase, 5 mM MgCl2 and 50 mM potassium phosphate buffer, pH 7.4. After a 60 min incubation at 37°C, reactions were terminated by adding one volume (0.5 mL) of ice-cold acetonitrile to precipitate proteins. After vortexing and centrifugation, the resulting supernatant was diluted further with an equal volume of water for LC-MS-MS analysis of GSH conjugates. Incubations for detection of mGST protein adducts of the substrates were performed similarly, except that the reaction (0.2 mL final volume) was performed without GSH and was terminated by adding LDS loading buffer (Invitrogen, Carlsbad, CA) and 75 mM DTT.
LC-MS-MS analysis of GSH conjugates
Samples were subjected to chromatographic separation on a 150 × 2.0 mm Luna 5μm C18 column (Phenomenex, Torrance, CA) and eluates were analyzed with a Thermo LCQ DecaXP ion trap mass spectrometer (ThermoElectron, San Jose, CA) operated in precursor ion monitoring mode to detect ions that fragment with loss of 129 Da, which corresponds to the loss of the glutamate residue (31). Elution solvents were 5% acetonitrile, 95% H2O and 0.5% acetic acid (solvent A) and 95% acetonitrile, 5% H2O and 0.5% acetic acid (solvent B). GSH conjugates were eluted with a segmented linear gradient programmed from 0–2 min at 95% A/5% B, then programmed from 5% to 85% B from 2–35 min, then held at 85% B from 35–45 min, and finally to 95% A from 46–56 min at a flow rate of 200 μL min−1. The GS conjugate of acetaminophen was detected at 4.6 min. MS-MS spectra of the M+H+ ions of the conjugate was obtained to confirm the identity of the GS-NAPQI conjugate.
Targeted LC-MS-MS analyses of mGST adducts of acetaminophen
Microsomal incubations with acetaminophen and SDS-PAGE separations of treated microsomal proteins and colloidal Coomassie staining were performed as described above. The prominent band at approximately 17 ± 5 kDa corresponding to mGST was cut and two bands from adjacent lanes representing replicate samples were combined for each analysis. Following in-gel tryptic digestion as described above, the peptides were evaporated in vacuo and then reconstituted in 100 μL of 10% acetonitrile, 0.1% formic acid. The samples (2 μL) were injected on a LTQ linear ion trap mass spectrometer equipped as described above. For reverse-phase LC, solvent A contained 0.1% formic acid in water, and solvent B contained 0.1% formic acid in acetonitrile. The peptides were desalted for 15 min with 2% solvent B then eluted with a linear gradient from 2% to 25% solvent B over 35 min, then to 90% B over 15 min at a flow rate of 0.7 μL min−1. For SRM analysis, the MH22+ to y9+ fragmentation transitions of the predicted adducts of the mGST peptide containing Cys50 (VFANPEDC*VAFGK) modified by NAPQI (m/z 773.7 → m/z 1114.4 ± 1.0) were recorded. MS-MS instrument parameters included isolation width 2.0, normal CID 35%, activation Q 0.25 and activation time 30 ms. For SIM analyses, MS-MS of the carboxamidomethylated cysteinyl (+57 Da) peptide (resulting from IAM treatment during workup) was monitored by collision-induced dissociation of the selected ion at m/z 727.6 (z = +2) as an internal standard, and MS-MS of the predicted Cys50 peptide adduct also was monitored to confirm the reliability of SRM results. The ratios of each adduct SRM peak area divided by the internal standard IAM adduct SIM peak area were calculated for the adduct at each time point of incubation. Because the mole fraction of Cys50 modified was low, the percentage of the IAM-modified Cys50 peptide in each sample was approximately constant. Spectra and chromatograms were analyzed with Thermo Xcalibur version 2.0 software.
RESULTS
Induction of ER stress by electrophiles in HEK293 cells
As in our previous work (23), we employed electrophile probes with two different alkylating chemistries to study the relationship between protein adduction and ER stress. The biotin-tagged thiol-reactive electrophiles IAB and BMCC (Figure 1) display reaction chemistries that are characteristic of many toxicologically relevant electrophiles. IAB reacts with protein thiols by an SN2 reaction mechanism typical of aliphatic epoxides, and alkyl halides and episulfonium ions, whereas BMCC reacts with thiols by a Michael addition mechanism typical of quinones and α,β-unsaturated carbonyls. We also employed the corresponding non-biotinylated analogs, IAM and NEM in our initial studies of ER stress in HEK 293 cells.
Stevens and colleagues demonstrated that IAM induces upregulation of hsp70 and grp78 expression and causes both apoptotic and necrotic cell death in renal epithelial cells (9, 32). However, sublethal concentrations of IAM induce protective induction of an ER stress response that blocks apoptosis by subsequent IAM treatments (10). To assess the relationship between protein alkylation and induction of ER stress, we treated HEK293 cells with the alkylating agents IAM and NEM and their biotinylated analogs IAB and BMCC. We also treated cells with the bona fide ER stress inducers DTT (an inhibitor of disulfide formation), thapsigargin (an inhibitor of Ca2+-ATPase) and tunicamycin (an inhibitor of protein N-glycosylation). Treatment with 25 μM IAM and 100 μM IAB as well as ER stress inducers substantially increased BiP protein expression in 24 hr (Figure 2). However, treatment with 25 μM NEM or 100 μM BMCC failed to induce BiP expression, suggesting that some protein adduction specific to IAM and IAB induces ER stress.
Figure 2.

Treatment of HEK293 cells with 25 μM IAM or 100 μM IAB induced BiP expression, as measured by western blotting (upper panel). Treatment with 25 μM NEM or 100 μM BMCC did not induce BiP expression (lower panel).
Protein targets of IAB and BMCC
Preliminary studies with microsomes prepared from HEK293 cells failed to yield reproducible preparations with a high enough specific content of ER proteins for proteomic analysis (data not shown). The choice of human liver microsomes for these studies was based on the abundance of ER protein targets in these preparations and their utility as a model system to study the bioactivation of model substrates to electrophiles (see below). Electrophile concentration and exposure time was determined empirically, with the objective of producing detectable protein labeling without saturating the available targets (23). Incubation with 100 μM IAB or BMCC produced time-dependent accumulation of adducts (Figure 3) and incubation for 30 min appeared to provide sufficient labeling that did not saturate the pool of protein targets. Visual inspection of the blots indicated clear differences in distribution of adducts formed by IAB and BMCC (Figure 3).
Figure 3.
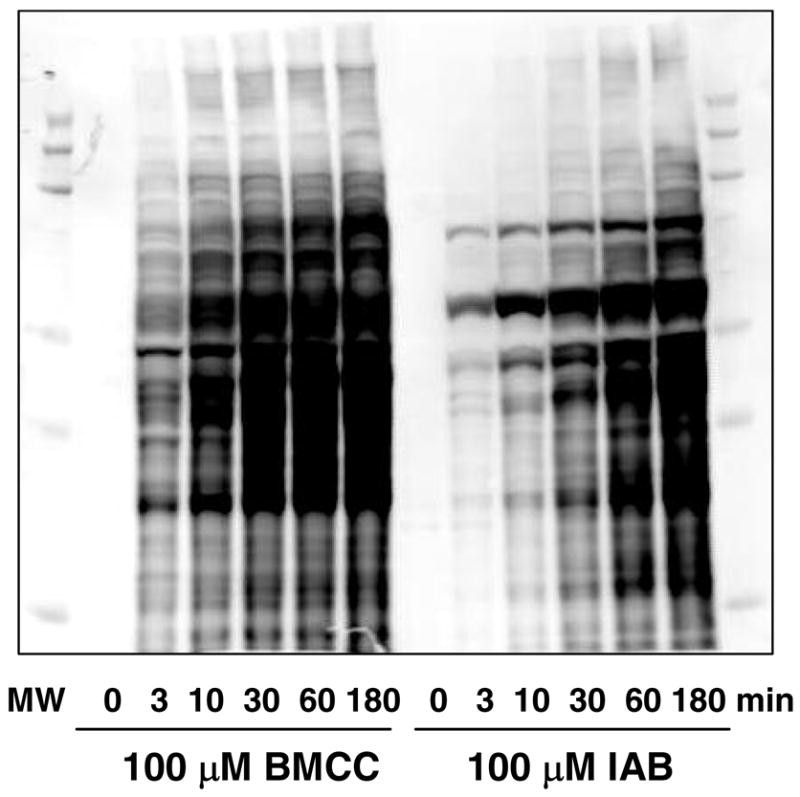
Time-dependent increase in covalent adduction of human liver microsomal proteins by 100 μM IAB and BMCC. After exposures to the electrophiles for the times indicated, samples of microsomal protein were analyzed by SDS-PAGE and western blotting with streptavidin. Note differences in adduction patterns produced by IAB and BMCC.
The major challenge in analyzing microsomal protein targets of electrophiles is the presence of many hydrophobic and membrane-associated proteins. We used the “GeLC-MS” approach, in which the proteins are resolved by 1D SDS-PAGE, the gel is then sliced into fractions and tryptic peptides are generated by in-gel digestion (33, 34). This method separates membrane proteins from detergents and other common contaminants of membrane preparations and enables efficient digestion even of hydrophobic membrane proteins. Adducted peptides from the in-gel digests were captured with streptavidin-conjugated sepharose beads and then subjected to LC-MS-MS analysis.
Peptide adducts were identified by database searching with Sequest (28), in which variable modifications to cysteines were specified as the expected adduct masses or their S-oxidized forms (+534/+550 Da for BMCC and +382/+398 for IAB). (We commonly observe S-oxidation of thioethers from in gel tryptic digests; the adducts contain oxidizable sulfur atoms in the biotin ring and the alkylated cysteine linkage.) Diagnostic b- and y-ion fragments allowed mapping of adducts to specific cysteines. Only peptide adducts that were detected in at least three of five experiments were accepted.
A total of 376 modified peptides derived from 263 proteins were identified and 243 of these were adducted by IAB and 223 by BMCC (Figure 4A). Several representative adduct MS-MS spectra are shown in Figures S1 and a complete list of the identified protein targets of IAB and BMCC are presented in Tables S1 and S2, respectively (Supporting Information). Approximately 36% of the identified IAB targets were annotated in the UniRef100 database as localized to the ER, whereas 26% and 20% are annotated as localized to cytoplasm and mitochondria, respectively. Similarly, 31% of BMCC adduct targets were annotated as ER proteins, whereas 25% are cytoplasmic and 22% mitochondrial. Smaller percentages (<5%) of the remaining protein targets carried nuclear, peroxisomal, lysosomal, plasma membrane or Golgi localization annotations. Among targets annotated as ER proteins, a total of 114 cysteines in 69 proteins were identified (Figure 4B). Covalent adduction of over 90% of the adducted proteins occurred at one or two cysteine residues (Figure 5), which is consistent with adduction frequency within protein sequences observed with cytoplasmic and nuclear proteins (23).
Figure 4.

Summary of protein and peptide targets of IAB and BMCC in human liver microsomes. Adducts were found in three of five independent experiments and represent all targets found in microsomes (A) or targets in proteins with ER subcellular annotation (B) according to PSORTII (see http://psort.ims.u-tokyo.ac.jp/).
Figure 5.

Number of adduct sites on identified human liver microsomal protein targets of PEO-IAB (filled bars) and BMCC (open bars).
In our previous studies of cytoplasmic and nuclear protein targets of electrophiles, target selectivity differed considerably between the electrophile structures studied (23). We observed the same dichotomy in target selectivity in microsomal proteins. Differences in electrophile selectivity were also grossly evident in the streptavidin blot analysis of adducted proteins (Figure 3). Only 90 peptides (19% of the total) in 70 proteins (21% of the total) were modified by both IAB and BMCC (Figure 4A, Table 1)2. Among proteins annotated as having ER localization,, 27 proteins were found to be consensus targets (Table 1). These included xenobiotic metabolizing enzymes, enzymes of lipid and sterol metabolism, chaperones and ion transporter proteins.
Table 1.
Consensus microsomal (or ER) protein targets of reactive electrophiles1.
| protein | Swiss-Prot ID | cysteine adducted2 | localization3 | function4 |
|---|---|---|---|---|
| 17-beta hydroxysteroid dehydrogenase | Q86W23 | C80, C94 | secreted, ER | metabolism, lipid |
| 17-beta hydroxysteroid dehydrogenase isoform 1 | Q86W22 | C217 | secreted, ER | metabolism, lipid |
| 3-ketoacyl-CoA thiolase, mitochondrial | P42765 | C179 | MT, ER | metabolism, lipid |
| 7-dehydrocholesterol reductase | Q9UBM7 | C380 | ER | metabolism, lipid |
| alcohol dehydrogenase IB (Class I), beta polypeptide | Q4ZGI9 | C47, C112, C241 | CYT, ER | metabolism protein folding, Ca |
| calreticulin | P27797 | C105 | ER | binding |
| carbonyl reductase [NADPH] 1 | P16152 | C226 | CYT, ER | metabolism |
| cathepsin B precursor | P07858 | C319 | lysosome, ER | proteolysis |
| cytochrome P450 2A6 | P11509 | C82 | ER | xenobiotic metabolism |
| cytochrome P450 2C9 | P11712 | C151 | ER | xenobiotic metabolism |
| cytochrome P450 2E1 | P05181 | C261, C268 | ER | xenobiotic metabolism |
| cytochrome P450-1A2 | Q9UK49 | C458 | ER | xenobiotic metabolism |
| DILV594 | Q6UX53 | C96 | CYT, ER | metabolism |
| dolichyl-diphosphooligosaccharide--protein glycosyltransfer | P04843 | C545 | ER | metabolism |
| flavin containing monooxygenase 3 isoform 2 variant | Q53FW5 | C197 | ER | xenobiotic metabolism |
| flavin containing monooxygenase 5 variant | Q53H53 | C248 | ER | metabolism proteolysis, defense |
| haptoglobin | P00738 | C309, C340 | secreted, ER | resonse |
| hypothetical protein PRO1855 | Q96AG4 | C48 | CYT, ER | unknown |
| liver carboxylesterase 1 | P23141 | C390 | ER | xenobiotic metabolism |
| long-chain-fatty-acid--CoA ligase 1 | P33121 | C55, C108 | ER | lipid metabolism |
| long-chain-fatty-acid--CoA ligase 5 | Q9ULC5 | C322, C610 | ER | lipid metabolism |
| membrane associated progesterone receptor component 2 | O15173 | C159 | ER | hormone receptor |
| microsomal glutathione S-transferase 1 | P10620 | C50 | ER | xenobiotic metabolism |
| mitochondrial carnitine/acylcarnitine carrier protein | O43772 | C136, C155 | MT, ER | transport signal transduction, |
| NAD(P) transhydrogenase | Q13423 | C936, C1073 | MT, ER | kinase |
| novel protein | Q5VT66 | C79 | Cyt, ER | unknown |
| oxidoreductase | O14756 | C177 | ER | metabolism |
| phosphatidylethanolamine-binding protein | P30086 | C132 | Cyt, ER | ATP/lipid binding signal transduction, |
| ras-related protein Rab-2A | P61019 | C21 | ER, Golgi | GTP-binding |
| sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | P16615 | C560 | ER peroxisomes, | calcium transport |
| serine-pyruvate aminotransferase | P21549 | C366 | ER | metabolism |
| signal recognition particle receptor beta subunit | Q9Y5M8 | C73, C246 | ER | receptor GTPase |
| solute carrier family 27 (Fatty acid transporter),member 5 | Q59H28 | C204 | ER | fatty acid transport |
| SPFH domain protein 2 | O94905 | C262 | ER | unknown |
| splice isoform 2 of O75947 | O75947-2 | C76 | Cyt, ER | ion transport |
| steroid dehydrogenase homolog | Q53GQ0 | C166 | ER | metabolism, lipid |
| sterol/retinol dehydrogenase | O75452 | C37, C60, C177 | ER | metabolism, lipid |
| sterol-4-alpha-carboxylate 3-dehydrogenase, decarboxylating | Q15738 | C86 | ER | cholesterol biosynthesis |
| succinate dehydrogenase cytochrome b560 subunit | Q99643 | C107 | MT, ER | electron transport |
| UDP-glucuronosyltransferase 1-4 | P22310 | C127 | ER | xenobiotic metabolism |
| very long-chain acyl-CoA synthetase homolog 2 | Q9Y2P5 | C390 | ER | metabolism, lipid |
| very-long-chain acyl-CoA synthetase | O14975 | C427 | ER | metabolism, lipid |
| vesicular integral-membrane protein VIP36 | Q12907 | C239 | ER | protein transport |
| voltage-dependent anion channel 1 | Q5FVE7 | C232 | MT, ER | transport |
Proteins adducted by both IAB and BMCC at the same cysteine residue and which are annotated as located in ER are listed. A complete list of protein targets of IAB and BMCC is in Supplemental Table 1.
Numbering of cysteine residues in the adducts reported here corresponds to the sequence position in the database entry. In many cases, numbering of certain residues in literature reports refers to posttranslationally cleaved forms of proteins, in which the sequence numbering is changed.
Protein subcellular localizations are based on PSORTII (see http://psort.ims.u-tokyo.ac.jp/).
Annotations for function and location are taken from Gene Ontology classifications (see http://www.godatabase.org).
Covalent adduction of BiP
A recent study by Liu et al. employed a biotinylated analog of a quinone methide metabolite of the selective estrogen receptor modulator raloxifene and detected covalent labeling of BiP in rat liver microsomes (35). Although their experimental design captured labeled proteins, rather than peptides, BiP appeared to be among a handful of the most prominent targets of this biotinylated electrophile. In our analyses, we were surprised that no BiP peptide adducts were detected for either IAB or BMCC. It is possible that some adducted peptides did not ionize efficiently or did not yield MS-MS spectra that matched to a BiP peptide sequence in the database. To further explore the possibility that BiP was adducted in our experiments, we incubated IAB- and BMCC-treated microsomal proteins with streptavidin beads to capture biotinylated proteins and then analyzed this captured protein fraction by SDS-PAGE and western blotting with anti-BiP antibodies (Figure 6). These analyses detected BiP in streptavidin-captured proteins from microsomes treated with IAB or BMCC, but not in untreated controls. Western blotting with anti-calreticulin in the same samples detected calreticulin (Figure 6), which was demonstrated by the LC-MS-MS analyses to be a target of both IAB and BMCC. This confirmed adduction of BiP by both electrophiles, despite the lack of detection of BiP-adducted peptides by LC-MS-MS.
Figure 6.

Detection of BiP and calreticulin adduction by streptavidin capture and western blotting. Human liver microsomes were treated with vehicle (CTL), IAB or BMCC as described under “Experimental Procedures”. Protein extracts then were incubated with streptavidin beads, washed to remove non-biotinylated proteins and then biotinylated proteins were eluted from the beads. The whole protein extracts (lanes 1, 4 and 7), unbound protein (lanes 2, 5 and 8) and proteins eluted with acetonitrile/acetic acid (lanes 3, 6 and 9) were analyzed by western blotting with anti-BiP (upper panel) and anti-calreticulin (lower panel). The presence of anti-BiP and anti-calreticulin immunoreactivity in lanes 6 and 9 indicates BMCC and IAB adduction of BiP and calreticulin, respectively. In contrast, eluted protein from untreated controls contained neither BiP nor calreticulin immunoreactivity (lane 3).
Evaluation of Cys50 of mGST as a target for acetaminophen
Because 20% of the identified cysteine targets reacted with both IAB and BMCC, we postulated that these “consensus” targets may be highly reactive toward other electrophiles. If so, adduction to these consensus targets could provide a quantitative index of reactive metabolite formation in microsomal systems. We performed a limited test of this hypothesis by analyzing the adduction of Cys50 of mGST, which was one of the consensus targets (see Table 1). Cys50 of mGST provides a functionally relevant target for alkylation, as this residue serves as a switch whose alkylation activates enzyme activity (36, 37). We chose acetaminophen, which is metabolized by microsomal CYP enzymes to the thiol-reactive quinoneimine NAPQI. Incubation of acetaminophen with human liver microsomes supplemented with an NADPH generating system and GSH resulted in formation of the expected GS-conjugate of NAPQI (Supplemental Figure S17). Targeted quantitative analysis was performed on the NAPQI-adducted Cys50-containing tryptic peptide isolated from the SDS-PAGE band corresponding to mGST. These analyses yielded an MS-MS spectrum confirming adduction of the NAPQI-adducted mGST Cys50 peptide (Figure 7A). The MS-MS transition from the doubly charged precursor to the y9+ product ion was used to quantify the formation of the NAPQI-Cys50 adduct over time and was normalized to the SIM signal for the IAM-modified peptide from unadducted mGST (Figure 7B). Adduct accumulation was linear for up to 1 hr (Figure 7C), thus indicating the ability of a specific protein adduct to serve as a quantitative indicator of microsomal covalent binding.
Figure 7.
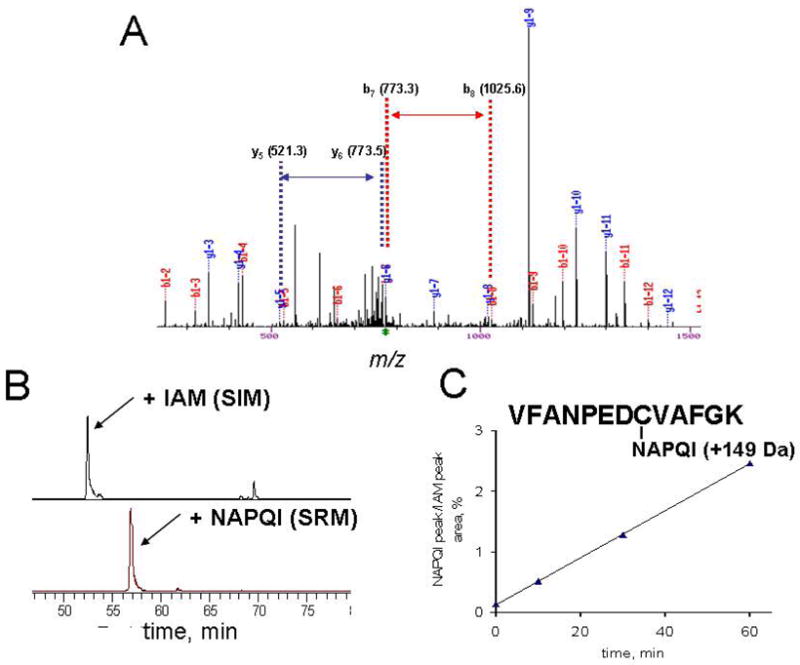
Detection and quantitation of mGST adduction at Cys50 by NAPQI during microsomal metabolism of acetaminophen. Targeted LC-MS-MS analysis of microsomal proteins harvested from microsomal incubations with acetaminophen revealed an M + 149 adduct at Cys50 (A). Selected reaction monitoring of the MS-MS transitions from the doubly-charged IAM-adducted or NAPQI-adducted Cys50 peptide ions to the corresponding y9 product ions (B) enabled quantitation of the NAPQI adduct as a function of incubation time (C).
DISCUSSION
The analysis of covalent binding of electrophiles has changed little in the past 30 years. The most common approach requires the synthesis of radiolabeled compounds, which represents a significant expense that limits implementation (3). The measurement of bound radiolabel indicates adduction of proteins of unknown identity and scope and is only a crude indicator of possible adverse effect. Indeed, both toxic and non-toxic compounds generate covalent adducts in microsomes and other test systems and bulk covalent binding measurements are unable to predict toxic versus non-toxic outcomes. Here we have applied model electrophile probes and LC-MS-MS-based proteomic analyses to identify microsomal protein targets for adduction. The data reveal a broad repertoire of microsomal targets, yet indicate differences that distinguish probes with different biological effects in intact cells.
The electrophile probes we used are similar or identical to those used in our previous studies (23). Whereas the iodoacetamide-like electrophile was used previously, here we used IAB, which is more cell-permeable than PEO-IAB. Other studies in our laboratory demonstrate significant differences in cellular stress responses elicited by IAB and BMCC. These compounds display different abilities to activate the transcription factor Nrf2 (38) and to inactivate protein phosphatase 2A in vitro (39). These probes enter cells and form covalent adducts with proteins in all cellular compartments2. Here we show that the iodoacetamido compound IAB induced ER stress characterized by induction of BiP, similar to the previously reported effect of its non-biotinylated congener IAM (9, 10). In contrast, both the N-alkylmaleimide BMCC and its non-biotinylated congener NEM failed to cause BiP induction. Interestingly, both IAB and BMCC adducted BiP itself (Figure 6), which suggests that BiP modification per se is not critical to the induction of ER stress.
The collection of microsomal protein targets modified by IAB and BMCC includes a total of 376 cysteines in 263 proteins. The fact that only a third of the proteins adducted are annotated as ER proteins probably reflects the heterogeneous, complex nature of microsomes, which are vesicles formed from multiple organelles during tissue disruption. This complexity of microsomal preparations was previously observed in shotgun proteome analyses (34).
As in our previous work (23), only cysteine adducts were detected, as our database searches specified IAB and BMCC adducts as variable modifications to cysteine residues. We note that adducts to other protein nucleophiles are possible (e.g., histidine imidazones, lysine α-amines, arginine guanidinium and protein N-termini), but we did not attempt to include these other modifications in the database searches, as these would have dramatically increased both search times and false-positive identification rates. Reactions with these other protein nucleophiles probably would compete poorly with cysteine alkylation, but could occur in some cases, particularly when noncovalent complexation of electrophiles juxtaposes them favorably with these nucleophiles.
Only about 20% of the protein targets identified in these studies were alkylated by both IAB and BMCC. The relatively modest overlap in protein targeting by the two electrophile probes is similar to that we observed in cytoplasmic and nuclear proteins (23) and probably reflects both differences in alkylation chemistry of the electrophiles (SN2 alkylation for IAB versus Michael addition for BMCC) and more subtle steric differences in the linker chains connecting the electrophile and biotin moieties. Consensus targets of both IAB and BMCC include proteins associated with well-established ER functions in vivo, including drug metabolism (CYP, GST and UGT enzymes), ion transport (SERCA2) and protein folding (calreticulin). Several previous reports indicated adduction of the chaperone protein PDI in microsomes (40) and in cell and tissue systems (11, 19, 41, 42) and with purified PDI proteins (43, 44). Our electrophile probes alkylated different PDI-like proteins. IAB alkylated thioredoxin domain containing protein 5 (Q8NBS9), whereas BMCC alkylated PDI A3 (P30101) (Supplemental Tables 1 and 2). It is important to note that targeting specificity in complex proteomes will differ depending on the electrophile studied, the analytical method and the experimental system studied. Whereas the previous studies with purified proteins identified adduction sites in PDI, the studies with complex proteomes only identified proteins that co-migrated with adduct immunoreactivity or radiolabel in 2D gel separations. Our approach both identified protein targets and mapped adducts to specific cysteine residues in the proteins.
Our observation that differences in covalent adduction targeting correlated with differences in ER stress induction is important, because it suggests that adduction specificity encodes biological effect. ER stress induction can be triggered by diverse agents that affect thiol-disulfide redox balance, protein glycosylation, S-nitrosation and alkylation damage (7–11). Generalized adduction of ER proteins is not sufficient to induce ER stress. Our data indicate that, although both IAB and BMCC adducted equivalent numbers of ER proteins, only IAB induced the ER stress response. We note also that IAB, but not BMCC induces apoptosis in HEK293 cells as the doses used in these studies3. Some targets adducted only by IAB may be mechanistically linked to the induction of ER stress and apoptosis, but the relationship between protein alkylation and these outcomes probably is not simple. Studies with additional compounds would be needed to establish predictive relationships between adduction selectivity and toxic or adaptive responses. Analyses of target-specific adduction in microsomes or other model systems may offer greater predictive utility than bulk covalent binding assays.
The microsomal proteins adducted by IAB and BMCC probably are targets for adduction by other electrophiles. If so, analysis of adducts to these specific targets could provide quantitative indices of electrophile-mediated damage. We tested this hypothesis with a preliminary study directed at one such target, Cys50 of mGST. After first establishing conditions for microsomal acetaminophen activation to NAPQI and the formation of the corresponding GS-conjugate, we targeted the mGST Cys50-containing peptide. NAPQI adduction of the target peptide accumulated linearly with time during microsomal acetaminophen metabolism (Figure 7C). Thus, individual protein adduction reactions can be quantified if suitable targets first can be identified. Beginning with the list of proteins adducted by IAB and BMCC, detection of other adducts to these proteins in MS-MS analyses can be done with the aid of novel software tools, such as SALSA (45, 46) and P-Mod (39, 47, 48).
In summary, our work shows that different target selectivities of the two electrophile probes correlated with different biological outcomes and that alkylation reactions of specific targets can be quantified. It thus appears possible that quantitative analyses of specific protein adduction reactions could be used to decipher mechanisms of toxicity and adaptation to stress. Moreover, analyses of protein adduction to a selected population of key targets could enable prediction of the adverse effects of electrophilic xenobiotic metabolites and of endogenous electrophiles formed by oxidative stress.
Supplementary Material
Acknowledgments
We thank Drs. F. P. Guengerich and M.V. Martin for supplying human liver microsomes used in this work and L. Manier for assistance with MS analyses. This work was supported by NIH Grants ES010056 and ES000267.
Footnotes
Abbreviations: ACN, acetonitrile; ATF6, activating transcription factor 6; BiP/GRP78, immunoglobulin heavy chain-binding protein/glucose-regulated protein of molecular weight 78 kDa; BMCC, 1-biotinamido-4-(4′-[maleimidoethylcyclohexane]-carboxamido)butane; DTT, dithiothreitol; EDTA, ethylene diaminetetraacedic acid; ER, endoplasmic reticulum; GSH, glutathione; mGST, microsomal glutathione S-transferase; HPLC, high performance liquid chromatography; IAB, N-iodoacetyl-N-biotinylhexylenediamine; IAM, iodoacetamide; LC-MS-MS, liquid chromatography tandem mass spectrometry; IRE1, inositol-requiring enzyme 1; MOPS, 3-(N-morpholino)propanesulfonic acid; MS, mass spectrometry; MS-MS, tandem mass spectrometry; NAPQI, N-acetyl-p-benzoquinoneimine; PBS, phosphate buffered saline; PERK, PKR-like ER protein kinase/pancreatic eIF2α (eukaryotic translation initiation factor 2, α subunit) kinase; SDS, sodium dodecyl sulfate; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TCEP, tris-carboxyethyl phosphine; TFA, trifluoroacetic acid.
Burnette, E.B. and Liebler, D.C., manuscript in preparation.
Wong, H.L., Orton, C.R. and Liebler, D.C., manuscript in preparation.
References
- 1.Gram TE. Separation of hepatic smooth and rough microsomes associated with drug-metabolizing enzymes. Methods Enzymol. 1974;31:225–237. doi: 10.1016/0076-6879(74)31024-5. [DOI] [PubMed] [Google Scholar]
- 2.White RE. High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery. Annu Rev Pharmacol Toxicol. 2000;40:133–157. doi: 10.1146/annurev.pharmtox.40.1.133. [DOI] [PubMed] [Google Scholar]
- 3.Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol. 2004;17:3–16. doi: 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- 4.Nelson SD. Mechanisms of the formation and disposition of reactive metabolites that can cause acute liver injury. Drug Metab Rev. 1995;27:147–177. doi: 10.3109/03602539509029821. [DOI] [PubMed] [Google Scholar]
- 5.Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- 6.Nelson SD, Pearson PG. Covalent and noncovalent interactions in acute lethal cell injury caused by chemicals. Annu Rev Pharmacol Toxicol. 1990;30:169–195. doi: 10.1146/annurev.pa.30.040190.001125. [DOI] [PubMed] [Google Scholar]
- 7.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Bowes RC, III, van de WB, Sillence C, Nagelkerke JF, Stevens JL. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J Biol Chem. 1997;272:21751–21759. doi: 10.1074/jbc.272.35.21751. [DOI] [PubMed] [Google Scholar]
- 10.van de Water B, Wang Y, Asmellash S, Liu H, Zhan Y, Miller E, Stevens JL. Distinct endoplasmic reticulum signaling pathways regulate apoptotic and necrotic cell death following iodoacetamide treatment. Chem Res Toxicol. 1999;12:943–951. doi: 10.1021/tx990054q. [DOI] [PubMed] [Google Scholar]
- 11.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 12.Satoh H, Gillette JR, Davies HW, Schulick RD, Pohl LR. Immunochemical evidence of trifluoroacetylated cytochrome P-450 in the liver of halothane-treated rats. Mol Pharmacol. 1985;28:468–474. [PubMed] [Google Scholar]
- 13.Witzmann FA, Jarnot BM, Parker DN, Clack JW. Modification of hepatic immunoglobulin heavy chain binding protein (BiP/Grp78) following exposure to structurally diverse peroxisome proliferators. Fundam Appl Toxicol. 1994;23:1–8. doi: 10.1006/faat.1994.1072. [DOI] [PubMed] [Google Scholar]
- 14.Butler LE, Thomassen D, Martin JL, Martin BM, Kenna JG, Pohl LR. The calcium-binding protein calreticulin is covalently modified in rat liver by a reactive metabolite of the inhalation anesthetic halothane. Chem Res Toxicol. 1992;5:406–410. doi: 10.1021/tx00027a014. [DOI] [PubMed] [Google Scholar]
- 15.Jewell WT, Miller MG. Identification of a carboxylesterase as the major protein bound by molinate. Toxicol Appl Pharmacol. 1998;149:226–234. doi: 10.1006/taap.1998.8381. [DOI] [PubMed] [Google Scholar]
- 16.Hargus SJ, Amouzedeh HR, Pumford NR, Myers TG, McCoy SC, Pohl LR. Metabolic activation and immunochemical localization of liver protein adducts of the nonsteroidal anti-inflammatory drug diclofenac. Chem Res Toxicol. 1994;7:575–582. doi: 10.1021/tx00040a014. [DOI] [PubMed] [Google Scholar]
- 17.Koen YM, Hanzlik RP. Identification of seven proteins in the endoplasmic reticulum as targets for reactive metabolites of bromobenzene. Chem Res Toxicol. 2002;15:699–706. doi: 10.1021/tx0101898. [DOI] [PubMed] [Google Scholar]
- 18.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 19.Lame MW, Jones AD, Wilson DW, Dunston SK, Segall HJ. Protein targets of monocrotaline pyrrole in pulmonary artery endothelial cells. J Biol Chem. 2000;275:29091–29099. doi: 10.1074/jbc.M001372200. [DOI] [PubMed] [Google Scholar]
- 20.Lin CY, Isbell MA, Morin D, Boland BC, Salemi MR, Jewell WT, Weir AJ, Fanucchi MV, Baker GL, Plopper CG, Buckpitt AR. Characterization of a structurally intact in situ lung model and comparison of naphthalene protein adducts generated in this model vs lung microsomes. Chem Res Toxicol. 2005;18:802–813. doi: 10.1021/tx049746r. [DOI] [PubMed] [Google Scholar]
- 21.Adam GC, Sorensen EJ, Cravatt BF. Chemical strategies for functional proteomics. Mol Cell Proteomics. 2002;1:781–790. doi: 10.1074/mcp.r200006-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Adam GC, Sorensen EJ, Cravatt BF. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat Biotechnol. 2002;20:805–809. doi: 10.1038/nbt714. [DOI] [PubMed] [Google Scholar]
- 23.Dennehy MK, Richards KAM, Wernke GW, Shyr Y, Liebler DC. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem Res Toxicol. 2006;19:20–29. doi: 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- 24.Guengerich FP, Martin MV. Purification of cytochrome P-450, NADPH-cytochrome P-450 reductase, and epoxide hydratase from a single preparation of rat liver microsomes. Arch Biochem Biophys. 1980;205:365–379. doi: 10.1016/0003-9861(80)90119-8. [DOI] [PubMed] [Google Scholar]
- 25.Guengerich FP. Analysis and characterization of enzymes and nucleic acids. In: Hayes AW, editor. Principles and Methods in Toxicology. Taylor and Francis; Philadelphia: 2001. pp. 1625–1687. [Google Scholar]
- 26.Ham AJ, Caprioli RM, Gross ML. The Encyclopedia of Mass Spectrometry, Volume 2 Biological Applications Part A: Peptides and Proteins. Elsevier Ltd; Kidlington, Oxford, UK: 2005. Proteolytic Digestion Protocols; pp. 10–17. [Google Scholar]
- 27.Licklider LJ, Thoreen CC, Peng J, Gygi SP. Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal Chem. 2002;74:3076–3083. doi: 10.1021/ac025529o. [DOI] [PubMed] [Google Scholar]
- 28.Eng JK, McCormack AL, Yates JR. An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in A Protein Database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 29.Tang W, Stearns RA, Wang RW, Chiu SH, Baillie TA. Roles of human hepatic cytochrome P450s 2C9 and 3A4 in the metabolic activation of diclofenac. Chem Res Toxicol. 1999;12:192–199. doi: 10.1021/tx9802217. [DOI] [PubMed] [Google Scholar]
- 30.Gan J, Harper TW, Hsueh MM, Qu Q, Humphreys WG. Dansyl glutathione as a trapping agent for the quantitative estimation and identification of reactive metabolites. Chem Res Toxicol. 2005;18:896–903. doi: 10.1021/tx0496791. [DOI] [PubMed] [Google Scholar]
- 31.Baillie TA. Advances in the application of mass spectrometry to studies of drug metabolism, pharmacokinetics, and toxicology. Int J Mass Spectrom Ion Proc 118/ 1992;119:289–314. [Google Scholar]
- 32.Liu H, Lightfoot R, Stevens JL. Activation of heat shock factor by alkylating agents is triggered by glutathione depletion and oxidation of protein thiols. J Biol Chem. 1996;271:4805–4812. [PubMed] [Google Scholar]
- 33.Simpson RJ, Connolly LM, Eddes JS, Pereira JJ, Moritz RL, Reid GE. Proteomic analysis of the human colon carcinoma cell line (LIM 1215): development of a membrane protein database. Electrophoresis. 2000;21:1707–1732. doi: 10.1002/(SICI)1522-2683(20000501)21:9<1707::AID-ELPS1707>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 34.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Li Q, Yang X, van Breemen RB, Bolton JL, Thatcher GR. Analysis of protein covalent modification by xenobiotics using a covert oxidatively activated tag: raloxifene proof-of-principle study. Chem Res Toxicol. 2005;18:1485–1496. doi: 10.1021/tx0501738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svensson R, Rinaldi R, Swedmark S, Morgenstern R. Reactivity of cysteine-49 and its influence on the activation of microsomal glutathione transferase 1: evidence for subunit interaction. Biochemistry. 2000;39:15144–15149. doi: 10.1021/bi001764u. [DOI] [PubMed] [Google Scholar]
- 37.Busenlehner LS, Codreanu SG, Holm PJ, Bhakat P, Hebert H, Morgenstern R, Armstrong RN. Stress sensor triggers conformational response of the integral membrane protein microsomal glutathione transferase 1. Biochemistry. 2004;43:11145–11152. doi: 10.1021/bi048716k. [DOI] [PubMed] [Google Scholar]
- 38.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 39.Codreanu SG, Adams DG, Dawson ES, Wadzinski BE, Liebler DC. Inhibition of protein phosphatase 2A activity by selective electrophile alkylation damage. Biochemistry. 2006;45:10020–10029. doi: 10.1021/bi060551n. [DOI] [PubMed] [Google Scholar]
- 40.Isbell MA, Morin D, Boland B, Buckpitt A, Salemi M, Presley J. Identification of proteins adducted by reactive naphthalene metabolites in vitro. Proteomics. 2005;5:4197–4204. doi: 10.1002/pmic.200401278. [DOI] [PubMed] [Google Scholar]
- 41.Lame MW, Jones AD, Wilson DW, Segall HJ. Protein targets of 1,4-benzoquinone and 1,4-naphthoquinone in human bronchial epithelial cells. Proteomics. 2003;3:479–495. doi: 10.1002/pmic.200390062. [DOI] [PubMed] [Google Scholar]
- 42.Shipkova M, Beck H, Voland A, Armstrong VW, Grone HJ, Oellerich M, Wieland E. Identification of protein targets for mycophenolic acid acyl glucuronide in rat liver and colon tissue. Proteomics. 2004;4:2728–2738. doi: 10.1002/pmic.200300836. [DOI] [PubMed] [Google Scholar]
- 43.Kaetzel RS, Stapels MD, Barofsky DF, Reed DJ. Alkylation of protein disulfide isomerase by the episulfonium ion derived from the glutathione conjugate of 1,2-dichloroethane and mass spectrometric characterization of the adducts. Arch Biochem Biophys. 2004;423:136–147. doi: 10.1016/j.abb.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 44.Carbone DL, Doorn JA, Kiebler Z, Petersen DR. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 45.Hansen BT, Jones JA, Mason DE, Liebler DC. SALSA: a pattern recognition algorithm to detect electrophile-adducted peptides by automated evaluation of CID spectra in LC-MS-MS analyses. Anal Chem. 2001;73:1676–1683. doi: 10.1021/ac001172h. [DOI] [PubMed] [Google Scholar]
- 46.Liebler DC, Hansen BT, Jones JA, Badghisi H, Mason DE. Mapping protein modifications with liquid chromatography-mass spectrometry and the SALSA algorithm. Adv Protein Chem. 2003;65:195–216. doi: 10.1016/s0065-3233(03)01020-9. [DOI] [PubMed] [Google Scholar]
- 47.Hansen BT, Davey SW, Ham AJ, Liebler DC. P-Mod: An Algorithm and Software To Map Modifications To Peptide Sequences Using Tandem MS Data. J Proteome Res. 2005;4:358–368. doi: 10.1021/pr0498234. [DOI] [PubMed] [Google Scholar]
- 48.Szapacs ME, Riggins JN, Zimmerman LJ, Liebler DC. Covalent adduction of human serum albumin by 4-hydroxy-2-nonenal: kinetic analysis of competing alkylation reactions. Biochemistry. 2006;45:10521–10528. doi: 10.1021/bi060535q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.