

Abstract
The purine riboswitch is one of a number of mRNA elements commonly found in the 5′-untranslated region capable of controlling expression in a cis-fashion via its ability to directly bind small molecule metabolites. Extensive biochemical and structural analysis of the nucleobase-binding domain of the riboswitch, referred to as the aptamer domain, has revealed that the mRNA recognizes its cognate ligand using an intricately folded three-way junction motif that completely encapsulates the ligand. High affinity binding of the purine nucleobase is facilitated by a distal loop-loop interaction that is conserved between both the adenine and guanine riboswitches. To understand the contribution of conserved nucleotides in both the three-way junction and the loop-loop interaction of this RNA we performed a detailed mutagenic survey of these elements in the context of an adenine-responsive variant of the xpt-pbuX guanine riboswitch from B. subtilis. The varying ability of these mutants to bind ligand as measured by isothermal titration calorimetry (ITC), uncovered the conserved nucleotides whose identity is required for purine binding. Crystallographic analysis of the bound form of five mutants and chemical probing of their free state demonstrate that the identity of several universally conserved nucleotides are not essential for formation of the RNA-ligand complex but rather for maintaining a binding-competent form of the free RNA. These data show that conservation patterns in riboswitches arise from a combination of formation of the ligand-bound complex, promoting an open form of the free RNA, and participating in the secondary structural switch with the expression platform.
Keywords: riboswitch, RNA, X-ray crystallography, isothermal titration calorimetry, RNA-ligand interactions
RNA-mediated regulation of gene expression, or riboregulation, plays a role in almost every facet of the storage, expression, and transmission of biological information. In recent years, a growing number of non-coding RNAs and regulatory elements have been discovered and characterized in all three domains of life (reviewed in (1–4)). In bacteria a common form of riboregulation is the riboswitch, a cis-acting element found in the 5′-untranslated region (5′-UTR) of mRNAs (reviewed in (5, 6)). Riboswitches control expression of the mRNA via two domains acting in concert: the aptamer domain and expression platform. The aptamer domain directly binds small molecule ligands and is responsive to intracellular ligand concentrations. Each type of riboswitch employs a unique secondary and tertiary structure to bind a specific ligand. Currently, there are over ten characterized classes of riboswitches recognizing a variety of metabolites including nucleobases (7, 8), amino acids (9–11), vitamin cofactors (12, 13), and metal ions (14). Ligand binding to the aptamer domain is subsequently communicated to an expression platform containing two mutually exclusive secondary structures that repress or activate expression at the transcriptional or translational levels (reviewed in (15)).
The most characterized class of riboswitches bind either the guanine or adenine nucleobase to regulate the expression of a number of purine biosynthetic and nucleobase transport genes in bacteria (7, 8, 16, 17). Structural analysis of the aptamer domain from the guanine and adenine riboswitches revealed an intricate fold in which a three-way junction motif serves as the binding site for the cognate ligand (18–20). Discrimination between guanine and adenine is achieved using a single pyrimidine residue in the junction (Y74, Figure 1a, b) that forms a Watson-Crick base pairing interaction with the ligand (Gua-C74 or Ade-U74); this single pyrimidine residue is the sole basis for the >20,000-fold specificity for guanine versus adenine (7, 18, 21). The ability of the RNA to bind ligand is facilitated by the presence of tertiary structural interactions between two loops (L2 and L3, Figure 1a, b, Supporting Figure 1a)) that globally organize the aptamer domain (22). The loop-loop interaction has been shown to form without ligand (7) and even in the absence of the three-way junction (23), clearly indicating that it represents an independently folding element within the aptamer domain.
Figure 1.
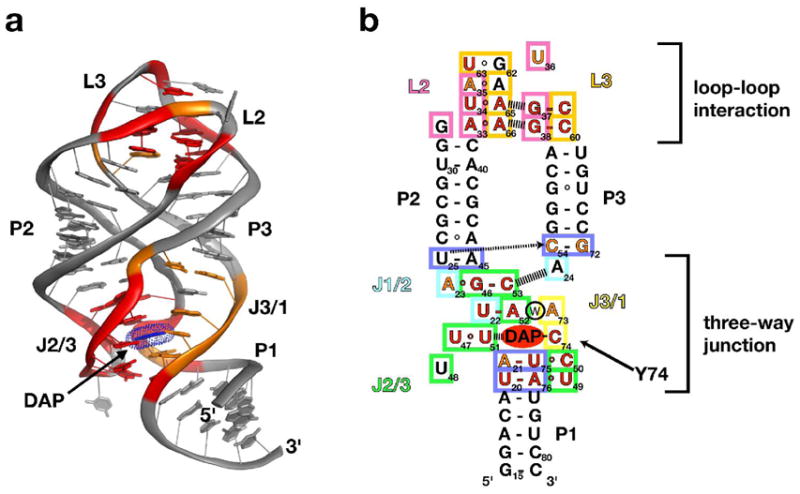
(a) Tertiary structure of GRA bound to 2,6-diaminopurine (blue), emphasizing the interactions of L2 and L3 and burial of 2,6-diaminopurine in the center of the three-way junction. (b) Secondary structure of the GRA RNA illustrating the interactions of residues in the thre e-way junction and loop-loop elements. Nucleotides that are >95% conserved in an alignment of 100 sequences found in Rfam 7.0 (31) are shown in red and those that are >80% conserved are in orange. Colored boxes correspond to the residue coloring in subsequent structure figures.
An essential feature of riboswitch function is communication of ligand binding to an expression platform that exerts the regulatory effect (24–26). This is commonly accomplished by sequestering important regulatory sequence elements within the P1 helix of the aptamer domain, a structural element that is stabilized upon ligand binding (Figure 1a). These regulatory sequences, found on the 3′-side of the P1 helix, are used in the formation of one of two mutually exclusive structures in the expression platform. The crystal structure and mechanistic studies of the guanine riboswitch revealed purine binding induces formation of tertiary interactions that buttress the P1 helix, specifically two base triples between J2/3 and the 3′-strand of P1 (7, 18, 21). Formation of this element of tertiary structure stabilizes the aptamer domain at the expense of a downstream antiterminator element, fating the expression platform to form a classic rho-independent terminator stem-loop structure that causes transcriptional termination. Moreover, regulation by riboswitches most often occurs co-transcriptionally, resulting in a process governed by a complex interplay between kinetic and thermodynamic parameters (27, 28). Structures of other riboswitches suggest that this is a general mechanism of the majority of ligand responsive riboswitches (29, 30).
Within the aptamer domain of the purine riboswitch there are a number of highly conserved (>90% across 100 sequences, Figure 1 and Supporting Table 1) nucleotides residing in the terminal loops and the three-way junction (7, 31). While crystal structures show the role of these residues in establishing the overall architecture and details of ligand recognition by the RNA, they do not illuminate the relative energetic contributions of individual nucleotide residues to folding and binding. Furthermore, a number of these conserved nucleotides have structural roles in which other nucleotides or base pairs appear to be able to substitute with little penalty. For instance, within the three-way junction there is a universally conserved Watson-Crick pairing between nucleotides U22 and A52 (18, 20) that takes part in a water-mediated base triple above the ligand in the binding pocket (Figure 1b). As this base-pair does not interact with other nucleotides in the binding pocket, other Watson-Crick pairs would likely be able to support ligand binding, yet do not occur in phylogeny.
In this paper, we have undertaken a detailed mutagenic survey of the purine riboswitch aptamer domain to ascertain the importance of conserved nucleotides to high affinity ligand binding. Single point mutations as well as base pair substitutions and transversions were introduced into the terminal loops, the three-way junction, and the base pairs adjacent to the junction of a C74U adenine-responsive mutant of the xpt-pbuX guanine riboswitch from B. subtilis. The ability of these RNAs to bind an adenine analog, 2,6-diaminopurine (DAP) was measured using isothermal titration calorimetry (ITC). The strength of this experimental technique is that it does not require modification or labeling of either the RNA or ligand and it is a true equilibrium technique. Crystal structures of five of these mutations show that three-dimensional architecture is not significantly altered. Instead, chemical probing analysis indicates that the free state of these RNAs is different, revealing that these nucleotides are important for promoting an active free RNA. As discussed, the results of these experiments reveal several important aspects to the sequence conservation of this RNA. In particular, we observe that certain nucleotides within the three-way junction are not critical for formation of the bound structure but rather are essential for ensuring that the free state of the RNA is receptive to ligand. Furthermore, we see that changes in the P1 helix adjacent to the three-way junction support ligand recognition with near wild-type affinity, suggesting that their conservation may be important in interdomain communication.
MATERIALS AND METHODS
RNA synthesis and purification
The parental RNA used in these studies, referred to as “GRA” because it is a naturally occurring guanine riboswitch (GR) that carries a C74U mutation rendering it adenine-responsive, binds adenine and its analogs with properties identical to that of the wild type adenine riboswitch (32). GRA was created and characterized as described in Gilbert et al (21). DNA sequences for transcribing GRA RNAs containing site-specific mutations were made using overlapping DNA oligonucleotides by PCR and cloned into pRAV12 (33) using standard molecular biological techniques (34). The sequences of all plasmids were verified prior to their use as templates for transcription. Synthesis by in vitro T7 RNA polymerase and subsequent purification of all RNAs used in this study was performed as described by Gilbert et al (21).
Isothermal Titration Calorimetry (ITC)
RNA was prepared for ITC by exhaustively dialyzing for 24 - 48 hours against 1 L of a buffer solution containing 50 mM K+-HEPES, pH 7.5, 100 mM KCl, and 10 mM MgCl2 at 4 °C. 2,6-diaminopurine (DAP; Sigma-Aldrich) was dissolved in the buffer that the RNA was dialyzed against to ensure an exact buffer match to minimize measuring a heat of dilution. The final concentration of DAP was approximately 8-fold higher than the RNA sample. Concentrations were determined using spectrophotometer readings at 260 nm for RNA and at 278 nm for DAP (ε278 = 10,200 M−1 cm−1). Prior to the titration, the RNA and DAP samples were degassed for 10 minutes at 25 °C to remove bubbles that could disrupt even heating. All ITC experimental parameters were set at 30 °C, a reference power of 5 μcal sec−1, an initial delay of 60 seconds and a titration of either 65 five μL (for tight binding mutants) or 32 ten μL injections (for weak binding mutants) at an injection rate of 0.5 μL sec−1 with individual injections spaced 180 seconds apart. RNA concentrations of ~20 μM were used for low affinity binding interactions and ~5 μM for high affinity interactions in order to ensure that the c value for each experiment was between 0.5 and 500 (Figure 2) (35, 36). The c value is the ratio of the concentration of the reactant in the sample cell, in our case the RNA, to the disassociation constant and represents an experimental concentration range in which useful data can be obtained (35, 36).
Figure 2.
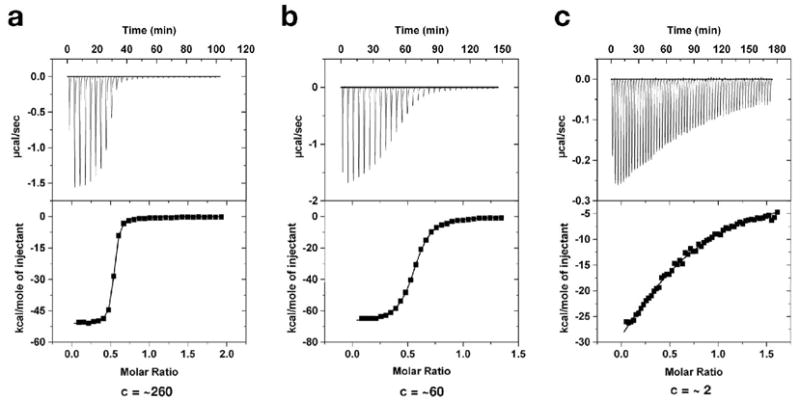
Representative data of 2,6-diaminopurine binding the (a) A35U, (b) C61U, and (c) G37A mutants of GRA RNA at 30 °C in a buffer containing 50 mM K+-HEPES, pH 7.5, 100 mM KCl and 10 mM MgCl2. The c-value (where c = Ka•[RNA]) for each of these experiments is 260, 60, and 2, respectively.
Data collected from each experiment were fit to a single-site binding model using Origin ITC software (Microcal Software Inc), from which values of n, Ka (in M−1), ΔH (in cal mol−1), and ΔS (in cal mol−1 K−1) were extrapolated using the equation
| (1) |
where q is the heat released, v is the known volume of the reaction, Ka is the association constant, and Li is the ligand concentration at the ith injection (36). The disassociation constant, Kd (in M), was calculated by taking the inverse of Ka, and the relationship
| (2) |
was used to calculate the change in free energy, ΔG. To compare changes of DAP binding to the mutant RNAs versus wild type, the difference in free energy values (ΔΔG) were calculated; these values directly compare the difference in free energy observed as a result of each mutation.
X-ray crystallography
Crystals were grown by the hanging drop/vapor diffusion method by mixing 2 μL of mutant:DAP complex with 2 μL mother liquor. Diffraction quality crystals grew to a maximal size within one week in mother liquor conditions containing 15–20% PEG 3K, 10 mM K+-HEPES, pH 7.5, 8–12 mM cobalt hexamine, 480–720 mM ammonium acetate, at 25 °C. Crystals were cryoprotected in mother liquor plus 30% 2-methyl-2,4-pentanediol for 5–10 minutes, mounted in a cryoloop (Hampton Research) and flash frozen in liquid nitrogen. Diffraction data was collected with an R-AXIS IV++ detector on an RU-200/Confocal blue optic source (Rigaku MSC). In most cases, clear reflections were observable to 1.9 Å unless otherwise indicated.
Collection data were indexed, integrated, and scaled using D*TREK (37) in the CrystalClear package (Rigaku MSC). Electron density maps of the mutant riboswitch:DAP complexes were calculated by molecular replacement in CNS (38) using the guanine riboswitch bound to hypoxanthine (PDB ID 1u8d) as the search model in which the solvent, ligand, and mutated bases removed. The initial 2Fo Fc maps showed clear electron density corresponding to the RNA and DAP ligand. Following several rounds of simulated annealing involving slow cooling from 5000 K in 25 K steps to minimize model bias (39), the ligand, mutated bases, cobalt hexammine, and a single acetate ion hydrogen bonding to G32 were built into the model and solvent added using automated water picking in CNS. The final model was obtained after several further rounds of energy minimization and B-factor refinement in CNS; final statistics for each model are listed in Table 2. The coordinates and structure factors have been deposited in the RCSB Protein Data Bank under the accession numbers 2EES, 2EET, 2EEU, 2EEV and 2EEW.
TABLE 2.
Crystallographic statistics for structures of mutants of GRA RNA.
| RNA mutant | A21U/U75A | A21G/U75C | U22A/A52U | U22C/A52G | U47C |
|---|---|---|---|---|---|
| Data Collection | |||||
| Spacegroup | C2 | C2 | C2 | C2 | C2 |
| a, b, c (Å) | 132.54, 35.96, 41.47 | 133.79, 35.28, 41.66 | 132.99, 35.24, 42.13 | 130.42, 35.04, 42.00 | 133.11, 35.26, 42.01 |
| β(°) | 91.31 | 90.66 | 90.42 | 89.86 | 90.83 |
| Resolution (Å) | 20-1.75 | 20-1.95 | 20-1.95 | 20-1.95 | 14.85-2.00 |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Completenessa (%) | 98.3 (85.2) | 92.8 (71.8) | 95.7 (82.0) | 97.6 (89.1) | 97.6 (99.6) |
| Measured reflections | 65,365 | 57,115 | 98,762 | 47,698 | 35,125 |
| Unique reflections | 19,129 | 13,407 | 13,890 | 13,749 | 13,088 |
| Average redundancy | 3.42 (2.81) | 4.26 (4.13) | 7.11 (6.21) | 3.47 (3.03) | 2.68 (2.57) |
| 〈I〉/〈σ(I)〉 | 15.9 (4.1) | 19.3 (6.0) | 14.8 (5.8) | 10.8 (3.3) | 10.1 (3.3) |
| Rmergeb | 0.048 (0.170) | 0.048 (0.150) | 0.067 (0.262) | 0.064 (0.295) | 0.057 (0.225) |
| Refinement | |||||
| Resolution: | 20-1.75 (1.86-1.75) | 20-1.95 (2.07-1.95) | 20-1.95 (2.02-1.95) | 20-1.95 (2.02-1.95) | 15-2.25 (2.33–2.25) |
| Number of reflections | |||||
| Working | 17,706 | 12,399 | 12,840 | 12,713 | 8,232 |
| Test set | 1,402 | 1,005 | 1,034 | 1,028 | 933 |
| Rxtalc (%) | 20.7 (30.0) | 21.3 (26.7) | 23.4 (27.1) | 21.0 (31.6) | 23.4 (26.4) |
| Rfree (%) | 24.1 (31.3) | 25.1 (27.6) | 27.7 (31.2) | 26.3 (34.7) | 28.5 (32.4) |
| r.m.s.d. bonds (Å) | 0.0211 | 0.00412 | 0.00421 | 0.00910 | 0.00445 |
| r.m.s.d. angles (°) | 2.073 | 0.882 | 0.983 | 1.38 | 0.827 |
| Luzzati coordinate error (Å) | 0.25 | 0.30 | 0.34 | 0.32 | 0.42 |
| Sigma-a coordinate error (Å) | 0.15 | 0.19 | 0.24 | 0.34 | 0.35 |
| Average B-factor (Å2) | 26.9 | 26.6 | 33.4 | 34.5 | 36.8 |
| PDB ID | 2EES | 2EET | 2EEU | 2EEV | 2EEW |
. Numbers in parenthesis correspond to the highest resolution shell.
. Rmerge=Σ|I−〈I〉|/ΣI, where I is the observed intensity and 〈I〉 is the average intensity of multiple measurements of symmetry related reflections.
. Rxtal = Σ|Fo − |Fc||/Σ|Fo|, Rxtal from the working set and Rfree from the test set.
Structure probing by chemical modification using SHAPE
Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) was performed as described by the Weeks laboratory (40). First, sequence was added to the 3′end of each mutant RNA by PCR. This sequence is complimentary to a 32P-labeled oligonucleotide used in subsequent reverse transcription reactions and contains in addition, sequence identified by Weeks, et. al. to facilitate optimal structural modification analysis (40). Briefly, 2 pmols of mutant RNAs in 12 μl of 0.5x TE buffer were heat denatured at 90 °C for 3 minutes followed by refolding on ice for 10 minutes. Folding buffer (333 mM HEPES, pH 8.0, 20 mM MgCl2, 333 mM NaCl) was added to the RNA to a final volume of 18 μl and a final MgCl2 concentration of 6 mM. Reactions were incubated at 30 °C for 20 minutes. Prior to incubation, the RNA sample was separated into two 9 μl reactions. A 130 mM N-methylisatoic anhydride (NMIA) (2.3 μg/ml) stock is prepared in anhydrous DMSO (Sigma) immediately before addition of the modifying reagent to the RNA sample. NMIA was added to each reaction in 1 μl amounts, and allowed to incubate at 30 °C for 84 minutes (five half-lives of the NMIA reagent in solution). Following modification, 3 μl of 32-P 5′-end labeled reverse transcription primer (~ 0.3 μM, 5′-GAACCGGACCGAAGCCCG) was added to the reaction and incubated at 65 °C for five minutes. A second incubation at 35 °C for 10 minutes is sufficient to promote primer annealing. Following an initial one minute incubation at 52 °C, 6 μl of enzyme mix (250 mM KCl, 167 mM Tris HCl, pH 8.3, 1.67 mM each dNTP, 16.7 mM DTT, 0.33 units Superscript III Reverse Transcriptase (Invitrogen, Inc.)) and allowed to incubate at 52 °C for five minutes. The reaction is stopped by the addition of 1 μl of 4 M NaOH, incubation at 90 °C for five minutes and finally, addition of 29 μl acid stop mix (4:25 (v/v) mixture of 1 M unbuffered Tris-HCl and stop dye (85% formamide, 0.5x TBE buffer, 50 mM EDTA, pH 8.0, with bromophenol blue and xylene cyanol)). Samples were loaded immediately onto a 12% denaturing PAGE gel and run at a constant 55 W for 4.5 hours or stored at −20 °C. Final gels were imaged using a Typhoon Scanner (Molecular Dynamics). The consistency of the RNA loaded was based upon the intensity of the band corresponding to U63, a nucleotide in L2 that was identified as being modified equally all RNAs tested.
Results
All mutations in this study were made in the context of the guanine riboswitch from B. subtilis that controls the xpt-pbuX operon (7). The aptamer domain of this riboswitch has been extensively characterized using genetic, biochemical, and structural methods (7, 18, 21). A single point mutation in this RNA, C74U, confers a complete specificity switch from guanine to adenine (7, 8). Crystallographic and biochemical analysis has demonstrated that this RNA (referred to as “GRA”, meaning guanine-riboswitch-to-adenine) faithfully mimics the behavior and specificity of naturally occurring adenine riboswitches (21, 32, 41). Using the GRA RNA as a model system for understanding ligand binding by the purine riboswitch has several distinct advantages. First, it binds with high affinity to 2,6 diaminopurine (DAP), an adenine analog that like guanine forms three hydrogen bonds with the specificity pyrimidine (Y74). Second, DAP is much more soluble than guanine (~5 mM vs. <50 μM). Finally, we can more readily measure the free energy differences between wild type GRA and potentially weak binding mutants by using a higher affinity ligand, maximizing the effective range of ΔΔG values. Thus, it is important to emphasize that all of the studies in this paper were performed on a natural guanine riboswitch containing a point mutation rendering it specific for adenine and its analogs. Our results are consistent with studies conducted using fully natural purine riboswitches, validating our approach (22, 23, 41).
Mutations in the loop-loop interaction
The loop-loop interaction between loops L2 and L3 is a central feature in both the adenine and guanine riboswitches and is essential for the purine riboswitch to productively bind nucleobases under physiological concentrations (18). Most of the nucleotides in these loops are strongly phylogenetically conserved (Figure 1, Supporting Table 1) (7, 31). The loops interact primarily through highly conserved residues forming two base quartets (Figure 1b, Supporting Figure 1a) each of which is comprised of a Watson-Crick pair and a non-canonical base pair buttressing the minor-groove side of the Watson-Crick pair (Figure 3) (18). Additional tertiary contacts are formed in the loop-loop interaction by two non-canonical base pairs above the quartets (A35•A64 and G62•U63, Supporting Figure 1). These pairs are less conserved to the extent that the side-by-side G62•U63 pair at the top of L3 is absent in the pbuE adenine riboswitch structure from V. vulnificus that was solved by X-ray crystallography (20).
Figure 3.
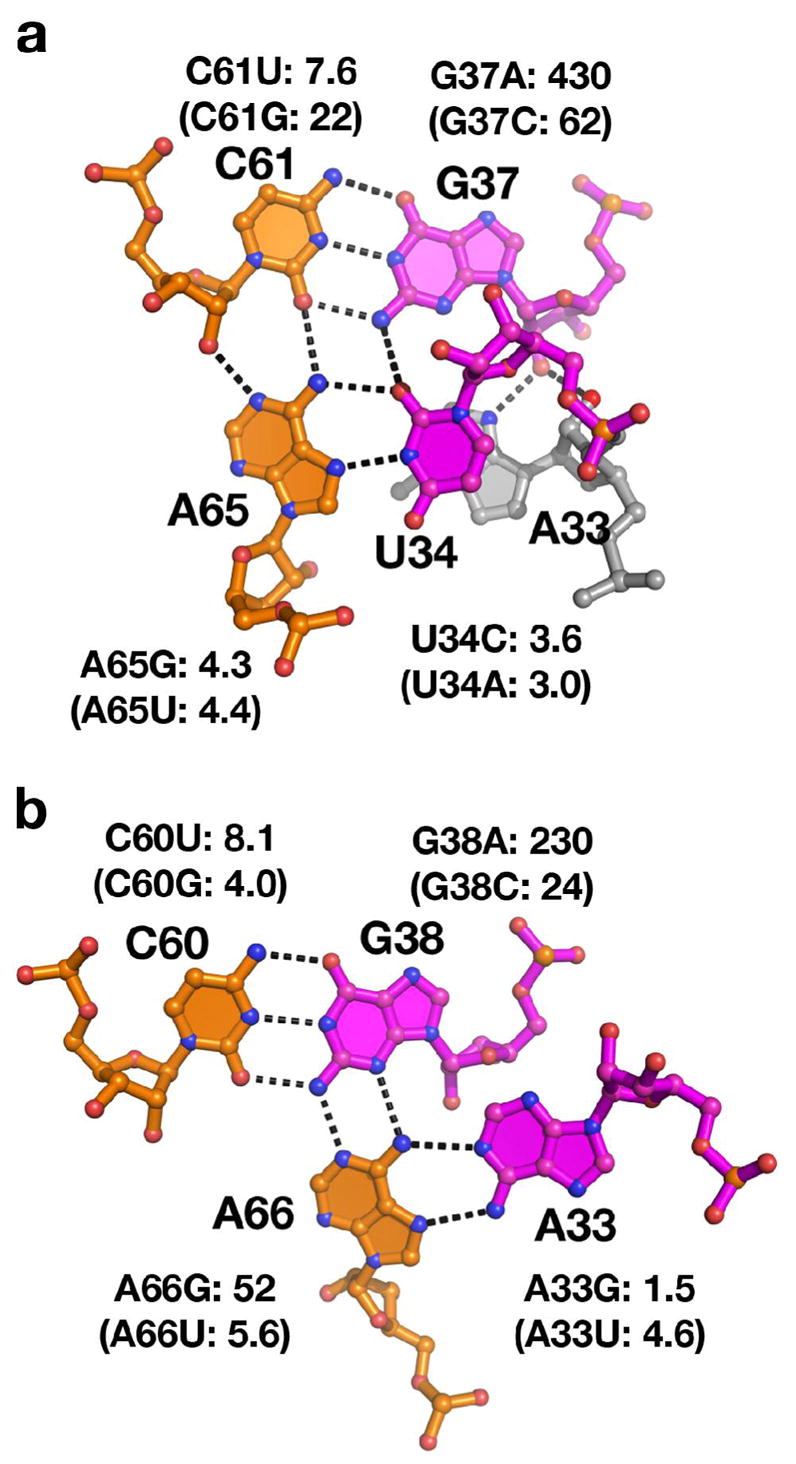
Base quadruple interactions between loop 2 (magenta) and 3 (orange) and the effect of mutations on each. The wild type base interactions from the xpt-pbuX guanine riboswitch are illustrated and the effect of mutations of each base are shown as KD,rel; measurements from a study by Lemay et al. (22) in parenthesis for comparison. (a) Structure of the C61-G37•(A65•U34) quadruple in which C61 and G37 form a standard Watson-Crick pair into whose minor groove face a non-standard trans Watson-Crick/Hoogsteen pair is docked. (b) Structure of the C60-G38•(A66•A33) quadruple which uses a similar architectural scheme in which a trans Watson-Crick/Hoogsteen pair is docked into the minor groove of a Watson-Crick pair.
To determine the relative importance of these bases in ligand recognition, we synthesized a series of RNAs containing single point mutations throughout the two loops and tested their ability to bind DAP by ITC. The mutations were chosen such that they would likely result in a minimal disruption of the loop-loop interaction, with the majority of the changes involving purine-to-purine and pyrimidine-to-pyrimidine changes (Table 1). This differs from the mutagenic strategy of the adenine riboswitch by Lemay et al (22), in which purines were converted to pyrimidines and vice versa. Despite the differences in both experimental techniques and mutagenic approach, the two sets of data are quite consistent.
TABLE 1.
Effects of mutations upon GRA RNA affinity for 2,6-diaminopurine.
| Mutation | KD, app (μM) |
Krel (KD,mut/KD,wt) |
ΔΔG
(kcal/mol) |
n |
|---|---|---|---|---|
| Wild type | ||||
| GR7(C74U) | 0.021 ± 0.003 | 1.0 | ||
| Loop 2 | ||||
| A33G | 0.032 ± 0.002 | 1.5 | 0.26 | 0.63 ± 0.02 |
| U34C | 0.075 ± 0.03 | 3.6 | 0.77 | 0.37 ± 0.20 |
| A35U | 0.019 ± 0.005 | 0.90 | −0.05 | 0.47 ± 0.05 |
| G37A | 9.0 ± 1.3 | 430 | 3.7 | 1a |
| G38A | 4.9 ± 1.3 | 230 | 3.3 | 0.56 ± 0.06 |
| Loop 3 | ||||
| C60U | 0.17 ± 0.01 | 8.1 | 1.3 | 0.46 ± 0.34 |
| C61U | 0.16 ± 0.05 | 7.6 | 1.2 | 0.46 ± 0.08 |
| U63A | 0.019 ± 0.01 | 0.90 | −0.05 | 0.56 ± 0.03 |
| A65G | 0.09 ± 0.05 | 4.3 | 0.88 | 0.55 ± 0.10 |
| A66G | 1.1 ± 1.0 | 52 | 2.4 | 0.48 ± 0.26 |
| P1 Helix – J2/3 | ||||
| U20C,A76G | 0.18 ± 0.04 | 8.6 | 1.3 | 0.28 ± 0.09 |
| U20A,A76U | 0.026 ± 0.002 | 1.2 | 0.13 | 0.48 ± 0.04 |
| U47C | 2.2 ± 0.85 | 105 | 2.8 | 1 |
| U49C | 0.38 ± 0.03 | 18 | 1.7 | 0.64 ± 0.04 |
| U49G | 2.1 ± 0.50 | 100 | 2.8 | 0.50 ± 0.05 |
| A21G,U75C | 0.076 ± 0.01 | 3.6 | 0.78 | 0.51 ± 0.06 |
| A21U,U75A | 0.047 ± 0.01 | 2.2 | 0.49 | 0.61 ± 0.06 |
| C50A | 3.1 ± 0.1 | 150 | 3.0 | 0.57 ± 0.15 |
| Upper 3WJ | ||||
| U25C,A45G | 0.023 ± 0.001 | 1.1 | 0.061 | 0.63 ± 0.04 |
| C54U,G72A | 0.066 ± 0.01 | 3.1 | 0.70 | 0.46 ± 0.11 |
| A23C | 37 ± 19 | 1760 | 4.5 | 1 |
| A24C | 2.2 ± 0.4 | 105 | 2.8 | 0.70 ± 0.04 |
| G46A,C53U | n.d.b | - | - | |
| U22C | n.d. | - | - | |
| U22C,A52G | 0.077 ± 0.03 | 3.7 | 0.79 | 0.77 ± 0.25 |
| U22A,A52U | 14 ± 2 | 680 | 3.9 | 1 |
| U22G,A52C | 18 ± 5 | 860 | 4.1 | 1 |
| A73G | n.d. | - | - | |
| A73C | 0.044 ± 0.01 | 2.2 | 0.49 | 0.54 ± 0.02 |
. In calculations of binding affinity, n=1 was a fixed parameter.
. No detectable binding.
In agreement with their relative lack of phylogenetic conservation, mutation of either of the two terminal non-canonical base pairs does not significantly affect ligand binding (Table 1). U63, which makes several hydrogen bonding contacts to the backbone of L2 (Supporting Figure 1a, b) and is highly conserved in phylogeny, can be changed to adenine without any adverse effect on binding. This mutation potentially pushes the top of L3 away from L2, along with abrogating all native hydrogen bonds. Likewise, mutation of A35 within the non-canonical pair A35•A64 immediately below the G62•U63 dinucleotide platform (Supporting Figure 1, Table 1) is not significantly deleterious to ligand binding, consistent with the lower degree of conservation of this nucleotide. Along with observations from a native adenine riboswitch (22), these data indicate that the hydrogen bonding network at the very top of the loop-loop interaction is not highly important for stabilizing and maintaining the active conformation of the RNA. This is also consistent with the fact that the structure of this region of the loop-loop interaction differs between the B. subtilis xpt-pbuX guanine and the pbuE adenine riboswitches (18, 20).
In contrast, mutations within the two quartets are significantly less tolerated (Table 1). Each quartet (Figure 3a, b) is composed of a standard Watson-Crick base pair between the two loops (G38-C60 and G37-C61) with a non-canonical base pair docked into its minor groove (the trans Watson-Crick/Hoogsteen A33•A66 pair and a trans Watson-Crick/Hoogsteen U34•A65, respectively) (42). A series of conservative mutations were made to seven of the eight nucleotides in the two quartets (Table 1). The most deleterious mutations involved changing G37 and G38 of the Watson-Crick pairs to adenosines, destroying the potential for stable pairing and thereby causing a >100 fold loss in DAP binding affinity. The loss in affinity was similar in magnitude to the binding affinity observed when the loops were deleted under identical conditions (Stoddard, et al., manuscript in preparation). The severity of these mutations unambiguously demonstrates that the high specificity and affinity of this RNA for purine nucleobases is strongly tied to its ability to form tertiary interactions distant from the ligand-binding pocket. Mutation of the complementary nucleotides C60 and C61 to uridines has a much more moderate effect (~10 fold loss in binding affinity), likely due to the ability to form wobble G-U pairs. Changes in the non-canonical pairs also have a less dramatic effect, ranging from almost no loss in affinity (A65G) to a ~50-fold loss (A66G). These data along with those of Lemay et al. (22) reveal that it is the two quartets of the loop-loop interaction, particularly the Watson-Crick pairing component, that are the principle contributors to the stability of this element of tertiary architecture.
The P1-J2/3 triples
Genetic regulation by riboswitches requires communication of ligand binding by the aptamer domain to the downstream expression platform. In the purine riboswitch, this has been proposed to be achieved through ligand-dependent formation of two base triples between the three-way junction and the P1 helix (Figure 4a, b) (18). These long-range interactions adjacent to the ligand-binding site stabilize the P1 helix, forcing the formation of a rho-independent transcription terminator in the expression platform. In phylogeny, most of the nucleotides that participate in this interaction are 100% conserved, although the A21-U75 pair of the P1 helix directly adjacent to the ligand binding pocket has a small tendency to switch to a U-A pair (Supporting Table 1). From a strictly structural perspective, it is not clear why there is such a high degree of conservation in these two sets of triples, as it would appear that the residues U49 and C50 that contact the minor groove side of the A21-U75 and U20-A76, respectively, could hydrogen bond in the same fashion with any Watson-Crick pairs in these positions. To investigate this, we made a series of mutations to both U49 and C50 on the J2/3 as well as the identity of the base pairs in the triples and characterized them by both ITC and X-ray crystallography.
Figure 4.
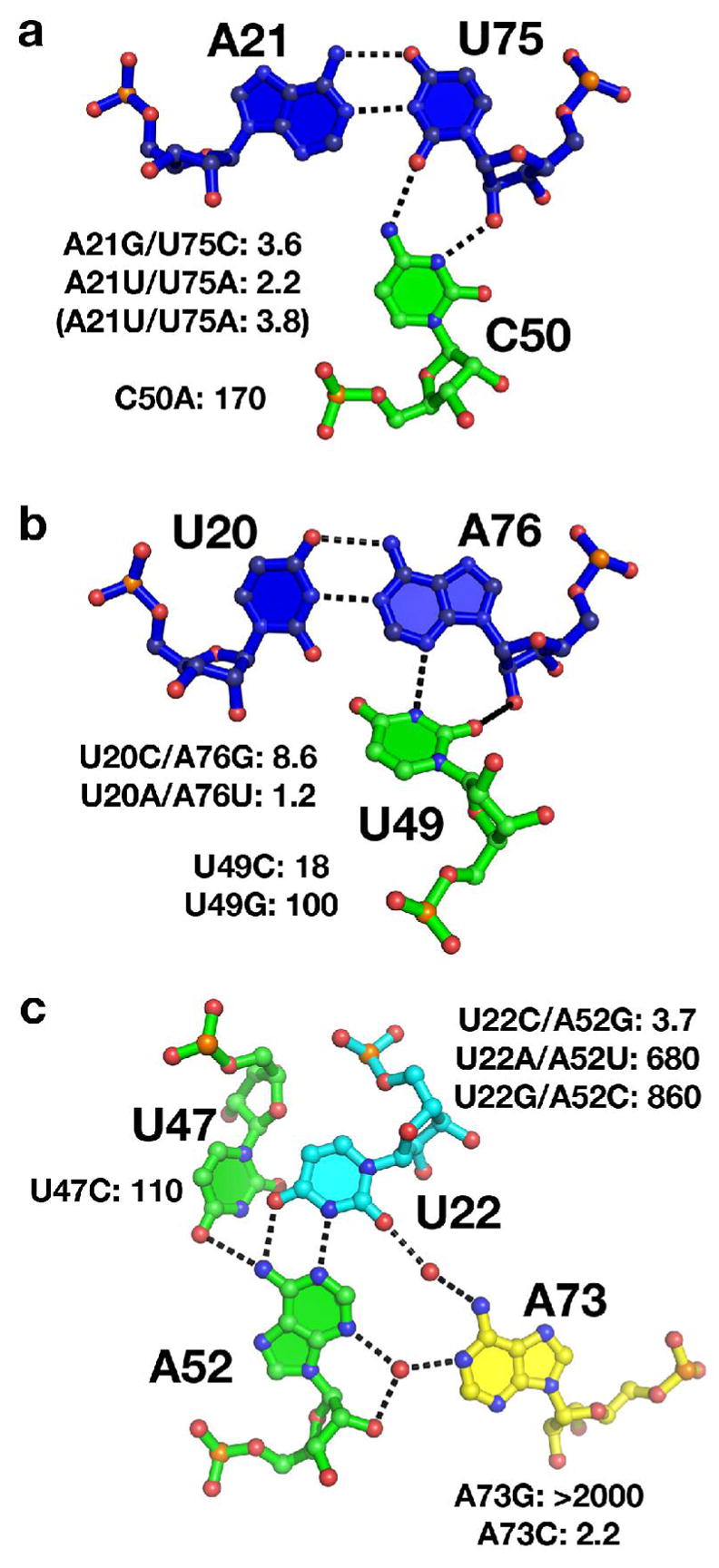
Base interactions around the ligand binding pocket; blue residues are part of the P1 helix, green are in J2/3, cyan in J1/2 and yellow in J3/1. The wild type base interactions from the xpt-pbuX guanine riboswitch are illustrated and the effect of mutations of each base are shown as KD,rel; measurements from a study by Lemay et al. (22) in parenthesis for comparison. (a) Minor groove triple at the top of the P1 helix immediately adjacent to the ligand-binding pocket. (b) The second minor groove triple formed between J2/3 and P1. (c) The water-mediated base triple that defines the top of the ligand binding pocket (the triple shown in (a) is the bottom). The red spheres represent well-ordered solvent that mediates the interaction between A73 and the U22-A52 base pair.
Despite their strong phylogenetic conservation, the two Watson-Crick pairs at the top of the P1 helix appear to be very tolerant to conservative base-pair substitutions (Table 1). Inversion of these pairs (A-U to U-A and vice versa) supports ligand binding with little change in affinity (Table 1). The presence of an additional functional group (N2 of guanosine) in the minor groove introduced through the conversion of the A-U or U-A pair to a G-C or C-G pair is only moderately disruptive to ligand binding. Therefore, the conservation of these pairs may serve additional functional roles besides promoting high affinity ligand binding (Figure 4a, b, Supporting Figure 2). Conversely, mutations made to either U49 or C50 on the J2/3 are highly disruptive to DAP binding. A conservative U49C mutation that preserves the pyrimidine residue and would support a C49(N2)-A76(O2′) hydrogen bond similar to the native structure has a greater effect on the binding affinity compared to any of the base-pair substitution mutants (Table 1). Replacement of either nucleotide U49 or C50 with a purine yields a strong decrease in affinity. These mutations clearly demonstrate that anchoring J2/3 to the P1 helix is crucial for stable ligand binding, consistent with a proposed model for ligand binding in which J2/3 acts as a flexible lid that encapsulates the purine ligand within the three-way junction (21, 43). Ligand-dependent formation of tertiary interactions involving the P1 helix appears to be a general feature of riboswitch regulation, as it is also observed in the thiamine pyrophosphate (44–46) and S-adenosylmethionine (47) riboswitch structures.
To determine whether alternative base pairs have any affect on the structure of the liganded RNA, we solved the crystal structures of two different mutants in the P1 helix (A21U,U75A and A21G,U75C) bound to DAP. These complexes crystallized under almost identical conditions and unit cell parameters as the wild type RNA bound with hypoxanthine and were solved by molecular replacement. Following several rounds of rebuilding, high temperature simulated annealing to reduce model bias and refinement (48), a simulated annealing omit map was calculated in which the nucleotides of the base triple were omitted. The models of these two structures and the corresponding maps clearly reflect that the alternative U21-A75 (Figure 5a) and G21-C75 (Figure 5b) pairs allow C50 to interact almost identically in each case. Superposition of the three structures using a maximum likelihood algorithm (THESEUS, (49, 50)) illustrates that the position of the C50 in the triple is identical within error in each RNA (Figure 5c). The identity of the Watson-Crick pairs at the 21–75 position, despite a high degree of conservation, does not play a vital role in the ability of the riboswitch to bind with high affinity to its cognate ligand nor form the fully folded structure.
Figure 5.
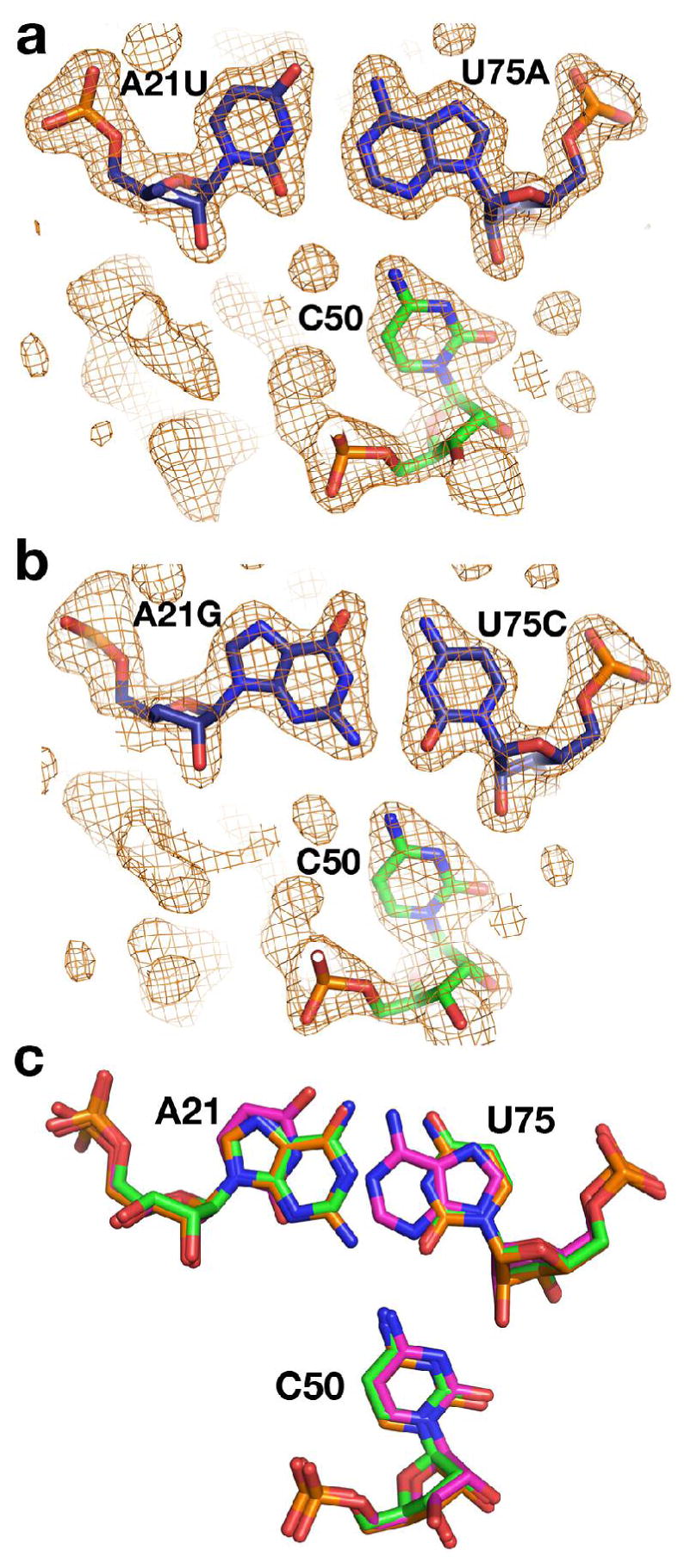
Crystal structures of RNAs containing mutations in the P1 helix. (a) 2Fo−Fc simulated annealing omit map contoured at 1 σ of the A21U,U75A mutant in which residues 21, 50 and 75 (shown as sticks) were omitted from the model used for map calculation. (b) 2Fo−Fc simulated annealing omit map contoured at 1 σ of the A21G,U75C mutant in which residues 21, 50 and 75 (shown as sticks) were omitted from the model used for map calculation. (c) Superposition of the wild type and mutants (superposition performed using all backbone atoms of each of the three structures), emphasizing that the positioning of the bases between the three RNAs is nearly identical.
The three-way junction
The three-way junction consists of a number of highly conserved base pairs and triples. At the top of this junction in both the guanine and adenine riboswitches is a single hydrogen bond interaction between the highly conserved C54-G72 pair at the base of P3 and the more variable U25-A45 pair at the base of P2. As expected from these structures, mutation of the U25-A45 to a C-G pair has no impact on binding, as this pair participates in the interaction via a non-bridging phosphate oxygen of U45 (Supporting Figure 2). Conversely, removal of the hydrogen bond by converting the C54-G72 pair to a U-A pair reveals a small loss in binding affinity that would be expected for the loss of this single interaction. Preferences for these two Watson-Crick pairs at the base of P2 and P3 are likely a result of optimal stacking interactions with more conserved nucleotides with the three-way junction (22), but only appear to have a small effect on the ability of the RNA to fold and bind ligand.
Two nearly invariant base triples, U22-A52•A73 and G46-C53•A23 are formed by the interaction of a single nucleotide (A73 and A23) with the minor groove face of a Watson-Crick pair (U22-A52 and G46-C53). The interaction between A73 and U22-A52 is water mediated while the interaction of A23C with the G46-C53 base pair is through direct hydrogen bonding. The latter is likely to be intolerant of almost any change made to it, as suggested by the 100% conservation across phylogeny. Mutations made to this triple, the single residue change A23C and the base pair identity change G46A,C43U mutations, were among the most deleterious of all tested (Table 1, Supporting Figure 2). On the other hand, it is not clear from the crystal structures of the wild type RNA why the Watson-Crick pair in the water-mediated triple (U22-A52) is invariant. Other base pairs should be able to support its indirect interaction with A73. Conversion of the U22-A52 pair to the other three Watson-Crick pairs revealed a clear pattern in the RNA’s 22–52 pair preference. While the U22C,A52G mutation was well tolerated, the U22G,A52C and U22A,A52G mutations exhibit significantly lower affinity for DAP. The aptamer has a clear preference for Y22-R52 pairs over R22-Y52 pairs. To determine if the R22-Y52 pair might significantly alter the structure of the three-way junction, we solved the crystal structures of the C22-G52 and A22-U52 base pair mutant GRA RNAs bound to DAP (Figure 6a, b). Interestingly, the well-tolerated C22-G52 pair has a larger impact upon the structure via a slight repositioning of both A73 and the Watson-Crick pair (Figure 6c) relative to the other two structures. The Y22-R52 preference observed for ligand binding affinity does not appear to significantly impact the fold of the ligand bound structure.
Figure 6.
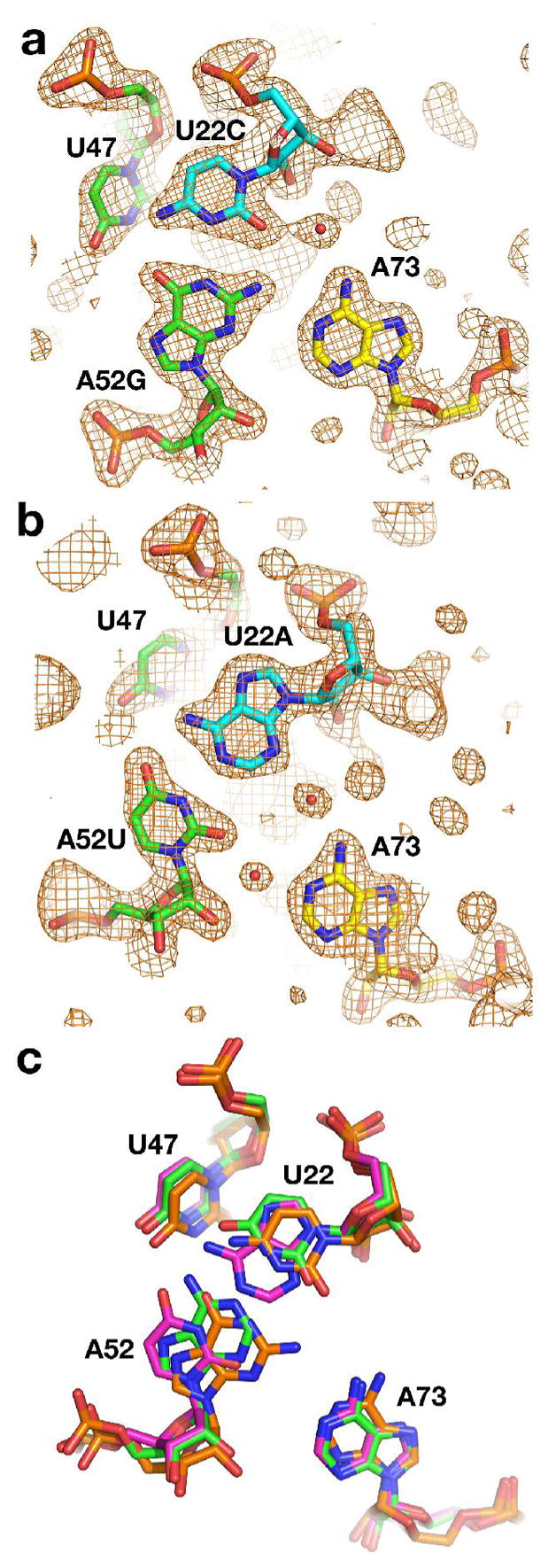
Crystal structures of RNAs containing mutations in the water-mediated base triple of the three way junction. (a) 2Fo−Fc simulated annealing omit map contoured at 1 σ of the U22C/A52G mutant in which residues 22, 47, 52 and 73 (shown as sticks) were omitted from the model used for map calculation. Residues in J1/2 are shown in cyan, those in J2/3 are in green and in J3/1, yellow. (b) 2Fo−Fc simulated annealing omit map contoured at 1 σ of the U22A/A52U mutant in which residues 22, 47, 52 and 73 (shown as sticks) were omitted from the model used for map calculation. (c) Superposition of the wild type and mutants with the superposition performed using all backbone atoms of each of the three structures. While the positioning of the wild type (green) and U22A/A52U mutants show nearly equivalent base positioning, the C22-G52 base pair (orange) is shifted towards A73, placing G52(N2) and A73(N6) in hydrogen bonding distance.
Instead of affecting the bound structure, R22-Y52 residues may impact the free state of the RNA by impairing its ability to productively interact with ligand. A purine nucleotide at position 22 has the potential to form an alternative R22-Y74 base pair, extending the P1 helix by one pair (Figure 7a). This interaction would have a dramatic impact on ligand binding, as we have proposed that the Watson-Crick face of Y74 serves as an initial docking site for the ligand (21). To determine if the G22-C52 or A22-U52 mutants have an altered structure of the free RNA, we used the chemical N-methylisatoic anhydride (NMIA) to probe the conformational flexibility of the backbone in the three-way junction (40). Regions of the RNA that are dynamic react with NMIA more readily, resulting in a reverse transcriptase stop in a sequencing gel (Supporting Figure 3). Wild-type GRA RNA displays a characteristic pattern of reactivity in the J1/2 and J2/3 in the absence of ligand, indicating that this region is conformationally flexible (Figure 7). In the presence of ligand, these regions of the RNA become less dynamic and thus unreactive to NMIA (Stoddard, et al., manuscript in preparation).
Figure 7.
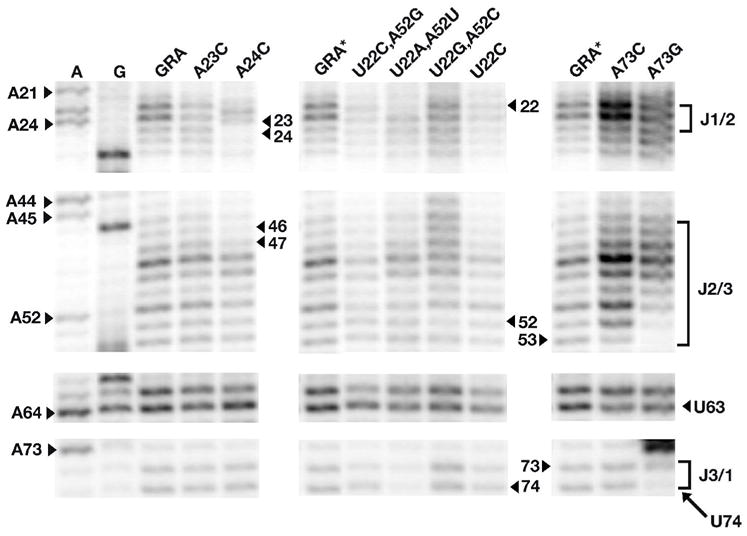
Probing the free state structure of three-way junction mutant GRA RNAs using N-methylisatoic anhydride (NMIA). Mutants are grouped according to their location in the three-way junction and shown in order of decreasing affinity for DAP (left to right). The lane corresponding to the wild type GRA shown at far left appears only once on the full sequencing gel (Supporting Figure 3) but is shown accompanying each set of mutants (GRA*) in order to illustrate the affect of each mutation; thus all three lanes labeled GRA are the identical lane from the gel. RNA was sequenced by inclusion of ddTTP or ddCTP in a reverse transcription reaction containing GRA to assign the nucleotide position of each modification (note that NMIA modification and the sequencing ladder are offset by one residue). Residues that are labeled correspond to the location of mutations and regions of possible alternative structure formation in the free state that prevents ligand binding in some mutants.
Mutations in the junction display differing degrees of changes in the pattern of NMIA reactivity in the free state. The mutant U22C, that impairs ligand binding by disrupting the U22-A52 Watson-Crick pair, shows a reactivity pattern nearly identical to GRA RNA. This is consistent with the mutation preventing the formation of a critical interaction in the bound state, rather than a disruption of the free state. Conversely, the reactivity patterns of both R22-Y52 mutants display altered patterns of reactivity in J1/2 and J2/3 (Figure 7), in contrast to crystal structure of the bound U22A,A52U that is identical to GRA. We hypothesize that an R22-Y52 pair affects the free state in a fashion that impairs productive ligand binding. In part, the potential for alternative pairing between R22 and Y74 (Supporting Figure 4) may be a contributing factor. This is supported by the observation that the reactivity pattern of the U22C,A52G mutant that cannot form alternative Watson-Crick pairs resembles wild type GRA.
Alternative pairing between J2/3 and J1/2 or J3/1 in the free state has significantly different affects upon productive ligand binding. A73G displays an altered pattern in NMIA reactivity with protection of nucleotides 52, 53, 73, and 74 (Figure 7), consistent with the formation of C53-G73 and A52-U74 base pairs (Supporting Figure 4). Furthermore, this RNA has a very strong intrinsic reverse transcriptase stop at nucleotide 72, consistent with the formation of additional stabilizing pairs in the junction (Supporting Figure 3). Similarly, the mutation A24C shows a reactivity pattern consistent with formation of alternative C24-G46 and A23-U47 pairs between J1/2 and J2/3 (Figure 7, Supporting Figure 4). Strikingly, while A73G cannot bind ligand, the A23C mutation is comparatively less deleterious. These data indicate two features of the free state of the RNA. First, nucleotides whose identities are seemingly unimportant in the context of the bound structure, can be critical for maintaining the free RNA in a conformation capable of productively binding ligand. Second, mispairing involving J3/1 is the most detrimental to binding, in support of the hypothesis that the availability of the Watson-Crick face of Y74 is the most critical feature for productive ligand binding.
One other nucleotide within J2/3, U47, appears to be critical for establishing the ligand-bound form of the three-way junction. This nucleotide forms extensive contacts with the J2/3 backbone as well as U51 and A52, suggesting that it is an important support element for the tight turn formed by J2/3 (21). Similar to mutation of the neighboring nucleotides U49 and C50, mutation of U47 to a cytosine decreases the binding affinity relative to wild type, but does support ligand binding (Table 1). Since cytosine at position 47 potentially abrogates hydrogen bonds to U51 and A52, we crystallized this mutant to determine if the J2/3 loop would form a significantly different conformation. Surprisingly, the mutant has an almost identical structure to that of the wild type RNA (Figure 8a) despite the fact that the position of C47 creates several unfavorable interactions with U51 and A52 (Figure 8b). While this mutation clearly destabilizes the ligand bound complex, it is accommodated sufficiently well to allow for the formation of a folded architecture almost identical to that of the wild type RNA.
Figure 8.
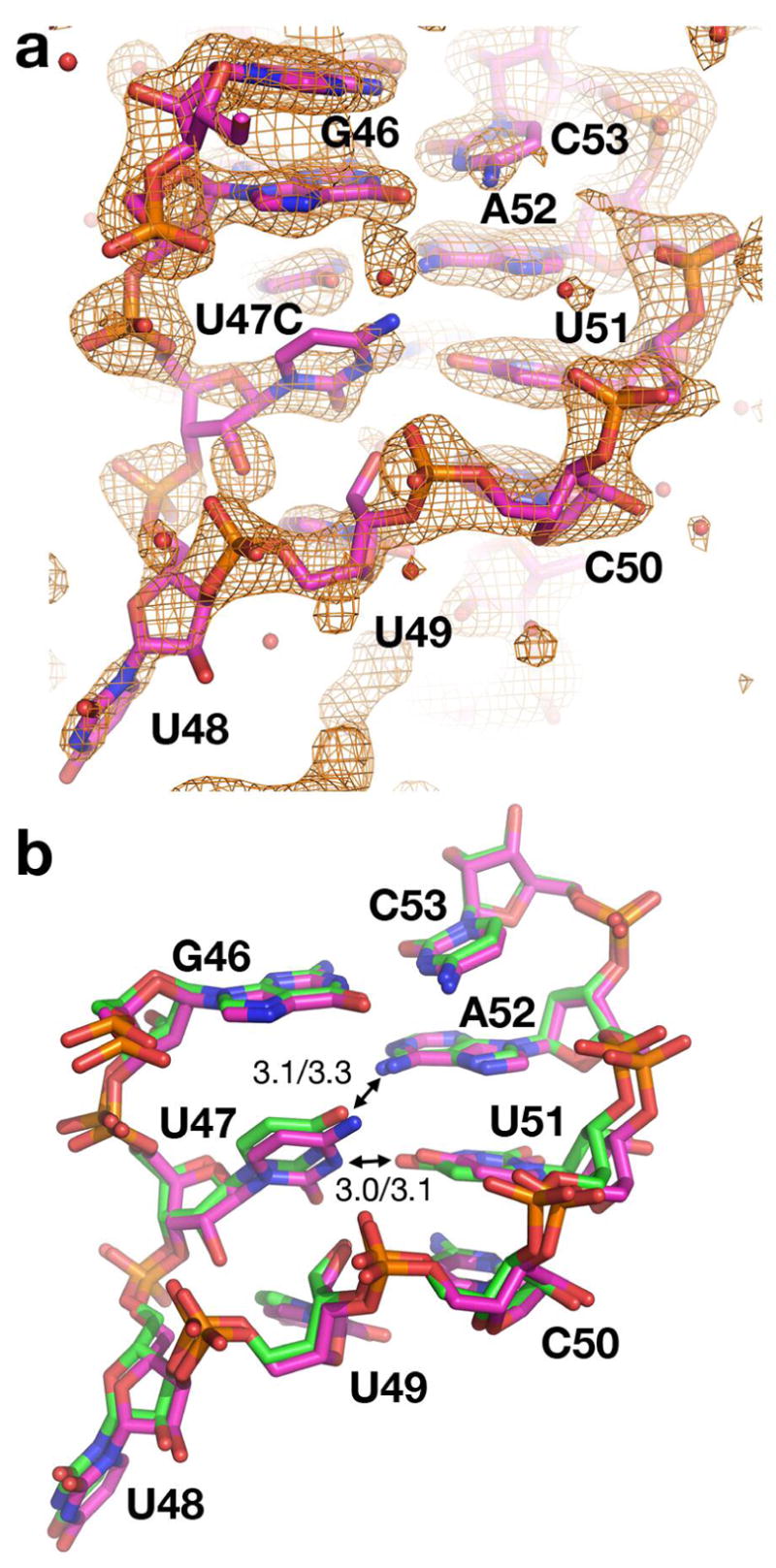
Structure of the U47C mutant. (a) 2Fo−Fc simulated annealing omit map contoured at 1 σ of the U47C mutant in which residue 47 was omitted from the model used for map calculation. Note the relatively poor definition of the ribose sugar and the base in the mutant. (b) Superposition of the wild type (green) and mutant (magenta) structures over all backbone atoms. The U47(N3)-U51(O2) and U47(O4)-A52(N6) hydrogen bonds in the wild type structure are shown with arrows and the interatomic distances (in Ångstroms) is denoted as the left value. The right value reflects the interatomic distances of the equivalent atoms in the U47C structure emphasizing that the cytosine packs against J2/3 in the same fashion despite having unfavorable interactions between these two sets of atoms.
In all five of the mutant structures, the base pair substitutions in the water-mediated triple and at the top of the P1 helix as well as the U47C mutant, are very close to the structure of the wild type complex. However, one mutant displays a clear difference in its degree of thermal motion (B-factor). While B-factor interpretation can be problematic, the fact that all five mutants crystallized under nearly identical conditions and yielded isomorphous crystals with the same intermolecular lattice contacts allows us to compare the thermal B-factors between structures (44, 51). In the highly ordered loop-loop interaction at the top of the P2 and P3 helices, the local B-factors in every structure are the same, reflecting a similar degree of order in each crystal (Figure 9). Even within the three-way junction and ligand-binding pocket very similar degrees of order are observed between structures (Figure 9, wt versus A21U,U75A). Only in the U47C mutant do the local B-factors deviate substantially from wild type. Nucleotides throughout the three-way junction and adjacent regions in P1, P2 and P3 of the U47C mutant structure exhibit higher B-factors and, thus, higher local thermal disorder. Therefore, U47 is indeed a critical nucleotide for stabilization of J2/3 against the three-way junction to form the liganded state.
Figure 9.
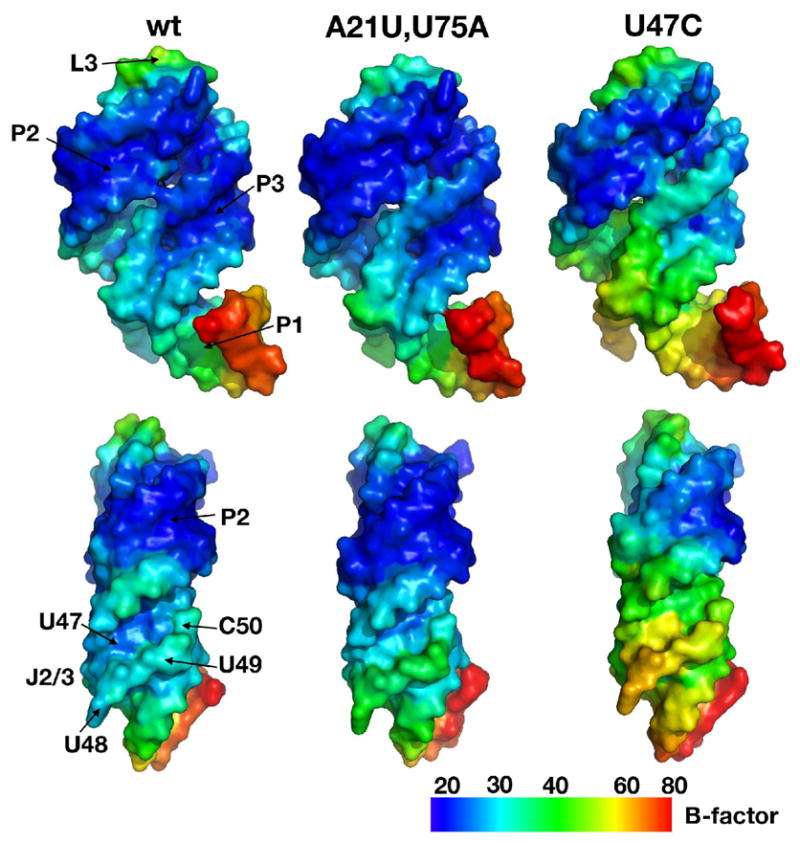
Surface representations of left, GRA RNA (“wt”, PDB ID 2B57); middle, the (A21U, U75A) mutant; right, the U47C mutant. The coloration of the surface represents the atomic B-factors for atoms in the RNA (bottom bar), with blue indicating low thermal disorder (B ~ 20–30) and red indicating high disorder in the lattice (B ~ 70–80). The bottom perspective represents a 90° counterclockwise rotation of the upper perspective.
Discussion
In the last several years, a number of structures of riboswitches bound to their cognate ligands have been solved, revealing some common themes (18, 20, 21, 32, 44–46). These include: (1) recognition of every functional group in the ligand resulting in almost complete encapsulation of the ligand within the binding pocket; (2) the use of complex tertiary structural elements to facilitate ligand binding; (3) long-range interactions important in P1 helix stabilization and gene regulation (reviewed in (52)). While these structures show how these RNAs specifically recognize their respective ligands, they pose several very significant questions, particularly how conserved elements in the aptamer domain may be important for other aspects of riboswitch function. In addition to ligand recognition, riboswitches must facilitate interdomain communication and maintain a ligand binding-competent form of the RNA in the unbound state. The purpose of this detailed mutagenic survey of the purine riboswitch is to probe the structure in a way that uncovers which nucleotides serve roles in these other functions.
The loop-loop interaction, while not directly involved in ligand recognition, is an important feature of the aptamer domain. Many of the nucleotides in both L2 and L3 are universally conserved within the purine riboswitch family (7) and under physiological ionic conditions this independently folding element of tertiary structure (22) is essential for ligand binding (18). The structures of the guanine and adenine riboswitches revealed that two sets of base quadruples are at the core of this interaction, each consisting of a Watson-Crick pair into whose minor groove face a non-canonical pair is docked (18, 20). Examination of our data, along with that of Lafontaine and coworkers who probed the pbuE adenine riboswitch (22), reveals several aspects of this element of tertiary structure and its relationship to ligand binding. First, the distal part of the loop-loop interaction is dispensable for ligand binding. Severe mutations in the dinucleotide platform (G62•U63) and the A35•A64 pair have little impact upon binding (Supporting Figure 1). This is consistent with the loose phylogenetic conservation of these nucleotides (7, 8, 31) and differences in the structures of this part of the interaction between the adenine and guanine riboswitches (20). In contrast, the quadruples are important for stable formation of the loop-loop interaction. By using the most conservative mutations possible, we observe that the most crucial components of this tertiary interaction are the G37-C61 and G38-C60 Watson-Crick pairs that comprise half the quadruple, and most particularly, the identities of G37 and G38. Changing either guanine to an adenine to potentially create an A•C mismatch causes a sharp decrease in DAP binding affinity (ΔΔG = 3.3 kcal/mol in each case), similar to the binding affinity for DAP of a loop-loop deletion mutant (Stoddard, et al., manuscript in preparation). Conversion of the cytosine residues to uracil results in a less dramatic loss in affinity (ΔΔG = 1.2 kcal/mol in each case). Presumably, this is because a G•U wobble is sufficient to promote the interaction. The identities of several nucleotides in the non-canonical pair component of the quadruple (A33, U34 and A65) are not nearly as important for ligand binding affinity. Therefore, Watson-Crick pairing between nucleotides 37–61 and 38–60 within the base quadruples is the primary stabilizing interaction that promotes formation of the loop-loop interaction. This is further supported in that changing either of the wild type G-C pairs to a C-G (22) or A-U (23) pair supports productive ligand binding, indicative of a stable loop-loop interaction.
The primary role of the loop-loop interaction appears to be preorganization of the global fold of the aptamer domain for ligand recognition (18, 43). Experiments done by in-line probing (7), NMR spectroscopy (23), and single molecule FRET (22) confirm that the structured loop-loop interaction is established prior to ligand binding. In contrast, the three-way junction remains locally disorganized in the absence of ligand (7, 18, 21), and only becomes structured in the presence of the purine nucleobase. This is supported by the fact that the ligand is encapsulated within the binding pocket (>95% solvent inaccessible in the bound state) as revealed by X-ray crystallography (18, 20). Thus, the three-way junction must remain in an “open” state receptive to ligand binding. Furthermore, the RNA must be resistant to misfolding in this loosely organized state. Alternative stable structures in the riboswitch would prevent ligand binding and consequently, loss of its ability to control gene expression.
A number of nucleotides in the three-way junction appear to have been selected to maintain the free RNA in a form capable of binding ligand. Residues A73, A24 and the Y22-R52 base pair have likely been conserved because alternative bases at these positions collapse the binding pocket by extension of one of the helices surrounding the three-way junction by at least a single Watson-Crick pair (Supporting Figure 4). Mutational studies made in the context of the pbuE adenine riboswitch (41) identified a similar tendency of residue 48 on the J2/3 (which is flipped out in the bound structure) to base pair with Y74 in J3/1 (which forms the Watson-Crick pair with the ligand) (41). Significantly, these studies reveal the importance of keeping the free state of the RNA in a form that allows Y74 to be accessed by the ligand to form a Watson-Crick pair. Thus, it is clear that a number of nucleotide positions within the three-way junction experience substantial selective pressure on their identity for the role they play in the free state of the RNA and not the bound.
In addition to residues in the three-way junction, nucleotides in the P1 helix also display a conservation pattern that is not directly related to high affinity ligand binding in the bound state. The Watson-Crick pairs in the P1 helix adjacent to the junction are likely conserved for the role they play in communication between the aptamer domain and expression platform. Most convincingly, the U20A,A76U mutation has no effect on ligand binding despite its universal conservation. Both the U20-A76 and A21-U75 pairs are tolerant to mutations that preserve pairing, although adenine-uracil pairs are preferred as the N2 of guanine creates a small amount of steric interference to base triple formation with U49 and C50. Clear sequence conservations in the P1 helix away from the junction (R19-Y77, Y18-R78, and U17-R79) (6, 7) that are not involved in the binding pocket or tertiary structure have arisen because nucleotides on the 3′-side of the P1 helix are directly involved in the secondary structural switch. Interestingly, the SAM-I riboswitch also displays similar conservation patterns of nucleotides within the P1 helix that are not directly involved in ligand binding (53). Phylogenetic conservation in this helix probably reflects thermodynamic and kinetic requirements for ligand dependent helix disruption and formation between the competing P1 helix and antiterminator stem-loop in the expression platform. Thus, the emerging picture is that the aptamer domains of riboswitches contain complex sequence requirements that involve formation of a stable ligand bound state, maintain a free state that can productively bind ligand, and secondary structural switching. The regulatory mechanism of riboswitches requires the finely tuned interplay between these three important sequence constraints on the RNA in order to efficiently control expression of its mRNA.
Supplementary Material
Table S1, phylogenetic conservation of key residues in the purine riboswitch; Figure S1, structure of the loop-loop interaction of the purine riboswitch and its non-canonical base pairs; Figure S2, structure of the three-way junction and several base interactions within it; Figure S3, sequencing gel of NMIA probing of mutant RNAs; Figure S4, proposed alternative base pairing in the three way junction. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank Steven Edwards for maintaining the X-ray facility at the Univ. Colorado, Boulder where all data was acquired, and members of the Batey laboratory for discussions and critical comments on the manuscript.
ABBREVIATIONS
- DAP
2,6-diaminopurine
- ITC
isothermal titration calorimetry
- R
purine
- Y
pyrimidine
- UTR
untranslated region
- GRA
xpt-pbuX guanine responsive riboswitch from B. subtilis carrying a C74U mutation
Footnotes
This work was supported by NIH Grant GM073850 and a Research Scholar Grant from the American Cancer Society to R.T.B.
References
- 1.Costa FF. Non-coding RNAs: Lost in translation? Gene. 2007;386:1–10. doi: 10.1016/j.gene.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 2.Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–3. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 3.Storz G, Altuvia S, Wassarman KM. An abundance of RNA regulators. Annu Rev Biochem. 2005;74:199–217. doi: 10.1146/annurev.biochem.74.082803.133136. [DOI] [PubMed] [Google Scholar]
- 4.Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J. A new frontier for molecular medicine: noncoding RNAs. Biochim Biophys Acta. 2005;1756:65–75. doi: 10.1016/j.bbcan.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Curr Opin Struct Biol. 2005;15:342–8. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Winkler WC. Riboswitches and the role of noncoding RNAs in bacterial metabolic control. Curr Opin Chem Biol. 2005;9:594–602. doi: 10.1016/j.cbpa.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003;113:577–86. doi: 10.1016/s0092-8674(03)00391-x. [DOI] [PubMed] [Google Scholar]
- 8.Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat Struct Mol Biol. 2004;11:29–35. doi: 10.1038/nsmb710. [DOI] [PubMed] [Google Scholar]
- 9.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–9. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 10.Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 2003;17:2688–97. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch? Nucleic Acids Res. 2003;31:6748–57. doi: 10.1093/nar/gkg900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winkler WC, Cohen-Chalamish S, Breaker RR. An mRNA structure that controls gene expression by binding FMN. Proc Natl Acad Sci U S A. 2002;99:15908–13. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nahvi A, Barrick JE, Breaker RR. Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res. 2004;32:143–50. doi: 10.1093/nar/gkh167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cromie MJ, Shi Y, Latifi T, Groisman EA. An RNA sensor for intracellular Mg(2+) Cell. 2006;125:71–84. doi: 10.1016/j.cell.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 15.Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem Sci. 2004;29:11–7. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Johansen LE, Nygaard P, Lassen C, Agerso Y, Saxild HH. Definition of a second Bacillus subtilis pur regulon comprising the pur and xpt-pbuX operons plus pbuG, nupG (yxjA), and pbuE (ydhL) J Bacteriol. 2003;185:5200–9. doi: 10.1128/JB.185.17.5200-5209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nygaard P, Saxild HH. The purine efflux pump PbuE in Bacillus subtilis modulates expression of the PurR and G-box (XptR) regulons by adjusting the purine base pool size. J Bacteriol. 2005;187:791–4. doi: 10.1128/JB.187.2.791-794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–5. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 19.Noeske J, Richter C, Grundl MA, Nasiri HR, Schwalbe H, Wohnert J. An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc Natl Acad Sci U S A. 2005;102:1372–7. doi: 10.1073/pnas.0406347102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol. 2004;11:1729–41. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert SD, Stoddard CD, Wise SJ, Batey RT. Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J Mol Biol. 2006;359:754–68. doi: 10.1016/j.jmb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Lemay JF, Penedo JC, Tremblay R, Lilley DM, Lafontaine DA. Folding of the adenine riboswitch. Chem Biol. 2006;13:857–68. doi: 10.1016/j.chembiol.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Noeske J, Buck J, Furtig B, Nasiri HR, Schwalbe H, Wohnert J. Interplay of ‘induced fit’ and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch. Nucleic Acids Res. 2006 doi: 10.1093/nar/gkl1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grundy FJ, Henkin TM. The S box regulon: a new global transcription termination control system for methionine and cysteine biosynthesis genes in gram-positive bacteria. Mol Microbiol. 1998;30:737–49. doi: 10.1046/j.1365-2958.1998.01105.x. [DOI] [PubMed] [Google Scholar]
- 25.Miranda-Rios J, Navarro M, Soberon M. A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc Natl Acad Sci U S A. 2001;98:9736–41. doi: 10.1073/pnas.161168098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002;9:1043. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 27.Wickiser JK, Cheah MT, Breaker RR, Crothers DM. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry. 2005;44:13404–14. doi: 10.1021/bi051008u. [DOI] [PubMed] [Google Scholar]
- 28.Wickiser JK, Winkler WC, Breaker RR, Crothers DM. The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol Cell. 2005;18:49–60. doi: 10.1016/j.molcel.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 29.Stoddard CD, Batey RT. Mix-and-match riboswitches. ACS Chem Biol. 2006;1:751–4. doi: 10.1021/cb600458w. [DOI] [PubMed] [Google Scholar]
- 30.Winkler WC, Dann CE., 3rd RNA allostery glimpsed. Nat Struct Mol Biol. 2006;13:569–71. doi: 10.1038/nsmb0706-569. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–4. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilbert SD, Mediatore SJ, Batey RT. Modified pyrimidines specifically bind the purine riboswitch. J Am Chem Soc. 2006;128:14214–5. doi: 10.1021/ja063645t. [DOI] [PubMed] [Google Scholar]
- 33.Kieft JS, Batey RT. A general method for rapid and nondenaturing purification of RNAs. RNA. 2004;10:988–95. doi: 10.1261/rna.7040604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook J, Russel DW. Molecular Cloning: A laboratory manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 35.Turnbull WB, Daranas AH. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125:14859–66. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 36.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–7. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 37.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 10):1718–25. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Hodel A, Kim SH, Brünger AT. Model Bias in Macromolecular Crystal-Structures. Acta Crystallogr A. 1992;48:851–858. [Google Scholar]
- 40.Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–6. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 41.Lemay JF, Lafontaine DA. Core requirements of the adenine riboswitch aptamer for ligand binding. RNA. 2007 doi: 10.1261/rna.142007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leontis NB, Westhof E. Conserved geometrical base-pairing patterns in RNA. Q Rev Biophys. 1998;31:399–455. doi: 10.1017/s0033583599003479. [DOI] [PubMed] [Google Scholar]
- 43.Gilbert SD, Batey RT. Riboswitches: fold and function. Chem Biol. 2006;13:805–7. doi: 10.1016/j.chembiol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Edwards TE, Ferre-D’Amare AR. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure. 2006;14:1459–68. doi: 10.1016/j.str.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–71. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thore S, Leibundgut M, Ban N. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science. 2006;312:1208–11. doi: 10.1126/science.1128451. [DOI] [PubMed] [Google Scholar]
- 47.Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–5. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT, Krukowski A, Erickson JW. Slow-cooling protocols for crystallographic refinement by simulated annealing. Acta Crystallogr A. 1990;46(Pt 7):585–93. doi: 10.1107/s0108767390002355. [DOI] [PubMed] [Google Scholar]
- 49.Theobald DL, Wuttke DS. Empirical Bayes hierarchical models for regularizing maximum likelihood estimation in the matrix Gaussian Procrustes problem. Proc Natl Acad Sci U S A. 2006;103:18521–7. doi: 10.1073/pnas.0508445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Theobald DL, Wuttke DS. THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics. 2006;22:2171–2. doi: 10.1093/bioinformatics/btl332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuriyan J, Weis WI. Rigid protein motion as a model for crystallographic temperature factors. Proc Natl Acad Sci U S A. 1991;88:2773–7. doi: 10.1073/pnas.88.7.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilbert SD, Montange RK, Stoddard CD, Batey RT. Structural Studies of the Purine and SAM Binding Riboswitches. Cold Spring Harbor Symp Quant Biol. 2007;LXXI doi: 10.1101/sqb.2006.71.015. In the press. [DOI] [PubMed] [Google Scholar]
- 53.Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat Struct Biol. 2003;10:701–7. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, phylogenetic conservation of key residues in the purine riboswitch; Figure S1, structure of the loop-loop interaction of the purine riboswitch and its non-canonical base pairs; Figure S2, structure of the three-way junction and several base interactions within it; Figure S3, sequencing gel of NMIA probing of mutant RNAs; Figure S4, proposed alternative base pairing in the three way junction. This material is available free of charge via the Internet at http://pubs.acs.org.