

Abstract
Ex-vivo generation of human insulin-producing cells is considered a promising approach to providing an abundant source of cells for beta-cell replacement therapy in diabetes. Expansion of adult beta-cells from the limited number of islet donors is an attractive prospect. However, while evidence supports the replicative capacity of both rodent and human beta-cells in vivo, attempts at expanding these cells in tissue culture result in loss of beta-cell phenotype, making it difficult to track their fate during continuous propagation and raising doubts about their therapeutic potential. Recent lineage-tracing studies demonstrate the ability of human beta-cells to survive and replicate to a significant degree in vitro. Beta-cell delamination out of the normal epithelial structure, a process that results in dedifferentiation, seems to be required for significant in-vitro proliferation. Therefore, ways must be found of inducing redifferentiation of the expanded cells ex vivo, or of restoring their function upon transplantation. Elucidation of the signaling pathways altered during beta-cell adaptation to growth in culture may provide clues to cell redifferentiation. In a recent study, we found that human beta-cell dedifferentiation and entrance into the cell cycle in vitro correlated with activation of the Notch pathway and downregulation of the cell cycle inhibitor p57. Inhibition of the Notch downstream target HES1 using short hairpin RNA reduced beta-cell dedifferentiation and replication, suggesting a potential target for inducing cell redifferentiation following expansion in culture. This review critically discusses the potential for using ex-vivo beta-cell replication and redifferentiation in cell replacement therapy in diabetes.
Keywords: diabetes, beta-cell replication, dedifferentiation, ex-vivo differentiation, Notch
Introduction
Ex-vivo development of human insulin-producing cells is considered a promising approach to generating an abundant source of cells for beta-cell replacement therapy in diabetes, which is currently limited by the shortage of organ donors. Both embryonic and adult tissue stem cells are being extensively investigated for their potential to adopt a beta-cell-like phenotype following in-vitro and/or in-vivo manipulations. To date, however, insulin-producing cells generated from stem cells have failed to produce high amounts of insulin and to regulate its secretion. The need to explore these alternative cell sources was evident from the difficulties experienced in significantly expanding functional human beta-cells in tissue culture. However, work in recent years has raised new hope that these efforts may yet prove successful.
Beta-cell replication in-vivo
Indirect evidence obtained from pancreas autopsy supports the ability of mature human beta-cells to proliferate in vivo, both in normal tissue maintenance [1] and in response to increasing demands for insulin, such as in pregnancy and obesity, or following beta-cell damage in type 1 or 2 diabetes [2]. A recent study in children (aged from 2 weeks to 21 years), combining data from both abdominal tomography and pancreas autopsy, reported that the increase in beta-cell mass in infancy is associated with beta-cell replication [3]. Excessive beta-cell replication is responsible for hyperinsulinism of infancy [4].
Since it is difficult to perform studies in humans, most of our knowledge about regulation of the beta-cell mass is derived from rodents. The increase in beta-cell mass in neonatal mice is largely due to beta-cell replication [5]. Lineage-tracing studies demonstrated that beta-cell replication is also the predominant mechanism to account for normal beta-cell turnover in adult mice [6, 7], as well as for islet regeneration following beta-cell ablation [8, 9]. Evidence suggests that replicative capacity is not restricted to a subpopulation of beta-cells; all mouse beta-cells seem to be equally capable of replication [10]. Other mechanisms, in addition to beta-cell replication, may play a role in islet regeneration, depending on the severity of damage. Xu et al. have recently shown that progenitor cells located in pancreatic ducts contribute to islet recovery following injury in adult mice [11]. This hypothesis is supported by lineage-tracing of mouse duct cells in a pancreas injury model [12].
Beta-cell replication in-vitro
Differentiated cells removed from their tissue context and placed in artificial conditions in tissue culture undergo multiple changes in gene expression and adapt to their new environment in a variety of ways. There are few examples of primary, untransformed, differentiated adult human cells which can be expanded in tissue culture without loss of phenotype. Peripheral blood leukocytes are a notable exception, probably because their in-vivo environment does not involve permanent attachment. Most cells, however, establish elaborate connections to neighboring cells and extracellular matrix (ECM) in their normal tissue location, which play key roles in regulating their phenotype and replication. When these connections are severed in vitro, cell phenotype is altered. Intact islets can be kept in suspension cultures for months without a significant decline in insulin production and secretion [13]. However, under these conditions, a significant degree of replication cannot be induced. Once attached to the culture dish surface, cells begin to migrate out of the islet structure and dedifferentiate within a period of several weeks. During this time, limited beta-cell replication can be stimulated by various agents.
Growth factors (e.g. growth hormone, placental lactogen, prolactin and glucagon-like peptide 1) and metabolites (particularly glucose) were shown to stimulate a small number of population doublings of rodent insulin-positive cells before cell phenotype was lost (see reference [14] for review). Attempts at culturing adult human islet cells resulted in a similar loss of beta-cell markers in the proliferating cells following a small number of cell-population doublings [15-17]. In addition, these cultures underwent senescence following >15 population doublings. In an effort to simulate the normal cell environment better, cell aggregation and ECM were employed. However, preservation of beta-cell function remained limited [18]. It is difficult therefore to assess beta-cell proliferation in vitro beyond the initial culture period, since the beta-cell phenotype is lost. This loss may reflect beta-cell dedifferentiation, or beta-cell death, accompanied by expansion of cells from a non-beta-cell origin.
Recent genetic lineage-tracing studies in cells cultured from transgenic mouse islets have made it possible, for the first time, to track the fate of mouse beta-cells in vitro [19-22]. These studies established that mouse beta-cells dedifferentiated and survived for a few weeks. Dedifferentiation was much faster (days) when the islets were dissociated into single cells, compared with culture of intact islets (weeks) [19]. However, no significant beta-cell proliferation could be detected in these studies. After several weeks in culture, the cell population was taken over by cells derived from a non-beta-cell origin. Thus, it was concluded that mouse beta-cells cannot proliferate in vitro under the culture conditions employed.
Lineage-tracing of cultured human beta-cells
To monitor the fate of cultured human beta-cells following their dedifferentiation we developed a lineage-tracing approach based on lentivirus vectors [23]. Using this method we found evidence for massive proliferation of cells derived from human beta-cells, unlike mouse beta-cells. Beta-cells dissociated from isolated human islets were specifically labeled with green fluorescent protein (GFP) expressed under the cytomegalovirus promoter. In this system, GFP expression was blocked by a loxP-flanked DNA fragment. Removal of the block using an insulin promoter-Cre recombinase activated GFP expression only in beta-cells. Label-positive, insulin-negative cells derived from beta-cells of 15 human donors aged 17-60 were shown to proliferate for a maximum of 16 population doublings (Figure 1).
Figure 1. In-vitro proliferation of cells derived from human beta-cells.
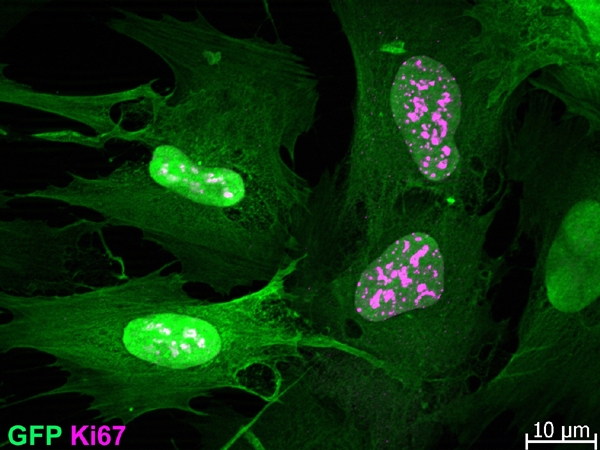
Isolated islets were dissociated and infected with an insulin promoter-Cre recombinase lentivirus and a reporter lentivirus containing a GFP gene downstream of a loxP-flanked stop fragment. Cell-specific removal of the stop fragment in beta-cells activated GFP expression. Dividing label-positive cells stained for Ki67 are shown at passage 10 (following about 70 days in culture).
We observed no age-related differences in the proliferation capacity of the cells within this age range. The approximate doubling time of dedifferentiated beta-cells was rather long, about 7 days. The doubling time of the GFP-negative cells present in the same culture was similar to that of the labeled cells, as evidenced by the fact that the fraction of cells derived from beta-cells remained stable throughout the culture period (about 40%). The proliferation of cells derived from beta-cells depended on soluble factor(s) secreted by the non-beta-cells present in the islet cell culture, as demonstrated by the finding that FACS-sorted GFP-positive cells proliferated poorly unless their culture medium was supplemented with medium conditioned by GFP-negative cells [23].
The latter finding is supported by the work of Parnaud et al., which showed that beta-cells separated from isolated human islets by labeling with Newport Green failed to proliferate [24]. This was in striking contrast to the massive replication capacity of similarly purified adult rat beta-cells cultured under the same conditions [24]. It should be noted, however, that Parnaud et al. sorted primary insulin-positive cells, while we sorted dedifferentiated beta-cells following proliferation in cell culture, making it difficult to compare the two studies. In addition, in the absence of a marker for dedifferentiated beta-cells, Parnaud et al. were unable to exclude the possibility that some of the insulin-negative cells that proliferated in their cultures were derived from beta-cells.
Analysis of mouse islet cells by our virus lineage-tracing method revealed a much lower proliferation of cells derived from mouse beta-cells, compared with human beta-cells, under similar culture conditions [23]. These findings confirmed the results obtained with transgenic mouse islets, and suggested that the culture conditions were more favorable for human, compared with mouse, beta-cell expansion.
Overall, these studies demonstrate a remarkable species difference with respect to beta-cell proliferation in vitro: while rat beta-cells replicate in the absence of support from other pancreatic cells, human dedifferentiated beta-cells seem to require soluble factor(s) released by non-beta-cells, and mouse dedifferentiated beta-cells cannot replicate even in the mixed culture. These findings indicate that caution is required when extrapolating conclusions obtained from studies with rodent beta-cells to humans.
Redifferentiation of cells expanded from human beta-cells
Our findings show that cells derived from adult human beta-cells can be expanded in tissue culture in sufficient numbers to provide all the cell needs for human beta-cell replacement at the current availability of human islet donors, provided that their lost phenotype can be restored. Despite studies on cell cycle regulation in beta-cells (see references 25 and 26 for reviews), our understanding of the relations between beta-cell replication and expression of differentiated functions is rather limited. It is unknown whether differentiated beta-cells in the human pancreas must undergo temporary dedifferentiation before re-entering the cell cycle, or whether those beta-cells involved in replication are not fully differentiated. Nevertheless, it appears that induction of significant replication in vitro requires cell delamination out of the normal epithelial structure, a process that results in dedifferentiation. Thus, even if beta-cell replication in vivo does not involve dedifferentiation, the latter seems inevitable for significant in-vitro proliferation. If the beta-cell phenotype can not be preserved during proliferation in culture, ways must be found of inducing ex-vivo redifferentiation of the expanded cells, or of restoring their function by cell re-introduction into the in-vivo environment upon transplantation.
A number of recent reports described ways of achieving varying levels of redifferentiation of cells expanded ex vivo from adult human islets. Gershengorn et al. [27] presented a protocol for expansion of adult human islet cells up to 1012-fold. In this procedure, intact islets were cultured, and cells migrating out of the islets were propagated in a simple serum-containing medium. The cell doubling time was about 60 h, and their morphology resembled that of fibroblasts. The cells were devoid of beta-cell markers. Instead they expressed a number of mesenchymal markers, such as vimentin. Serum withdrawal from the expanded cells resulted in a low level of insulin expression. This work postulated that beta-cells underwent epithelium-to-mesenchyme transition (EMT) upon entering into the cell cycle, and a reverse mesenchyme-to-epithelium transition (MET) following serum withdrawal. However, there was no direct evidence for these transitions.
Subsequent attempts to reproduce cell redifferentiation in vitro in serum-free medium varied: while one group reported successful induction of insulin mRNA [28], another group failed to reproduce this result [29]. A study employing somewhat different culture conditions also reported a redifferentiating effect of serum withdrawal in cells expanded from adult human islets. However the effect was inconsistent among cells from different donors [30].
In contrast to these expansion protocols, which cultured intact islets, our group used a protocol involving dissociation of the primary islets into a single-cell suspension [31]. This resulted in rapid cell dedifferentiation within the first week in culture, followed by induction of replication in most cells. As described above, these cells differed in some of their properties from those expanded by the Gershengorn group. Their doubling time was longer (about 7 days) and their overall expansion rate was lower, only <105. These cells were shown by lineage tracing to consist on average of about 40% cells derived from beta-cells [23]. Serum-free medium failed to induce redifferentiation in these cultures [31]. However, treatment with the EGF-family member betacellulin restored varying levels of insulin production and secretion in cells from different donors, ranging from normal insulin content to no effect [31]. We failed to identify any obvious cause of these variations, such as donor age, sex, or health status, nor the quality of islet isolation or shipment. Our preliminary results showed that the expanded cells were capable of differentiation in vivo into insulin-producing cells (unpublished results).
Subsequent work from the Gershengorn group abandoned the EMT hypothesis and suggested instead that the expanded cells, termed human Islet Progenitor Cells (hIPC), were derived from mesenchymal stem cells (MSC) normally present in the islets. hIPCs expanded in vitro were shown to express MSC markers and differentiate in vitro into mesodermal cell types, such as adipocytes and osteocytes [32]. The presence of MSC in human islet preparations was supported by another group [28]. However, the presence of MSC in islets in vivo has not been confirmed, and their occurrence in islet cultures may result from contaminating duct tissue [33]. While their differentiation in vitro into insulin-producing cells was not efficient, hIPCs formed insulin-producing cells when transplanted under the renal capsule of immunodeficient mice [32]. The investigators postulated that these cells represented a specialized type of MSC with a capacity to differentiate into pancreatic endocrine cells.
This hypothesis was supported by the finding that the insulin gene in these cells, although not expressed, was organized in an open chromatin structure [34]. Preliminary results from studies in our laboratory using the lineage-tracing approach showed that cells expanded from beta-cells expressed a number of mesenchymal and MSC markers (unpublished results). These findings support the occurrence of EMT in cultured beta-cells but do not exclude the possibility that other cells in these cultures are generated by expansion of MSC found in the islet preparation.
Gao et al. [35] presented evidence for dedifferentiation of cultured human islet cells into CK19-positive cells with a duct-cell-like phenotype. These cells could be expanded in vitro, although their endocrine differentiation capacity was very limited. In the absence of a rigorous lineage-tracing study, it was hard to confirm that these cells originated from beta-cells. It is possible that these cells were derived from duct cells contaminating the islet preparations. Such cells have been shown to have a limited proliferation capacity in vitro and to differentiate efficiently into insulin-producing cells [36-38].
Signaling pathways involved in ex-vivo human beta-cell dedifferentiation and replication
An attractive approach to identifying molecular targets for redifferentiation of cells expanded from adult human beta-cells involves elucidation of the signaling pathways altered during the adaptation of these cells to growth in culture. In a recent study, we found that human beta-cell dedifferentiation and entrance into the cell cycle in vitro correlated with activation of the Notch pathway and dowregulation of the cell cycle inhibitor p57 [39]. Using lineage-labeled cells we showed that the Notch intracellular domain (NICD) and its downstream target HES1 appeared in the nuclei of cells derived from beta-cells which lost insulin expression. Inhibition of HES1 upregulation using small hairpin RNA (shRNA) resulted in higher levels of p57 in beta-cells, compared with cells treated with a nontarget shRNA, and a diminished beta-cell proliferation [39]. Moreover, inhibition of HES1 upregulation reduced beta-cell dedifferentiation, as manifested in higher levels of insulin and the beta-cell transcription factors PDX1 and NeuroD1, although it did not totally prevent cell dedifferentiation. These findings suggest that partial cell dedifferentiation is independent of HES1 activity and cell replication. However, induction of advanced dedifferentiation and cell replication requires HES1 upregulation (Figure 2).
Figure 2. Proposed model for beta-cell dedifferentiation, replication, and redifferentiation.
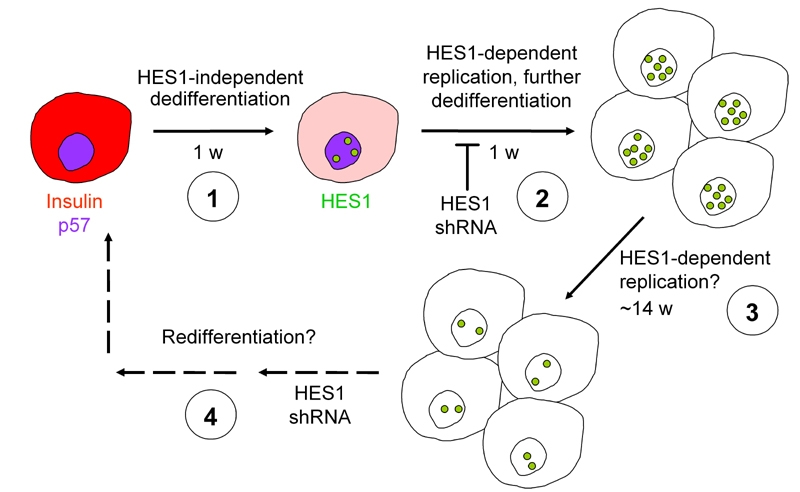
1. During the initial days in culture insulin expression declines, while HES1 expression is induced. 2. HES1 induction blocks p57 expression, induces beta cell replication, and causes further dedifferentiation. HES1 shRNA prevents these events. 3. HES1 levels decline; cell replication continues for about 14 additional weeks until cells senesce. 4. HES1 shRNA, along with other effectors, may be useful in induction of cell redifferentiation following expansion.
This interpretation is supported by the finding that the majority of decrease in insulin mRNA in cultured human beta-cells occurs during the first week in culture, preceding the peak in HES1 mRNA levels. It is therefore possible that loss of most of the insulin content is a precondition for beta-cell entrance into the cell cycle in vitro. The findings emphasize the role of components of the Notch pathway in the transition of quiescent beta-cells into a dedifferentiated, proliferative state in vitro, and demonstrate a negative correlation between replication and maintenance of differentiated function in cultured beta-cells. These findings suggest that significant beta-cell expansion inevitably involves dedifferentiation and will require the development of ways for cell redifferentiation following expansion. Components of the Notch pathway may represent molecular targets for induction of redifferentiation in the expanded cells.
In a recent publication, Ikonomou et al. [40] reported the involvement of activated beta-catenin signaling in hIPC proliferation in vitro. However, since the source of these cells has not been correlated to beta-cells, it is difficult to propose a role for beta-catenin in the replication of dedifferentiated beta-cells by extrapolating from these results.
Conclusion
While cells derived from adult human islet beta-cells can be significantly expanded in culture, an efficient method for in-vitro redifferentiation has not yet been identified. Nevertheless, the potential of these cells to redifferentiate into insulin-producing cells in vivo and reverse hyperglycemia is suggested by preliminary transplantation studies. It is possible that the dedifferentiated cells maintain sufficient epigenetic modifications that allow them to redifferentiate if provided with appropriate conditions. Thus, the search is on for an efficient in-vitro redifferentiation protocol. Alternatively, if it is eventually concluded that full redifferentiation of these cells can only be achieved in vivo, their therapeutic potential will have to be carefully evaluated against the risks of transplanting undifferentiated cells. This risk is expected to be far smaller than that involved in the transplantation of cells derived from embryonic stem cells, since the residual replicative potential of cells derived from human islets is very limited.
Acknowledgments
This work in my laboratory is supported by the Juvenile Diabetes Research Foundation, Israel Science Foundation, and the European Union BetaCellTherapy Consortium.
References
- 1.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta-cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 2.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 3.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 2000;49:1325–1333. doi: 10.2337/diabetes.49.8.1325. [DOI] [PubMed] [Google Scholar]
- 5.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 7.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117:2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cano DA, Rulifson IC, Heiser PW, Swigart LB, Pelengaris S, German M, Evan GI, Bluestone JA, Hebrok M. Regulated beta-cell regeneration in the adult mouse pancreas. Diabetes. 2008;57:958–966. doi: 10.2337/db07-0913. [DOI] [PubMed] [Google Scholar]
- 10.Brennand K, Huangfu D, Melton D. All beta cells contribute equally to islet growth and maintenance. PLoS Biol. 2007;5:1520–1529. doi: 10.1371/journal.pbio.0050163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 12.Bonner-Weir S, Inada A, Yatoh S, Li WC, Aye T, Toschi E, Sharma A. Transdifferentiation of pancreatic ductal cells to endocrine beta-cells. Biochem Soc Trans. 2008;36:353–356. doi: 10.1042/BST0360353. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen JH, Brunstedt J, Andersson A, Frimodt-Møller C. Preservation of beta cell function in adult human pancreatic islets for several months in vitro. Diabetologia. 1979;16:97–100. doi: 10.1007/BF01225457. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen JH, Galsgaard ED, Møldrup A, Friedrichsen BN, Billestrup N, Hansen JA, Lee YC, Carlsson C. Regulation of beta-cell mass by hormones and growth factors. Diabetes. 2001;50(Suppl 1):S25–S29. doi: 10.2337/diabetes.50.2007.s25. [DOI] [PubMed] [Google Scholar]
- 15.Hayek A, Beattie GM, Cirulli V, Lopez AD, Ricordi C, Rubin JS. Growth factor/matrix-induced proliferation of human adult beta-cells. Diabetes. 1995;44:1458–1460. doi: 10.2337/diab.44.12.1458. [DOI] [PubMed] [Google Scholar]
- 16.Beattie GM, Cirulli V, Lopez AD, Hayek A. Ex vivo expansion of human pancreatic endocrine cells. J Clin Endocrinol Metab. 1997;82:1852–1856. doi: 10.1210/jcem.82.6.4009. [DOI] [PubMed] [Google Scholar]
- 17.Beattie GM, Itkin-Ansari P, Cirulli V, Leibowitz G, Lopez AD, Bossie S, Mally MI, Levine F, Hayek A. Sustained proliferation of PDX-1+ cells derived from human islets. Diabetes. 1999;48:1013–1019. doi: 10.2337/diabetes.48.5.1013. [DOI] [PubMed] [Google Scholar]
- 18.Beattie GM, Montgomery AM, Lopez AD, Hao E, Perez B, Just ML, Lakey JR, Hart ME, Hayek A. A novel approach to increase human islet cell mass while preserving beta-cell function. Diabetes. 2002;51:3435–3439. doi: 10.2337/diabetes.51.12.3435. [DOI] [PubMed] [Google Scholar]
- 19.Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes. 2007;56:1299–1304. doi: 10.2337/db06-1654. [DOI] [PubMed] [Google Scholar]
- 20.Chase LG, Ulloa-Montoya F, Kidder BL, Verfaillie CM. Islet-derived fibroblast-like cells are not derived via epithelial-mesenchymal transition from Pdx-1 or insulin-positive cells. Diabetes. 2007;56:3–7. doi: 10.2337/db06-1165. [DOI] [PubMed] [Google Scholar]
- 21.Atouf F, Park CH, Pechhold K, Ta M, Choi Y, Lumelsky NL. No evidence for mouse pancreatic beta-cell epithelial-mesenchymal transition in vitro. Diabetes. 2007;56:699–702. doi: 10.2337/db06-1446. [DOI] [PubMed] [Google Scholar]
- 22.Morton RA, Geras-Raaka E, Wilson LM, Raaka BM, Gershengorn MC. Endocrine precursor cells from mouse islets are not generated by epithelial-to-mesenchymal transition of mature beta cells. Mol Cell Endocrinol. 2007;270:87–93. doi: 10.1016/j.mce.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russ HA, Bar Y, Ravassard P, Efrat S. In vitro proliferation of cells derived from adult human beta cells revealed by cell-lineage tracing. Diabetes. 2008;57:1575–1583. doi: 10.2337/db07-1283. [DOI] [PubMed] [Google Scholar]
- 24.Parnaud G, Bosco D, Berney T, Pattou F, Kerr-Conte J, Donath MY, Bruun C, Mandrup-Poulsen T, Billestrup N, Halban PA. Proliferation of sorted human and rat beta cells. Diabetologia. 2008;51:91–100. doi: 10.1007/s00125-007-0855-1. [DOI] [PubMed] [Google Scholar]
- 25.Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic beta-cell proliferation. Annu Rev Cell Dev Biol. 2006;22:311–338. doi: 10.1146/annurev.cellbio.22.010305.104425. [DOI] [PubMed] [Google Scholar]
- 26.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocaña A, Vasavada R, Stewart AF. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006;27:356–370. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- 27.Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–2264. doi: 10.1126/science.1101968. [DOI] [PubMed] [Google Scholar]
- 28.Gallo R, Gambelli F, Gava B, Sasdelli F, Tellone V, Masini M, Marchetti P, Dotta F, Sorrentino V. Generation and expansion of multipotent mesenchymal progenitor cells from cultured human pancreatic islets. Cell Death Differ. 2007;14:1860–1871. doi: 10.1038/sj.cdd.4402199. [DOI] [PubMed] [Google Scholar]
- 29.Kayali AG, Flores LE, Lopez AD, Kutlu B, Baetge E, Kitamura R, Hao E, Beattie GM, Hayek A. Limited capacity of human adult islets expanded in vitro to redifferentiate into insulin-producing beta-cells. Diabetes. 2007;56:703–708. doi: 10.2337/db06-1545. [DOI] [PubMed] [Google Scholar]
- 30.Lechner A, Nolan AL, Blacken RA, Habener JF. Redifferentiation of insulin-secreting cells after in vitro expansion of adult human pancreatic islet tissue. Biochem Biophys Res Commun. 2006;327:581–588. doi: 10.1016/j.bbrc.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 31.Ouziel-Yahalom L, Zalzman M, Anker-Kitai L, Knoller S, Bar Y, Glandt M, Herold K, Efrat S. Expansion and redifferentiation of adult human pancreatic islet cells. Biochem Biophys Res Commun. 2006;341:291–298. doi: 10.1016/j.bbrc.2005.12.187. [DOI] [PubMed] [Google Scholar]
- 32.Davani B, Ikonomou L, Raaka BM, Geras-Raaka E, Morton RA, Marcus-Samuels B, Gershengorn MC. Human islet-derived precursor cells are mesenchymal stromal cells that differentiate and mature to hormone-expressing cells in vivo. Stem Cells. 2007;25:3215–3222. doi: 10.1634/stemcells.2007-0323. [DOI] [PubMed] [Google Scholar]
- 33.Seeberger KL, Dufour JM, Shapiro AM, Lakey JR, Rajotte RV, Korbutt GS. Expansion of mesenchymal stem cells from human pancreatic ductal epithelium. Lab Invest. 2006;86:141–153. doi: 10.1038/labinvest.3700377. [DOI] [PubMed] [Google Scholar]
- 34.Mutskov V, Raaka BM, Felsenfeld G, Gershengorn MC. The human insulin gene displays transcriptionally active epigenetic marks in islet-derived mesenchymal precursor cells in the absence of insulin expression. Stem Cells. 2007;25:3223–3233. doi: 10.1634/stemcells.2007-0325. [DOI] [PubMed] [Google Scholar]
- 35.Gao R, Ustinov J, Korsgren O, Otonkoski T. In vitro neogenesis of human islets reflects the plasticity of differentiated human pancreatic cells. Diabetologia. 2005;48:2296–2304. doi: 10.1007/s00125-005-1935-8. [DOI] [PubMed] [Google Scholar]
- 36.Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A. 2000;97:7999–8004. doi: 10.1073/pnas.97.14.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yatoh S, Dodge R, Akashi T, Omer A, Sharma A, Weir GC, Bonner-Weir S. Differentiation of affinity-purified human pancreatic duct cells to beta-cells. Diabetes. 2007;56(7):1802–1809. doi: 10.2337/db06-1670. [DOI] [PubMed] [Google Scholar]
- 38.Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet beta-cells from pancreatic duct cells and an increase in functional beta-cell mass. J Clin Endocrinol Metab. 2005;90:3401–3409. doi: 10.1210/jc.2004-0761. [DOI] [PubMed] [Google Scholar]
- 39.Bar Y, Russ HA, Knoller S, Ouziel-Yahalom L, Efrat S. HES1 is involved in adaptation of adult human beta cells to proliferation in vitro. Diabetes. 2008 doi: 10.2337/db07-1323. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikonomou L, Geras-Raaka E, Raaka BM, Gershengorn MC. Beta-catenin signalling in mesenchymal islet-derived precursor cells. Cell Prolif. 2008;41:474–491. doi: 10.1111/j.1365-2184.2008.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]