

Abstract
Factor VIII consists of a heavy chain (A1A2B domains) and light chain (A3C1C2 domains), whereas the contiguous A1A2 domains are separate subunits in the cofactor, factor VIIIa. The intrinsic instability of the cofactor results from weak affinity interactions of the A2 subunit within factor VIIIa. The charged residues Glu272, Asp519, Glu665, and Glu1984 appear buried at the interface of the A2 domain with either the A1 or A3 domain, and thus may impact protein stability. To determine the effects of these residues on procofactor/cofactor stability, these residues were individually replaced with either Ala or Val, and stable BHK cell lines expressing the B-domainless proteins were prepared. Specific activity and thrombin generation parameters for 7 of the 8 variants were more than 80% the wild-type value. Factor VIII activity at 52°C to 60°C and the decay of factor VIIIa activity after thrombin activation were monitored. Six of the 7 variants showing wild-type-like activity demonstrated enhanced stability, with the Glu1984Val variant showing a 2-fold increase in thermostability and an approximately 4- to 8-fold increase in stability of factor VIIIa. These results indicate that replacement of buried charged residues is an effective alternative to covalent modification in increasing factor VIII (VIIIa) stability.
Introduction
Factor VIII, a plasma protein that is decreased or defective in patients with hemophilia A, circulates as a noncovalent, metal ion-dependent heterodimer. This procofactor form of the protein consists of a heavy chain (HC) composed of A1(a1)A2(a2)B domains and a light chain (LC) composed of (a3)A3C1C2 domains, with the lower case “a” representing short (∼ 30-40 residue) segments rich in acidic residues (for review, see Fay1). Factor VIII is activated by proteolytic cleavages at the a1A2, a2B, and a3A3 junctions catalyzed by thrombin or factor Xa. The product of this reaction, factor VIIIa, is a heterotrimer composed of subunits designated A1, A2, and A3C1C2 that functions as a cofactor for the serine protease factor IXa in the membrane-dependent conversion of zymogen factor X to the serine protease, factor Xa (for review, see Fay1)
Reconstitution studies have shown that the factor VIII heterodimeric structure is supported by both electrostatic and hydrophobic interactions,2,3 and the interchain affinity is further strengthened by factor VIII binding von Willebrand factor.2,4 Metal ions also contribute to the interchain affinity and activity parameters.5 Calcium is required to yield the active factor VIII conformation. Mutagenesis studies mapped a calcium-binding site to a segment rich in acidic residues within the A1 domain (residues 110-126) and identified specific residues within this region prominent in the coordination of the ion.6 A recent intermediate resolution X-ray structure7 confirmed this calcium-binding site as well as suggested a second potential site within the A2 domain. This structure also showed occupancy of the 2 type 1 copper ion sites within the A1 and A3 domains. Earlier functional studies have shown that copper ions facilitate the association of HC and LC to form the heterodimer, increasing the interchain affinity by several-fold at physiologic pH.8–10
The instability of factor VIIIa results from weak electrostatic interactions between the A2 subunit and the A1/A3C1C2 dimer8,11 and leads to dampening of factor Xase activity.12,13 Limited information is available regarding the association of the A2 subunit in factor VIIIa, and residues in both the A1 and A3 domains appear to make contributions to the retention of this subunit. Several factor VIII point mutations have been shown to facilitate the dissociation of A2 relative to wild type (WT), and these residues localize to either the A1-A2 domain interface14,15 or the A2-A3 domain interface.16 These factor VIII variants demonstrate a characteristic 1-stage/2-stage assay discrepancy,17,18 with significant reductions in activity values determined by the latter assay as a result of increased rates of A2 subunit dissociation.
Examination of the ceruloplasmin-based homology model for the A domains of factor VIII19 suggests an extended interface between the A2 domain and the A1 and A3 domains with multiple potential contacts contributing to binding interactions. In a recent study, we examined hydrogen-bonding interactions at this interface after mutation of selected charged/polar residues spatially separated by less than 2.8 Å.20 Approximately half of the residues examined showed loss of function as judged by increased rates of factor VIII decay at 55°C and/or rates for factor VIIIa decay relative to WT, suggesting that multiple residues at the A1A2 and A2A3 domain interfaces contribute to the stabilization of factor VIII. However, this model also predicts that several charged residues at the interface do not contribute to hydrogen-bonding interactions based on distance separations of more than 2.8 Å. As such, these residues may be destabilizing to factor VIII structure and/or may facilitate the dissociation of the A2 subunit after activation of the factor VIII procofactor. In this report, we individually mutated these charged residues to hydrophobic ones (Ala or Val) with the aim of increasing the buried hydrophobic area and reducing the buried hydrophilic area to enhance interdomain binding affinity.21 Stability parameters were assessed after the activity of the factor VIII variants at elevated temperature and time courses for the decay of factor VIIIa activity resulting from A2 subunit dissociation. Results from these studies demonstrated that a number of mutations yielded increased stability parameters consistent with the elimination of destabilizing forces probably the result of buried charge at the A2 domain interface.
Methods
Reagents
Recombinant factor VIII (Kogenate) was a generous gift from Dr Lisa Regan of Bayer (Berkeley, CA). Phospholipid vesicles containing 20% phosphatidylcholine, 40% phosphatidylethanolamine, and 40% phosphatidylserine were prepared using octylglucoside as described previously.22 The reagents α-thrombin, factor VIIa, factor IXaβ, factor X, and factor Xa (Enzyme Research Laboratories, South Bend, IN), hirudin, and phospholipids (DiaPharma, West Chester, OH), the chromogenic Xa substrate, Pefachrome Xa (Pefa-5523, CH3OCO-D-CHA-Gly-Arg-pNA·AcOH; Centerchem, Norwalk, CT), recombinant human tissue factor (rTF), Innovin (Dade Behring, Deerfield, IL), fluorogenic substrate, Z-Gly-Gly-Arg-AMC (Calbiochem, San Diego, CA), and thrombin calibrator (Diagnostica Stago, Parsippany, NJ) were purchased from the indicated vendors.
Construction, expression, and purification of WT and variant factor VIII
Ala and Val mutants at charged residues (Glu272, Asp519, Glu665, and Glu1984), and WT factor VIII forms were individually constructed as a B-domainless factor VIII, lacking residues Gln744-Ser1637 in the B-domain.23 The cloning and expression constructs were generous gifts from Dr Pete Lollar and John Healey. Recombinant WT and variant factor VIII forms were stably expressed in BHK cells and purified as described previously.6 After transfection, there were no significant differences in the amounts of factor VIII secretion among the variants. Protein yields for the variants ranged from more than 10 to approximately 100 μg from two 750-cm2 culture flasks, with purity from approximately 85% to more than 95% as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The primary contaminant in the factor VIII preparations was albumin and at the concentrations present in the factor VIII showed no effect on stability of activity parameters. Factor VIII concentration was measured using an enzyme-linked immunosorbent assay (ELISA), and factor VIII activity was determined by a one-stage clotting assay and a 2-stage chromogenic factor Xa generation assay as described below in the so-named sections.
SDS-PAGE and Western blotting
Factor VIII proteins (0.77 μg for gel staining and 0.34 μg for Western blot) were electrophoresed on 8% polyacrylamide gel at constant voltage (100 V). Gels were stained with Gelcode Blue (Thermo Electron Corporation, Waltham, MA) or transferred to a polyvinylidene fluoride membrane and probed with biotinylated anti-A2 antibody (R8B12, Green Mountain Antibodies, Burlington, VT) followed by incubation with peroxidase-conjugated streptoavidin (Calbiochem). The chemifluorescence substrate (ECF substrate; GE Healthcare, Little Chalfont, United Kingdom) was reacted and the fluorescence signal scanned using a phosphoimager (Storm 860; GE Healthcare). The density of single chain factor VIII form (170 kDa) and heavy chain (HC, 90 kDa) were quantified using ImageQuant software (GE Healthcare), and the amount ratios were calculated.
ELISA
A sandwich ELISA was performed to measure the concentration of factor VIII proteins as previously described24 using purified commercial recombinant factor VIII (Kogenate, Bayer) as a standard. Factor VIII capture used the anti-C2 antibody (ESH-8; American Diagnostica, Stamford, CT), and a biotinylated R8B12 antibody was used for factor VIII detection.
One-stage clotting assay
One-stage clotting assays were performed using substrate plasma chemically depleted of factor VIII25 and assayed using a Diagnostica Stago clotting instrument. Plasma was incubated with activated partial thromboplastin time reagent (bioMérieux, Durham, NC) for 6 minutes at 37°C after which a dilution of factor VIII was added to the cuvette. After 1 minute, the mixture was recalcified, and clotting time was determined and compared with a pooled normal plasma standard.
Two-stage chromogenic factor Xa generation assay
The rate of conversion of factor X to factor Xa was monitored in a purified system26 according to methods previously described.5,27 Factor VIII (1 nM) in buffer containing 20 mM N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES), pH 7.2, 0.1 M NaCl, 0.01% Tween 20, 0.01% bovine serum albumin (BSA), 5 mM CaCl2, and 10 μM phosphatidylserine/phosphatidylcholine/phosphatidylethanolamine (PSPCPE) vesicles (buffer A) was activated with 20 nM α-thrombin for 1 minute. The reaction was stopped by adding hirudin (10 U/mL), and the resulting factor VIIIa was reacted with factor IXa (40 nM) for 1 minute. Factor X (300 nM) was added to initiate reactions, which were quenched after 1 minute by the addition of 50 mM ethylenediaminetetraacetic acid. Factor Xa generated was determined after reaction with the chromogenic substrate Pefachrome Xa (0.46 mM final concentration). All reactions were run at 23°C.
Thrombin generation assay
The amount of thrombin generated in plasma was measured by calibrated automated thrombography.28,29 In a 96-well plate, 80 μL factor VIII–deficient plasma (< 1% residual activity, platelet-poor) from a patient with severe hemophilia A lacking factor VIII inhibitor (George King Bio-Medical, Overland Park, KS) was mixed with factor VIII samples (20 μL; 6 nM) in HEPES-BSA buffer (20 mM HEPES, pH 7.35, 0.15 M NaCl, 6% BSA) containing 3 pM rTF (the concentration of rTF stock was determined by factor Xa generation assay using known concentrations of factor VIIa), PSPCPE vesicles (24 μM), or 20 μL thrombin calibrator (630 nM), and reactions were immediately started by mixing with 20 μL fluorogenic substrate (2.5 mM, Z-Gly-Gly-Arg-AMC) in HEPES-BSA buffer, including 0.1 M CaCl2. All reagents were prewarmed at 37°C. Final concentrations of reagents were 1 nM (factor VIII), 0.5 pM (rTF), 4 μM (PSPCPE vesicles), 433 μM (fluorogenic substrate), 13.3 mM CalCl2, and 105 nM (thrombin calibrator). The development of a fluorescent signal at 37°C was monitored at 8-second intervals using a microplate fluorometer (SpectraMax Gemini; MDS Analytical Technologies, Sunnyvale, CA) with a 355 nm (excitation)/460 nm (emission) filter set. Fluorescent signals were corrected by the reference signal from the thrombin calibrator samples,28 and actual thrombin generation (in nM) was calculated as previously described.29
Factor VIII activity at elevated temperature
WT factor VIII or factor VIII variants (4 nM) in buffer A were incubated at 52°C to 60°C. Aliquots were removed at the indicated times, and residual factor VIII activity was determined using a 2-stage chromogenic factor Xa generation assay.
Factor VIIIa activity time course
WT and mutant factor VIII (4 nM) in buffer A containing 10 μM PSPCPE vesicles were activated by 20 nM of thrombin for 1 minute at 23°C. Reactions were immediately quenched by hirudin (10 U/mL), aliquots removed at the indicated times, and activity determined using the factor Xa generation assay after addition of factor IXa (40 nM) and factor X (300 nM). For decay measurements run in the presence of factor IXa, factor IXa (40 nM) was added to reactions before thrombin addition.
Factor VIII stability in plasma
WT or variant factor VIII (1 nM) was added to factor VIII–deficient plasma (< 1% residual activity) from a patient with severe hemophilia A lacking factor VIII inhibitor (George King Bio-Medical). Plasma was supplemented with 0.02% NaN3 to prevent the growth of microorganisms, and samples were incubated at 37°C. Aliquots were removed at the indicated times, and residual activity was determined by a one-stage clotting assay.
Data analysis
Factor VIIIa activity values as a function of time were fitted to a single exponential decay curve by nonlinear least squares regression using the equation,
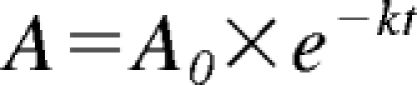 |
where A is residual factor VIIIa activity (nM/min/nM factor VIII), A0 is the initial activity, k is the apparent rate constant, and t is the time (minutes) of reaction of either factor VIII at elevated temperature (for factor VIII decay experiments) or after thrombin activation was quenched (for factor VIIIa decay measurements). Nonlinear least-squares regression analysis was performed using Kaleidagraph (Abelbeck/Synergy, Reading, PA). Comparison of average values was performed by the Student t test. The factor VIII A domain-modeled structure19 was analyzed using Swiss PDB Viewer to identify charged residues that were located at the A2 domain interface and showed little potential for hydrogen bonding interactions based on a threshold of more than 2.8 Å separating the polar atoms of the complementary domains.30
Results
Identification of target residues and generation of point mutants
Using the ceruloplasmin-based homology model19 for the A domains of factor VIII, we identified 4 charged residues, Glu272, Asp519, Glu665, and Glu1984, that appeared to be buried at the interface of the A2 domain with either the A1 domain (Glu272 and Asp519) or the A3 domain (Glu665, and Glu1984), which in addition, did not appear to contribute to H-bonding interactions based on spatial separations of more than 2.8 Å with potential bonding neighbors. These residues were mutated to either Ala or Val to eliminate charge as well as provide for potential hydrophobic interactions with similar side chains from other buried residues. Factor VIII variants were prepared as B-domainless factor VIII in stable-expressing BHK cell lines.
Factor VIII is expressed as a mixture of single chain and heterodimer forms (Figure 1). Protein purity ranged from approximately 85% to more than 95% as judged by SDS-PAGE (Figure 1A). Western blotting using an anti-A2 domain antibody was used to quantitate the stoichiometry of the single chain and heterodimer forms (Figure 1B). This value was near unity for WT and was somewhat lower and variable for the factor VIII variants.
Figure 1.

SDS-PAGE and Western blot analysis of factor VIII mutants and WT factor VIII. (A) Purified WT and mutant factor VIII proteins (0.77 μg) after SDS-PAGE on 8% polyacrylamide gels were visualized by GelCode. (B) Purified WT and mutant factor VIII proteins (0.34 μg) were electrophoresed on 8% polyacrylamide gels, transferred to PVDF membranes, and probed by biotinylated R8B12 antibody. Bands were visualized by chemifluorescence. WT (lane 1), Glu272Ala (lane 2), Glu272Val (lane 3), Asp519Ala (lane 4), Asp519Val (lane 5), Glu665Ala (lane 6), Glu665Val (lane 7), Glu1984Ala (lane 8), and Glu1984Val (lane 9). MW indicates molecular weight marker; sFVIII, single chain form factor VIII; HC, heavy chain; LC, light chain. An apparent stoichiometry ratio of single chain form to heterodimer of WT and mutant factor VIII forms were 0.96 (WT), 0.64 (Glu272Ala), 0.92 (Glu272Val), 0.74 (Asp519Ala), 0.8 (Asp519Val), 0.64 (Glu665Ala), 0.63 (Glu665Val), 0.91 (Glue1984Ala), and 0.5 (Glu1984Val).
Purified proteins were assessed for specific activity using both 1- and 2-stage assays (Figure 2A) and thrombin generation parameters (Figure 2B-D). All but the Glu272Ala variant yielded specific activity values that were at least 80% that of WT, suggesting the remaining mutations had little if any effect on factor VIII cofactor function. Thrombin generation performed at low rTF concentration (0.5 pM) and a physiologic concentration (1 nM) factor VIII yielded results that paralleled the specific activity values. Parameter values shown in Figure 2D indicated the peak value and endogenous thrombin potential (ETP) for the Glu272Ala were reduced compared with WT, whereas all other parameter values for the remaining variants ranged from more than 80% to 110% the WT value.
Figure 2.

Specific activity of factor VIII mutants relative to WT factor VIII and thrombin generation assays. (A) Specific Activity. Activity values were determined using a 1-stage clotting assay ( ) and 2-stage chromogenic factor Xa generation assay (■). (B,C) Thrombogram of factor VIII proteins. WT (
) and 2-stage chromogenic factor Xa generation assay (■). (B,C) Thrombogram of factor VIII proteins. WT ( ), Glu272Ala (□), Glu272Val (■), Asp519Ala (○), Asp519Val (●), Glu665Ala (△), Glu665Val (▲), Glu1984Ala (◇), and Glu1984Val (♦). (D) Parameter values obtained from thrombin generation assays. Thrombograms show the average values of triplicated samples. The parameter values were expressed as values (%) relative to WT. The actual values for WT were 7.5 plus or minus 0.5 minutes (lag time), 13.7 plus or minus 0.3 minutes (peak time), 157.3 plus or minus 14.7 nM (peak value), and 979.8 plus or minus 37.9 nM/min (ETP). Lag time (□), peak time (), peak value (■), and ETP (▧). Error bars represent SD values averaged from 3 separate determinations.
), Glu272Ala (□), Glu272Val (■), Asp519Ala (○), Asp519Val (●), Glu665Ala (△), Glu665Val (▲), Glu1984Ala (◇), and Glu1984Val (♦). (D) Parameter values obtained from thrombin generation assays. Thrombograms show the average values of triplicated samples. The parameter values were expressed as values (%) relative to WT. The actual values for WT were 7.5 plus or minus 0.5 minutes (lag time), 13.7 plus or minus 0.3 minutes (peak time), 157.3 plus or minus 14.7 nM (peak value), and 979.8 plus or minus 37.9 nM/min (ETP). Lag time (□), peak time (), peak value (■), and ETP (▧). Error bars represent SD values averaged from 3 separate determinations.
Thermostability of factor VIII variants
The purified factor VIII mutant proteins were assessed for stability at elevated temperatures as judged by rates of activity loss. Factor VIII (4 nM) was incubated at 52°C to 60°C and at the indicated times an aliquot was removed, cooled to room temperature, reacted with thrombin, and residual cofactor activity was measured using a factor Xa generation assay. Results shown in Figure 3A illustrate the time course for activity decay of the factor VIII WT and variants at 55°C. This temperature was chosen based on an earlier study3 showing near-complete activity loss within 1 hour for WT factor VIII. The WT protein lost 50% activity in approximately 15 minutes. We observed that the Glu272Ala and −Val variants displayed reduced stability as judged by somewhat faster activity decay, and this property may be related to the reduced specific activities observed for mutations at this site. On the other hand, Ala and Val replacements for Asp519, Glu665, and Glu1984 all showed improved stability at the elevated temperature with variants possessing mutations at the 2 former sites retaining 50% activity through approximately 20 to 25 minutes, whereas mutations at the latter site yielded variants that maintained this activity level through more than 30 minutes. Comparison of the decay rate values from the fitted curves (Table 1) indicated that factor VIII thermal stability was improved approximately 2-fold for the Glu1984 variants relative to WT with mutation to Val appearing somewhat preferred to Ala.
Figure 3.

Activity decay of WT and mutant factor VIII. Factor VIII (4 nM) was incubated at various temperatures (52°C-60°C); and at the indicated times, aliquots were removed and assayed for activity by factor Xa generation assays. Data were fitted by nonlinear least squares regression, and decay rates were obtained. Each point represents the value averaged from 3 separate determinations. Results are shown for WT (, ×), Glu272Ala (□), Glu272Val (■), Asp519Ala (○), Asp519Val (●), Glu665Ala (△), Glu665Val (▲), Glu1984Ala (◇), Glu1984Val (♦), and full-length Kogenate factor VIII ( ). (A) Representative factor VIII decay curves after 55°C incubation. (B) Plots of factor VIII decay rate at various temperatures. (Inset) Magnified view of the decay results incubated at 52°C to 55°C.
). (A) Representative factor VIII decay curves after 55°C incubation. (B) Plots of factor VIII decay rate at various temperatures. (Inset) Magnified view of the decay results incubated at 52°C to 55°C.
Table 1.
Factor VIII and VIIIa decay rates
| Factor VIII decay |
Factor VIIIa decay |
|||
|---|---|---|---|---|
| Thermostability at 55°C, min−1 | Plasma stability, h−1 | Factor IXa absent, min−1 | Factor IXa present, min−1 | |
| WT | 0.0471 (1.00) | 0.0178 (1.00) | 0.0836 (1.00) | 0.0154 (1.00) |
| E272A | 0.0542 (1.15) | ND | 0.1638 (1.95) | 0.0163 (1.06) |
| E272V | 0.0602 (1.28) | ND | 0.2271 (2.72) | 0.0159 (1.03) |
| D519A | 0.0336 (0.71)* | 0.0066 (0.37)* | 0.0556 (0.66)* | 0.0063 (0.41)* |
| D519V | 0.0262 (0.56)* | 0.0184 (1.03) | 0.0642 (0.77)* | 0.0068 (0.44)* |
| E665A | 0.0359 (0.76)* | 0.0149 (0.84)† | 0.0520 (0.62)* | 0.0078 (0.51)* |
| E665V | 0.0309 (0.66)* | 0.0047 (0.26)* | 0.0160 (0.19)* | 0.0052 (0.34)* |
| E1984A | 0.0240 (0.51)* | 0.0080 (0.45)* | 0.0241 (0.29)* | 0.0027 (0.18)* |
| E1984V | 0.0211 (0.45)* | 0.0078 (0.44)* | 0.0217 (0.26)* | 0.0019 (0.13)* |
Values are means. SDs for rate decay values are estimated based upon least squares curve fitting and are within approximately 10% of mean values for thermostability and factor VIIIa decay measurements and within approximately 15% of mean values for the plasma stability measurements. Values in parentheses are relative to the WT value. A single letter code is used to designate the amino acid residues, E (Glu), D (Asp), A (Ala), and V (Val).
ND indicates not determined.
P < .001 compared with the rate of WT (Student t test).
P < .05 compared with the rate of WT (Student t test).
Results assessing a range of temperatures (Figure 3B) indicated that both the Ala and Val variants of Asp519, Glu665, and Glu1984 consistently showed reductions of decay rate up to 2-fold compared with WT at all temperatures tested. However, the presence of both single-chain and heterodimer forms in somewhat various ratios may impact these decay rate results should one form show greater stability. A control experiment using Kogenate factor VIII, which is essentially all in the heterodimer form,5 yielded decay rates that were approximately 2-fold that of WT (Figure 3B), consistent with the heterodimer form showing less stability to elevated temperature than single-chain factor VIII. Thus, the decay rates measured are apparent because of heterogeneity of single-chain and 2-chain content in the various factor VIII forms. However, given that all the variants possessed less relative single-chain factor VIII compared with the WT (Figure 1B), these data indicated that decay rate values for these variants underestimate the increase in stability between the mutants and WT.
Factor VIII stability in plasma at 37°C
To test the effects of the mutations on factor VIII stability under more native conditions, we incubated a near physiologic concentration of the proteins (1 nM) in (anticoagulated) factor VIII–deficient plasma from a hemophilia A patient free from factor VIII inhibitor activity at 37°C for up to 4 days. Residual factor VIII was assayed daily using a one-stage clotting assay. Activity of the WT factor VIII was reduced to approximately 50% after 2 days as was that of the Asp519Val variant, whereas the Glu665Ala variant showed a modest (∼ 15%) reduction in the rate of activity decay (Figure 4; Table 1). However, the activity values for the Asp519Ala, Glu665Val, and both Glu1984 variants were more than or equal to 50% the initial value at day 4. The results obtained from the plasma incubation, in large part, parallel those from the incubations performed at elevated temperature with the Glu665Val variant and the 2 Glu1984 variants demonstrating significant increases in stability under the 2 reaction conditions as judged by retention of function. Whereas both Asp519 variants showed improved stability at elevated temperature, only the Ala variant showed improvement in the plasma assay.
Figure 4.

Activity decay of factor VIII in plasma at 37°C. Factor VIII (1 nM) was incubated at 37°C in factor VIII–deficient plasma and at the indicated times aliquots were removed and assayed using the one-stage clotting assays. Results are shown for WT (, ×), Asp519Ala (○), Asp519Val (●), Glu665Ala (△), Glu665Val (▲), Glu1984Ala (◇), and Glu1984Val (♦). Data were fitted by nonlinear least squares regression, and each point represents the value averaged from 3 separate determinations.
Factor VIIIa decay rates
These results indicate that mutations consistent with replacing buried charged residues with hydrophobic residues in general yielded factor VIII protein showing enhanced stability. Because these mutations are at or near the interface of the A2 domain with A1 or A3, we predicted they could positively impact the lability of factor VIIIa by reducing rates for dissociation of the A2 subunit. We assessed rates of loss of factor VIIIa activity resulting from this mechanism under 2 conditions. In the first, the WT and factor VIII variants were activated with thrombin and at indicated times the remaining cofactor activity was determined after addition of factor IXa and factor X and monitoring rates of factor Xa generation. In the second method, the assay described herein was modified to include addition of factor IXa before factor VIII activation to allow for immediate formation of factor Xase. Incorporation of factor VIIIa in the factor Xase complex has been shown to partially stabilize cofactor activity by reducing its decay rate as much as 10-fold by a mechanism consistent with factor IXa tethering the A2 and A3C1C2 subunits with Xase.13
Results obtained in the absence and presence of added factor IXa are shown in Figure 5A and 5B, respectively. In the absence of factor IXa, WT factor VIIIa lost 50% of its activity in approximately 8 minutes (Figure 5A), whereas this level of activity persisted for approximately 40 minutes when factor IXa was included during factor VIII activation (Figure 5B). Decay rate values are shown in Table 1 and indicate a more than 5-fold stabilization of cofactor activity by formation of factor Xase. Evaluation of the variants revealed that both Glu272Ala and −Val forms possessed 2- and 3-fold increased rates of decay, respectively, in the absence of factor IXa compared with the WT control. These results suggested a weakened intersubunit affinity with either mutation, possibly the result of loss of a relatively weak affinity bonding interaction involving the acidic side chain. In the presence of factor IXa, decay rates for the 2 variants were essentially indistinguishable from that of WT, suggesting that inclusion of factor IXa eliminated any detrimental interaction generated by the mutations at this residue.
Figure 5.

Activity decay of WT and mutant factor VIIIa in the absence and presence of factor IXa. (A) Thrombin-activated factor VIIIa (4 nM) was incubated at 23°C, aliquots were taken at indicated time points, and activity was measured by factor Xa generation assay. (B) Activity decay of WT and mutant factor VIIIa in the presence of factor IXa. Factor VIII (4 nM) was activated with thrombin in the presence of 40 nM factor IXa, aliquots were taken at indicated time points, and activity was measured by factor Xa generation assay. Results are shown for WT (, ×), Glu272Ala (□), Glu272Val (■), Asp519Ala (○), Asp519Val (●), Glu665Ala (△), Glu665Val (▲), Glu1984Ala (◇), and Glu1984Val (♦). Data were fitted by nonlinear least squares regression, and each point represents the value averaged from 3 separate determinations.
Mutations at the other 3 sites (Asp519, Glu665, and Glu1984) all resulted in reductions in factor VIIIa decay rates with the degree of reduction variable depending on the specific residue changed and in one case, the replacement residue. Mutations at Asp519 yielded approximately 30% reductions in decay rates that were similar for both the Ala and Val variants when factor IXa was absent. Rates for activity decay of these variants were decreased more than 2-fold in the presence of factor IXa, suggesting a synergy of the mutations with the stabilizing effects of binding the enzyme. Whereas the Glu665Ala variant showed similar values to the 2 Asp519 variants, the Glu665Val variant showed 5-fold and 3-fold reductions in decay rates in the absence and presence of factor IXa, respectively, suggesting that replacement with the larger hydrophobic residue yielded a more favorable interaction with neighboring residues for A2 subunit retention. Finally, both Glu1984 variants showed approximately 4-fold reductions in factor VIIIa decay compared with WT in the absence of factor IXa, and 5- to 8-fold reductions when factor IXa was present. The significance of this enhanced stability is observed in Figure 5B, which shows more than 90% factor VIIIa activity remaining after 40 minutes in factor Xase composed of either Glu1984 variant. The similarity in responses with either Ala or Val at Glu1984 suggested that both residues were well tolerated with perhaps a slightly stronger intersubunit affinity achieved in the presence of Val. Overall, these results demonstrate significant enhancement in factor VIIIa stability resulting from improved A2 subunit retention after selective replacement of charged with hydrophobic residues.
Discussion
In the current study, we show that substitution of selected charged residues with hydrophobic ones at sites predicted to interface the A2 domain resulted in a variable, but general, increase in the stability of factor VIII. This stability was assessed after activity retention at elevated temperature as well as by reduction in the rate of A2 subunit dissociation in the cofactor. The rationale for this study was based, in large part, on results obtained in our recent analysis of 30 residues localized to the factor VIII A2 domain interface that, based on spatial separations of less than 2.8 Å, could potentially form hydrogen-bonding partners.20 In that study, charged/polar residues were mutated to Ala (or Phe for Tyr residues), recombinant proteins stably expressed and rates of loss of activity were measured using similar methods to those used in the present study. Fourteen of the 30 residues examined showed more than 2-fold increases in rates of factor VIII decay at 55°C and/or rates for factor VIIIa decay relative to WT, suggesting that multiple residues at the A1A2 and A2A3 domain interfaces contribute to the stabilization of factor VIII. Interestingly, 2 acidic residues that were examined in that earlier study, Asp302 and Glu287, yielded modest (< 2-fold) enhancement in stability in both the procofactor and active cofactor forms when mutated to Ala. These results suggested that these acidic side chains did not contribute to stabilizing hydrogen-bonding interactions but rather were somewhat detrimental to factor VIII structure as assessed by functional stability.
The present paper focuses on creating novel hydrophobic interactions to produce a gain of function. The 4 acidic residues examined in the present study are conserved in human, canine, porcine, mouse, rabbit, and bat factor VIII, whereas Glu665 is Ala and Glu1984 is Thr in rat factor VIII.31 We now show that 3 of these residues (Asp519, Glu665, and Glu1984), when replaced with Ala and/or Val, resulted in enhancements in protein stability. Only one acidic residue evaluated yielded results that were detrimental to activity when mutated. Mutation at Glu272 to Ala yielded a low specific activity factor VIII form with reduced thrombin generation parameters values, and both Ala and Val replacements possessed moderately decreased thermostability and 2- to 3-fold higher rates of A2 subunit dissociation in the cofactor form compared with WT. From these observations, we suggest that Glu272 may indeed participate in bonding interaction(s) with neighboring residues, and subsequent mutations at this site disrupt these interactions. This conclusion is consistent with examination of the hemophilia A database32,33 that lists Lys (charge reversal) or Gly (small side chain) at position 272 as a moderate/mild phenotype with reduced factor VIII antigen. The latter observation is consistent with the mutations yielding increased plasma instability. However, we noted no significant effects on levels of expression in cell culture after mutations at this site to Ala or Val. Conversely, no mutations at Asp519, Glu665, and Glu1984 are listed in the database. Proteins tend to fold so that the charged or polar moieties remain solvent exposed whereas hydrophobic groups are buried.34 Therefore, based on the observed gain-of-function mutations at Asp519, Glu665, and Glu1984 when these residues are replaced with hydrophobic ones, we speculate that these charged residues are buried at the A2 domain interfaces. Furthermore, these results suggest that these acidic residues do not contribute to electrostatic bonding interactions and are probably destabilizing to protein structure and/or subunit interactions.
We used mutagenesis using either Ala or Val to replace the charged acidic residues. Because both residues are hydrophobic, they would tend to stabilize other hydrophobic contacts at the interface. Furthermore, the Val side chain is larger than Ala, so comparison of effects on activity after replacement at a given site may offer some insights into residue packing and volume at that site. For example, we observed that replacement of Glu1984 with either residue yielded similar results, suggesting that both were well tolerated at that site, whereas Glu665Val showed a 3-fold reduced factor VIIIa decay rate compared with Glu665Ala, suggesting the larger volume side chain of Val was better tolerated in the putative hydrophobic binding pocket.
Overall, the results of this study as well as our prior evaluation of hydrogen bonding interactions20 contribute to our understanding of factor VIII A domain structure, which is limited to models derived from homology with a high resolution structure for ceruloplasmin19 and from a recent, intermediate resolution structure (3.75 Å) of human factor VIII.7 Whereas the latter structure does not allow for assignments of hydrogen bonding interactions (< 2.8 Å), the authors of that study indicate that the A domains of factor VIII can be superimposed onto those of ceruloplasmin with a high degree of accuracy. The ceruloplasmin model suggests that Asp302 and Glu287 could contribute hydrogen bonding interactions, whereas our recent stability studies20 suggest these acidic side chains are buried in a hydrophobic environment. Conversely, results from the present study suggest that Glu272 probably contributes a hydrogen bonding interaction at the A2 domain interface because loss of this charge reduces factor VIII (VIIIa) stability. The remaining 3 acidic residues we evaluated in this report appear to be buried at the interface as predicted by the model, in that no polar atom from a neighboring residue on a complementary domain appears to localize near the carboxylic groups of these residues (Figure 6). Rather, we note that these moieties appear to be proximal with hydrophobic groups. For example, the model predicts that the carboxyl oxygen of Asp519 and methyl carbon of Thr275 are separated by approximately 4.2 Å, the carboxyl oxygen of Glu665 and methyl carbon of Val1982 are separated by approximately 8.1 Å, and the carboxyl oxygen of Glu1984 and methyl carbon of Val663 are separated by approximately 6.2 Å (Figure 6).
Figure 6.

Residues surrounding Asp519, Glu272, Glu1984, and Glu665. Factor VIII surface models of indicated regions based on the A domain homology model19 are drawn by Swiss PDB viewer; A1 domain (residues 1-336), A2 domain (residues 373-711), and A3 domain, (residues 1690-2332). Hydrogen, carbon, oxygen, sulfur, and nitrogen are colored cyan, white, red, yellow, and blue, respectively. There are no possible hydrogen acceptor or donor from the residues near the residues Asp519 (A), Glu272 (B), Glu1 984 (C), and Glu665 (D). (Inset) Factor VIII surface model of individual domains are drawn by Swiss PDB viewer and colored as yellow (A1), transparent blue (A2), red (A3), green (C1), and gray (C2). White dots indicate the location of side chain atoms of the indicated residues (Asp519, Glu272, Glu1984, and Glu665) as shown in the panels A, B, C, and D, respectively.
Factor VIII variants demonstrating enhanced stability and reduced rates of cofactor activity decay represent positive attributes for a therapeutic preparation. The former property could allow for increased yields of active protein during its purification and formulation, resulting in overall higher specific activity values. These reagents may also possess a longer circulating half-life relative to WT, exclusive of various cellular clearance mechanisms. Two groups have reported on factor VIII variants where cofactor activity has been stabilized by reducing/eliminating the rate of A2 subunit dissociation. In both cases, mutations were used to covalently link the A2 domain to other regions of the molecule. In one case, an inactivation-resistant factor VIII was prepared by linking the A2 domain to a segment of B-domain contiguous with the A3C1C2 domains and lacking thrombin cleavage sites that would release either the A2 domain or B-domain fragment after procofactor activation.35 In a second case, selected residues in the A3 and A2 domains that were in close proximity were replaced with Cys residues so as to form disulfide bridges between the 2 domains such that A2 would remain covalent with A3 after thrombin activation.36,37 The latter mutants also demonstrated augmented activity in thrombin generation assays, although the reaction conditions used in these studies used a sub-physiologic (< 0.5 nM) concentration of factor VIII. Our results using a physiologic factor VIII level (1 nM) showed little difference between WT and the variants demonstrating higher stability, although the Glu272Ala yielded reduced thrombin generation parameters consistent with its lower specific activity. The failure to observe a significant difference with the high stability variants may reflect differences in reaction conditions and/or that these mutations do not covalently bridge the A2 domain and the rates for factor VIIIa decay are not sufficiently reduced.
Results in the current study demonstrate several-fold decreases in rates for cofactor inactivation after single point mutations to convert acidic residues to hydrophobic ones. In contrast to the mutations yielding covalent alterations, the mutations reported in the present study occur at interfaces where the altered residues are probably buried and not surface exposed, and do not alter covalent interactions within the protein. Furthermore, preliminary results show that activated protein C-catalyzed cleavages at Arg336 and Arg562 in the cofactor forms of the Glu1984Val and −Ala variants occur at similar rates as observed for the WT cofactor (J.D. and P.J.F., unpublished observations, February 11, 2008), suggesting that down-regulation of these higher stability variants would proceed via the protein C pathway. Work is in progress to assess combinations of the point mutations described in this report to determine whether additive or synergistic effects will result in further enhancements in factor VIII (VIIIa) stability.
Acknowledgments
The authors thank Lisa M. Regan of Bayer Corporation for the gifts of recombinant human factor VIII, Pete Lollar and John Healey for the factor VIII cloning and expression vectors, and Qian Zhou for excellent technical assistance.
This work was supported by National Institutes of Health grants HL38199 and HL76213 and an AHA predoctoral fellowship (F.V.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.W. designed and performed research, analyzed data, and wrote the paper; F.V. and J.D. performed research; and P.J.F. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Philip J. Fay, Department of Biochemistry and Biophysics, University of Rochester School of Medicine, 601 Elmwood Avenue, Rochester, NY 14642; e-mail: philip_fay@urmc.rochester.edu.
References
- 1.Fay PJ. Activation of factor VIII and mechanisms of cofactor action. Blood Rev. 2004;18:1–15. doi: 10.1016/s0268-960x(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 2.Fay PJ. Reconstitution of human factor VIII from isolated subunits. Arch Biochem Biophys. 1988;262:525–531. doi: 10.1016/0003-9861(88)90404-3. [DOI] [PubMed] [Google Scholar]
- 3.Ansong C, Miles SM, Fay PJ. Factor VIII A1 domain residues 97-105 represent a light chain-interactive site. Biochemistry. 2006;45:13140–13149. doi: 10.1021/bi061202w. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman RJ, Pipe SW. Regulation of factor VIII expression and activity by von Willebrand factor. Thromb Haemost. 1999;82:201–208. [PubMed] [Google Scholar]
- 5.Wakabayashi H, Koszelak ME, Mastri M, Fay PJ. Metal ion-independent association of factor VIII subunits and the roles of calcium and copper ions for cofactor activity and inter-subunit affinity. Biochemistry. 2001;40:10293–10300. doi: 10.1021/bi010353q. [DOI] [PubMed] [Google Scholar]
- 6.Wakabayashi H, Freas J, Zhou Q, Fay PJ. Residues 110-126 in the A1 domain of factor VIII contain a Ca2+ binding site required for cofactor activity. J Biol Chem. 2004;279:12677–12684. doi: 10.1074/jbc.M311042200. [DOI] [PubMed] [Google Scholar]
- 7.Shen BW, Spiegel PC, Chang CH, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111:1240–1247. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fay PJ, Haidaris PJ, Smudzin TM. Human factor VIIIa subunit structure: reconstruction of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991;266:8957–8962. [PubMed] [Google Scholar]
- 9.Wakabayashi H, Zhou Q, Nogami K, et al. pH-dependent association of factor VIII chains: enhancement of affinity at physiological pH by Cu2+. Biochim Biophys Acta. 2006;1764:1094–1101. doi: 10.1016/j.bbapap.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansong C, Fay PJ. Factor VIII A3 domain residues 1954-1961 represent an A1 domain-interactive site. Biochemistry. 2005;44:8850–8857. doi: 10.1021/bi050145o. [DOI] [PubMed] [Google Scholar]
- 11.Lollar P, Parker CG. pH-dependent denaturation of thrombin-activated porcine factor VIII. J Biol Chem. 1990;265:1688–1692. [PubMed] [Google Scholar]
- 12.Lollar P, Parker ET, Fay PJ. Coagulant properties of hybrid human/porcine factor VIII molecules. J Biol Chem. 1992;267:23652–23657. [PubMed] [Google Scholar]
- 13.Fay PJ, Beattie TL, Regan LM, O'Brien LM, Kaufman RJ. Model for the factor VIIIa-dependent decay of the intrinsic factor Xase: role of subunit dissociation and factor IXa-catalyzed proteolysis. J Biol Chem. 1996;271:6027–6032. doi: 10.1074/jbc.271.11.6027. [DOI] [PubMed] [Google Scholar]
- 14.Pipe SW, Eickhorst AN, McKinley SH, Saenko EL, Kaufman RJ. Mild hemophilia A caused by increased rate of factor VIII A2 subunit dissociation: evidence for nonproteolytic inactivation of factor VIIIa in vivo. Blood. 1999;93:176–183. [PubMed] [Google Scholar]
- 15.Pipe SW, Saenko EL, Eickhorst AN, Kemball-Cook G, Kaufman RJ. Hemophilia A mutations associated with 1-stage/2-stage activity discrepancy disrupt protein-protein interactions within the triplicated A domains of thrombin-activated factor VIIIa. Blood. 2001;97:685–691. doi: 10.1182/blood.v97.3.685. [DOI] [PubMed] [Google Scholar]
- 16.Hakeos WH, Miao H, Sirachainan N, et al. Hemophilia A mutations within the factor VIII A2-A3 subunit interface destabilize factor VIIIa and cause one-stage/two-stage activity discrepancy. Thromb Haemost. 2002;88:781–787. [PubMed] [Google Scholar]
- 17.Duncan EM, Duncan BM, Tunbridge LJ, Lloyd JV. Familial discrepancy between the one-stage and two-stage factor VIII methods in a subgroup of patients with haemophilia A. Br J Haematol. 1994;87:846–848. doi: 10.1111/j.1365-2141.1994.tb06749.x. [DOI] [PubMed] [Google Scholar]
- 18.Rudzki Z, Duncan EM, Casey GJ, Neumann M, Favaloro EJ, Lloyd JV. Mutations in a subgroup of patients with mild haemophilia A and a familial discrepancy between the one-stage and two-stage factor VIII:C methods. Br J Haematol. 1996;94:400–406. doi: 10.1046/j.1365-2141.1996.d01-1792.x. [DOI] [PubMed] [Google Scholar]
- 19.Pemberton S, Lindley P, Zaitsev V, Card G, Tuddenham EG, Kemball-Cook G. A molecular model for the triplicated A domains of human factor VIII based on the crystal structure of human ceruloplasmin. Blood. 1997;89:2413–2421. [PubMed] [Google Scholar]
- 20.Wakabayashi H, Fay PJ. Identification of residues contributing to A2 domain-dependent structural stability in factor VIII and factor VIIIa. J Biol Chem. 2008;283:11645–11651. doi: 10.1074/jbc.M710252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sammond DW, Eletr ZM, Purbeck C, Kimple RJ, Siderovski DP, Kuhlman B. Structure-based protocol for identifying mutations that enhance protein-protein binding affinities. J Mol Biol. 2007;371:1392–1404. doi: 10.1016/j.jmb.2007.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimms LT, Zampighi G, Nozaki Y, Tanford C, Reynolds JA. Phospholipid vesicle formation and transmembrane protein incorporation using octyl glucoside. Biochemistry. 1981;20:833–840. doi: 10.1021/bi00507a028. [DOI] [PubMed] [Google Scholar]
- 23.Doering C, Parker ET, Healey JF, Craddock HN, Barrow RT, Lollar P. Expression and characterization of recombinant murine factor VIII. Thromb Haemost. 2002;88:450–458. [PubMed] [Google Scholar]
- 24.Wakabayashi H, Su YC, Ahmad SS, Walsh PN, Fay PJ. A Glu113Ala mutation within a factor VIII Ca(2+)-binding site enhances cofactor interactions in factor Xase. Biochemistry. 2005;44:10298–10304. doi: 10.1021/bi050638t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Over J. Methodology of the one-stage assay of Factor VIII (VIII:C). Scand J Haematol Suppl. 1984;41:13–24. doi: 10.1111/j.1600-0609.1984.tb02764.x. [DOI] [PubMed] [Google Scholar]
- 26.Lollar P, Fay PJ, Fass DN. Factor VIII and factor VIIIa. Methods Enzymol. 1993;222:128–143. doi: 10.1016/0076-6879(93)22010-d. [DOI] [PubMed] [Google Scholar]
- 27.Wakabayashi H, Schmidt KM, Fay PJ. Ca(2+) binding to both the heavy and light chains of factor VIII is required for cofactor activity. Biochemistry. 2002;41:8485–8492. doi: 10.1021/bi025589o. [DOI] [PubMed] [Google Scholar]
- 28.Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- 29.Hemker HC, Beguin S. Thrombin generation in plasma: its assessment via the endogenous thrombin potential. Thromb Haemost. 1995;74:134–138. [PubMed] [Google Scholar]
- 30.Weiner SJ, Kollman PA, Case DA, et al. A new force field for molecular mechanical simulation of nucleic acids proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 31.Swiss Institute of Bioinformatics. [Accessed July 2, 2008]; UniProtKB/Swiss-Prot Release 55.5 of June 10, 2008, UniProtKB/TrEMBL Release 38.5 of June 10, 2008. http://ca.expasy.org/sprot.
- 32.Kemball-Cook G, Tuddenham EG, Wacey AI. The factor VIII Structure and Mutation Resource Site: HAMSTeRS version 4. Nucleic Acids Res. 1998;26:216–219. doi: 10.1093/nar/26.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kemball-Cook G. [Accessed July 2, 2008];MRC Clinical Sciences Centre, Haemophilia A Mutation Database. http://europium.csc.mrc.ac.uk.
- 34.Pace CN, Shirley BA, McNutt M, Gajiwala K. Forces contributing to the conformational stability of proteins. FASEB J. 1996;10:75–83. doi: 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- 35.Pipe SW, Kaufman RJ. Characterization of a genetically engineered inactivation-resistant coagulation factor VIIIa. Proc Natl Acad Sci U S A. 1997;94:11851–11856. doi: 10.1073/pnas.94.22.11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gale AJ, Pellequer JL. An engineered interdomain disulfide bond stabilizes human blood coagulation factor VIIIa. J Thromb Haemost. 2003;1:1966–1971. doi: 10.1046/j.1538-7836.2003.00348.x. [DOI] [PubMed] [Google Scholar]
- 37.Radtke KP, Griffin JH, Riceberg J, Gale AJ. Disulfide bond-stabilized factor VIII has prolonged factor VIIIa activity and improved potency in whole blood clotting assays. J Thromb Haemost. 2007;5:102–108. doi: 10.1111/j.1538-7836.2006.02283.x. [DOI] [PubMed] [Google Scholar]