

Abstract
Plants have substantially higher gene duplication rates compared with most other eukaryotes. These plant gene duplicates are mostly derived from whole genome and/or tandem duplications. Earlier studies have shown that a large number of duplicate genes are retained over a long evolutionary time, and there is a clear functional bias in retention. However, the influence of duplication mechanism, particularly tandem duplication, on duplicate retention has not been thoroughly investigated. We have defined orthologous groups (OGs) between Arabidopsis (Arabidopsis thaliana) and three other land plants to examine the functional bias of retained duplicate genes during vascular plant evolution. Based on analysis of Gene Ontology categories, it is clear that genes in OGs that expanded via tandem duplication tend to be involved in responses to environmental stimuli, while those that expanded via nontandem mechanisms tend to have intracellular regulatory roles. Using Arabidopsis stress expression data, we further demonstrated that tandem duplicates in expanded OGs are significantly enriched in genes that are up-regulated by biotic stress conditions. In addition, tandem duplication of genes in an OG tends to be highly asymmetric. That is, expansion of OGs with tandem genes in one organismal lineage tends to be coupled with losses in the other. This is consistent with the notion that these tandem genes have experienced lineage-specific selection. In contrast, OGs with genes duplicated via nontandem mechanisms tend to experience convergent expansion, in which similar numbers of genes are gained in parallel. Our study demonstrates that the expansion of gene families and the retention of duplicates in plants exhibit substantial functional biases that are strongly influenced by the mechanism of duplication. In particular, genes involved in stress responses have an elevated probability of retention in a single-lineage fashion following tandem duplication, suggesting that these tandem duplicates are likely important for adaptive evolution to rapidly changing environments.
Plant genomes contain a higher proportion of recently duplicated genes compared with most other eukaryotes (Lockton and Gaut, 2005). These duplicates are mostly derived from segmental, whole genome, and tandem duplication events (Arabidopsis Genome Initiative, 2000; Goff et al., 2002; Tuskan et al., 2006). Based on analyses of gene duplication timing and synteny, it is believed that three rounds of whole genome duplication (WGD) likely occurred in the Arabidopsis (Arabidopsis thaliana) lineage after its split from the rice (Oryza sativa) lineage approximately 150 million years ago (Vision et al., 2000; Simillion et al., 2002; Raes et al., 2003; Blanc and Wolfe, 2004; Paterson et al., 2004; Tuskan et al., 2006). Genome rearrangement after WGD and additional small-scale duplication events likely contributed to the segmental structure of duplicated blocks. Although the rate of observed ancient WGD in the Arabidopsis lineage is low (approximately one event per 50 million years), the effect of each WGD is large because all genes are doubled in a single event. In addition to segmental duplication and WGD, tandem duplication, which produces duplicates that are located in close proximity, has contributed significantly to the expansion of plant gene families (Zhang and Gaut, 2003; Rizzon et al., 2006). In contrast to WGD, tandem duplications have occurred much more frequently and are responsible for much of the gene copy number and allelic variation within a population (Fortna et al., 2004; Rostoks et al., 2005; Clark et al., 2007). Although each tandem duplication event only affects a small number of genes, tandemly duplicated genes constitute approximately 14% of all duplicates in Arabidopsis (Rizzon et al., 2006).
Several evolutionary and population genetic models of duplicate gene fate have been proposed that provide the theoretical and mechanistic explanations for gene retention (Ohno, 1970; Walsh, 1995; Force et al., 1999; Lynch and Force, 2000). While the typical fate of duplicates is rather rapid gene loss, genome-wide analyses of several eukaryotic genomes and gene families indicate that a substantial number of duplicated genes are retained (Moore and Purugganan, 2003, 2005; Blanc and Wolfe, 2004). In addition, there is a significant bias in the functions of retained genes in different species (Blanc and Wolfe, 2004; Seoighe and Gehring, 2004; Maere et al., 2005; Shiu et al., 2005, 2006; Wapinski et al., 2007). Since duplication mechanisms vary greatly in terms of scale and frequency, one intriguing question is if duplicates generated by different mechanisms differed significantly in their functions during vascular plant evolution. It is thought that tandem duplicates tend to be involved in stress responses (Parniske et al., 1997; Michelmore and Meyers, 1998; Lucht et al., 2002; Kovalchuk et al., 2003; Leister, 2004; Shiu et al., 2004; Maere et al., 2005; Mondragon-Palomino and Gaut, 2005; Rizzon et al., 2006). For example, it has been shown that duplicate genes associated with membranes and/or involved in stress responses are overrepresented among tandem duplicates, while those involved in nucleic acid-binding functions are overrepresented among nontandem duplicates (Rizzon et al., 2006). Since the studies on functional bias so far have mainly been concerned with the averaged behavior of duplicate genes, it is unclear if these patterns will remain consistent if one considers functional bias in the context of lineage-specific expansion (Lespinet et al., 2002). That is, when examining the functional bias of duplicated genes, in addition to considering whether genes have apparent paralogs, it is informative to distinguish the time frames during which the duplication events occurred. In addition, previous studies have not addressed whether the bias in the retention of duplicated genes depends on the type of stress condition (e.g. biotic or abiotic) and/or the nature of the stress response (e.g. up-regulation or down-regulation). It is possible that the relationship between tandem duplicates and stress responses only holds for certain types of stresses.
To study patterns of functional bias among genes derived from lineage-specific expansion events, we first classified genes from Arabidopsis, poplar (Populus trichocarpa), rice, and the moss Physcomitrella patens into orthologous groups (OGs) and determined the degree of expansion for each OG. After identifying genes in expanded versus nonexpanded OGs, we used Arabidopsis Gene Ontology (GO) annotations to examine the functional bias of retained duplicates in the Arabidopsis lineage. To better understand the relationship between the nature or types of stress responses (such as different biotic and abiotic conditions) and duplication mechanisms (tandem versus nontandem), we examined an Arabidopsis expression data set containing 15 abiotic and biotic stress treatments and identified stress conditions enriched in retained genes duplicated via tandem or nontandem mechanisms. Finally, we compared patterns of lineage-specific expansion and functional bias among tandem and nontandem genes to determine how duplication mechanism contributed to gene family expansion during land plant evolution.
RESULTS AND DISCUSSION
Rate of Lineage-Specific Expansion
Plants have substantially higher rates of gene duplication than other organisms. This, together with substantial functional bias in gene retention, has contributed to dramatic differences in the degree of lineage-specific expansion among plant gene families. To address the question if the rate of gene gains was constant throughout the evolutionary history of land plants, protein-coding genes from Arabidopsis, moss, rice, and poplar were classified into similarity clusters (referred to as gene families; see “Materials and Methods”). Among 14,745 gene families, 5,060 are shared among all four plant species. A gene tree was generated for each shared family (see “Materials and Methods”). The gene tree and the species tree of these four plants were reconciled for estimating ancestral gene numbers. These ancestral gene numbers were then used to determine gene-gain events in the lineage leading to Arabidopsis (Fig. 1A). The rates of gene gain (total gain during a time period divided by the estimated duration) are not constant over the three time periods we examined (branches 1, 2, and 3 in Fig. 1A). The gain rates for branches linked together by older ancestral nodes are smaller than those linked by “younger” branches. For example, the gain rate in branch 1 (14.1–28.2 gains per million years) is approximately four times slower than that in branch 3 (44.3–53.2 gains per million years).
Figure 1.

Rates of gene gain and functional overrepresentation in the Arabidopsis lineage. A, For each branch (time period), numbers of gene gains were estimated from the differences between the extant and/or ancestral gene numbers between the two nodes defining the branch. The rates of gene gain were inferred based on the divergence time estimates (Wolfe et al., 1989; Heckman et al., 2001; Tuskan et al., 2006; Rensing et al., 2008). MYA, Million years ago. B, GO categories with an overrepresented number of genes in expanded OGs in the Arabidopsis lineage. In the “specific” row, categories that are specific to a particular OG type are shown. In the “common” row, overrepresented categories that are consistent among all three OG types are shown.
One explanation for this gain rate difference is that, early in vascular plant evolution, the duplication rate was substantially lower. If this is the case, one may expect plant lineages that diverged earlier in the vascular plant lineage to have low duplication rates. However, it is estimated that Ceratopteris richardii, a fern in the lineage that split from the flowering plant lineage early in vascular plant evolution, likely has a higher proportion of paralogs than the flowering plants (Nakazato et al., 2006). Therefore, we speculate that the duplication rate early in vascular plant evolution may have been similar to that of the present day. Another explanation is that even though many genes were fixed and retained, a large number of them did not survive in the long run. This explanation is consistent with the observed gradual decay of paralog synonymous substitution rates of several eukaryotes over time (Lynch and Conery, 2000). More detailed analysis regarding gene birth and death in plant gene families is necessary to address the issue of duplicate longevity further.
Functional Bias of Lineage-Specific Retention in the Arabidopsis Lineage
In addition to differences in the rates of gene gain over the course of vascular plant evolution, it is likely that the functional bias in duplicate retention may differ depending on the time period examined, due to differences in organismal complexity and environment. To determine if the patterns of functional biases differ depending on the timing of gene duplication, we evaluated the representation of duplicate genes in expanded versus nonexpanded OGs in various functional categories. Here, duplicates in expanded OGs are genes with elevated rates of retention/duplication compared with genes in nonexpanded OGs. By examining OGs, the ancestral gene numbers and subsequent gains can be estimated; therefore, all sorts of classifications, in particular functional categories of genes, can be compared in the same evolutionary period.
Ideally, we would evaluate the functional categories for each internal or external node in the four-plant phylogeny to directly determine the functional bias of genes in expanded OGs. However, this is problematic due to the difficulty in inferring ancestral functions. Although we have gene-gain data for all four plant species, we focused on the comparison of Arabidopsis with other plants in a pair-wise fashion for two reasons. First, the functional annotation data for other plant species are either absent or not as comprehensive as those for Arabidopsis. Second, functional annotation criteria may differ between species and influence the interpretation of results. It should be noted that this approach resulted in analyzing functional biases in a somewhat nonindependent fashion, since some of the branches overlap.
We assessed if there is a functional bias among genes in expanded OGs by determining the GO categories with overrepresented numbers of genes in OGs that have expanded in the Arabidopsis lineage (Supplemental Fig. S1). For each shared family, we identified three “types” of OGs (Fig. 1): Arabidopsis-poplar (A-P), Arabidopsis-rice (A-R), and Arabidopsis-moss (A-M). Each OG represents the presence of one ancestral gene from the progenitor of the species pair and all duplicates generated and retained after speciation. Currently, orthology is determined either by reconciling species and gene trees (referred to as tree-based; Chen et al., 2000) or by applying iterative search algorithms on a sequence similarity matrix (referred to as similarity-based; Remm et al., 2001). Since both approaches have their merits and caveats (Chen et al., 2007), we applied both to define OGs and generated two “sets” of OGs (tree- and similarity-based, not to be confused with OG types) for each species pair. The overlap between these two approaches is approximately 80% for all species pairs, indicating that gene membership for most OGs generated by these two approaches is consistent (Supplemental Table S1). Nonetheless, to ensure that our analyses were not biased by the method of OG inference, the results for both approaches are shown to highlight consistent trends. It should be noted that the annotation quality for poplar and moss is lower than that for rice and Arabidopsis. In addition to the annotation quality issue, uncertainty in phylogenetic reconstruction also likely reduces the accuracy of the orthology inference. However, we expect that such errors in orthology assignment will be global, which will reduce the relevant biological signals in subsequent analyses but will not necessarily bias our results in a particular direction.
Based on the three types of OGs defined (A-P, A-R, and A-M) encompassing different periods in the evolution of the Arabidopsis lineage, we could compare the overrepresented functional categories and determine which categories are specific to a time period. The number of gene gains in an OG is defined as the number of Arabidopsis genes in an OG minus one (because each OG indicates the presence of an ancestral gene). In each functional category, the number of gene gains was compared with the number of expected gene gains using the χ2 test (see “Materials and Methods”). For example, if functional category X is overrepresented among A-M type but not A-P or A-R type OGs, ancestral genes with function X are regarded as expanded in the branch after the Arabidopsis-moss split but before the divergence between the Arabidopsis and the rice lineages (branch 1 in Fig. 1B). Interestingly, genes related to biotic and abiotic stress responses and stress signaling networks, as well as a number of other functional categories, are found in OGs that have expanded in all three types of OGs (Fig. 1B). This finding indicates that OGs containing genes mediating stress responses expanded at significantly higher rates during the early period of vascular plant evolution. These OGs have continued to expand in the relatively recent evolutionary history of the Arabidopsis lineage.
Functional Bias, Duplication Mechanisms, and Signaling Networks
Based on published reports, it is anticipated that tandem duplication will be more closely associated with stress-related genes than nontandem duplication (Parniske et al., 1997; Michelmore and Meyers, 1998; Lucht et al., 2002; Kovalchuk et al., 2003; Leister, 2004; Shiu et al., 2004; Maere et al., 2005; Mondragon-Palomino and Gaut, 2005; Rizzon et al., 2006). One open question is if this relationship exists for genes in lineage-specifically expanded OGs. To determine how genes in OGs that expanded via tandem and nontandem mechanisms differ in their involvement in stress and responses to environmental stimuli during vascular plant evolution, we compared GO categories with an overrepresented number of genes in OGs (A-M, A-R, and A-P) that expanded via tandem or other duplication mechanisms (Supplemental Fig. S2). It should be noted that genome rearrangements may have occurred that disrupt the relationships between relatively ancient tandem duplicates. Therefore, in this study, we likely missed older tandem duplicates. Nonetheless, OGs containing genes in categories such as response to biotic stimulus, defense response, response to toxin, various transport functions, glycosinolate metabolism, and phosphorous metabolism (phosphorylation) have expanded significantly due to tandem duplications (Fig. 2A, red circles; Supplemental Fig. S3). In addition, cellular component categories related to the extracellular compartment and cell surface tend to be enriched in genes that are in OGs that expanded via tandem duplication mechanisms (Fig. 2B, red circles). In contrast, genes in OGs expanded via nontandem mechanisms tend to be found in functional categories related to intracellular components, regulation of metabolism, hormone metabolism, transcriptional regulation, cell communication, and response to hormone stimulus (Fig. 2, blue circles; Supplemental Fig. S3). Most functional categories enriched in genes that are in OGs that expanded via tandem or nontandem mechanisms are consistent among the three types of OGs.
Figure 2.

GO categories with overrepresented numbers of tandem or nontandem duplicates in expanded OGs. A, Biological process categories. The arrowheads point to subcategories. Categories with significantly (χ2 test, false discovery rate adjusted P < 0.05) more genes in OGs expanded via tandem and nontandem mechanisms are shown in red and blue circles, respectively. B, Cellular component categories. Only categories discussed in the text are shown. For more detail, see Supplemental Figures S2 and S3. Color coding is the same as in A. C, Selected molecular function categories with overrepresented numbers of genes that are responsive to biotic stress conditions. The GO category descriptions are shown on the right. There are six columns for each stress condition. The columns represent types/sets of OGs in the following order: similarity-based A-M, tree-based A-M, similarity-based A-R, tree-based A-R, similarity-based A-P, and tree-based A-P. Color coding is the same as in A.
Signaling networks can be partitioned into three major layers: transducers, transcriptional regulators, and regulatory targets (Doebley and Lukens, 1998). By partitioning functional categories into these three layers, differences between genes in OGs that expanded via tandem and nontandem mechanisms become more apparent. Transducers are composed of proteins involved in signal production, perception, transmission, and modification such as phosphorylation. Based on our analysis of functional categories, most genes that serve as transducers of signaling cascades (cell communication, signal transduction) are in OGs that expanded via nontandem mechanisms (Fig. 2A, blue circles). One exception involves genes in the phosphorylation categories. A large number of these genes belong to the receptor-like kinase family, many of which are cell surface receptors (Shiu and Bleecker, 2001). Many receptor-like kinases are in OGs that have expanded significantly due to tandem duplication, and there is experimental evidence for their involvement in the perception of biotic stimuli (Shiu et al., 2004). We found that if the transducer layer is further partitioned into cell surface and intracellular transducers, genes in OGs expanded via tandem mechanisms are enriched in cell surface transducer categories (Fig. 2, red circles), while nontandem duplicates are enriched in intracellular transducer categories (Fig. 2, blue circles).
Assuming that OGs that have expanded via nontandem mechanisms are mostly derived from whole genome duplications, our finding is consistent with earlier reports that transcriptional regulation categories have overrepresented numbers of duplicates from polyploidization (Blanc and Wolfe, 2004; Seoighe and Gehring, 2004; Maere et al., 2005) and have an overall higher retention rate compared with Arabidopsis genes in general (Shiu et al., 2005). The regulatory target layer can be seen as the effector layer, or more generally the response layer, since many of the regulatory target genes encode products mediating the response to stimuli. Among stimuli-responsive categories, hormonal response categories tend to be enriched in genes in OGs expanded via nontandem mechanisms, while response to external stimuli categories are overrepresented in genes in OGs expanded via tandem duplication.
In this study, we focus on the properties of lineage-specific expanded OGs instead of the properties of paralogous genes, as in previous studies (Maere et al., 2005; Rizzon et al., 2006). The earlier work by Maere et al. (2005) contrasted the decay rate differences among genes of various functions in the context of small-scale and large-scale duplication events. Here, the small-scale events are likely predominantly tandem duplication events. Nonetheless, despite the differences in methodology and our focus on expansion instead of duplication, we reach a very similar conclusion regarding overrepresentation of tandem genes in stress response functional categories.
Stress Responsiveness Categories and Retention of Duplicates Generated via Tandem and Nontandem Mechanisms
Based on the analysis of functional categories with overrepresented numbers of genes in expanded OGs, one of the most notable differences between tandem and nontandem duplicates is their involvement in the response to environmental stimuli and biotic stress. However, it remains an open question if this is a property of stress genes in general or genes involved in certain types of stress conditions. To address this question, we examined the expansion patterns of stress-responsive genes in the Arabidopsis lineage using the AtGenExpress stress expression data set (Kilian et al., 2007). We focused on seven abiotic and eight biotic stress conditions (Table I). For each stress response, up- and down-regulated genes were defined by comparing intensities of stress-treated samples with those of controls (false discovery rate corrected P < 0.05). These differentially regulated genes are referred to as “stress-responsive” genes, while genes with no significant change in either direction are regarded as “nonresponsive.” Genes up-regulated under all of the abiotic and biotic stress conditions (except heat) are members of OGs that have expanded significantly throughout all three periods of the Arabidopsis lineage evolution (the Expansion [Exp] columns for A-M, A-R, and A-P in Table I). However, there is no clear trend for down-regulated genes (the Exp columns in Table I). These findings indicate that expansion of OGs containing genes that are up-regulated by most stress conditions, both biotic and abiotic, has occurred continuously over the course of vascular plant evolution.
Table I.
Overrepresentation of stress-responsive genes in OGs that expanded in the Arabidopsis lineage and the major contributing duplication mechanisms
| Statistical Test | Up-Regulation
|
Down-Regulation
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A-M
|
A-R
|
A-P
|
A-M
|
A-R
|
A-P
|
|||||||
| Expa | T/Nb | Exp | T/N | Exp | T/N | Exp | T/N | Exp | T/N | Exp | T/N | |
| Abiotic stress conditionsc | ||||||||||||
| UV-B | + | T | + | T | + | T | + | N | ||||
| Wounding | + | T | + | + | + | + | ||||||
| Drought | + | + | T | + | + | + | ||||||
| Cold4C | + | N | + | N | + | N | ||||||
| Heat | + | N | + | N | ||||||||
| Salt | + | + | + | |||||||||
| Osmotic | + | + | + | |||||||||
| Biotic stress conditionsc | ||||||||||||
| Flg22 | + | T | + | T | + | + | + | |||||
| GST-NPP1 | + | T | + | T | + | T | ||||||
| HrcC- | + | T | + | T | + | T | ||||||
| P. infestans | + | T | + | T | + | T | ||||||
| Psph | + | T | + | T | + | T | ||||||
| HrpZ | + | + | + | |||||||||
| AvrRpm1 | + | + | + | |||||||||
| DC3000 | + | + | + | |||||||||
A + sign indicates that the ratio of stress-responsive genes in expanded OGs versus nonexpanded OGs is significantly higher (P ≤ 0.05) than for nonresponsive genes.
T/N, Tandem versus nontandem; T, the genes responsive for a given stress condition are significantly enriched in tandem relative to nontandem genes; N, significant enrichment of nontandem genes (q < 0.05).
Condition names follow those from AtGenExpress.
To determine whether stress-responsive genes found in expanded OGs tend to be derived from tandem or nontandem duplications, we asked if there is a relative enrichment of stress-responsive genes in OGs that expanded via tandem or nontandem duplication for each stress condition (the Tandem [T]/Nontandem [N] columns in Table I). Up-regulated genes in general belong to OGs with higher rates of expansion compared with the OGs containing nonresponsive genes. However, there are clear differences in how duplication mechanism contributed to the expansion of OGs containing stress-responsive genes. Genes in OGs that expanded via tandem duplication are more likely to be up-regulated under biotic stresses than those in OGs that expanded via nontandem duplication (the T/N columns for up-regulation in Table I), which is consistent with the GO-based analysis. In contrast, genes up-regulated by abiotic stress and down-regulated genes in general belong to OGs that are equally likely to have expanded via either tandem or nontandem mechanisms (Table I).
Interestingly, the fact that biotic stress-responsive genes tend to be derived from tandem duplication does not affect the relationship between functional partitioning in signaling networks and duplication mechanism, as postulated in the previous section. For example, among genes up-regulated by biotic stress, molecular function categories, including DNA binding and transcription regulator activity, are still enriched in nontandem duplicates (Fig. 2C). Taken together, these findings indicate that expansion of biotic stress response genes has occurred more often via tandem duplication than expected. However, the position of a gene in the cellular signaling network has an overriding influence on the predominant duplication mechanisms that contribute to lineage-specific expansion.
Influence of Duplication Mechanism on Patterns of Expansion at the OG Level
Our study examines the functional bias of retained duplicates in the Arabidopsis lineage and shows that genes in OGs that expanded via tandem and nontandem duplication mechanisms are related to stress responses and intracellular regulatory roles, respectively. Since different plant lineages have very different life histories, the nature of selection pressure imposed by their environmental conditions is expected to be diverse. Therefore, if expansion of certain OGs with tandem duplicates were involved in adaptive responses to the environment specific to one species, their orthologous genes in another species will tend not to be retained. To test this prediction, we first evaluated if the number of OGs with a particular lineage-specific gene number combination (e.g. for an OG with two Arabidopsis and three moss genes; Fig. 3A) is overrepresented or underrepresented (Fig. 3). Overrepresentation or underrepresentation of a gene number combination was evaluated using the log ratios between observed and expected numbers of gains generated based on the power distribution of branch size in a particular lineage. Interestingly, we found that the gene number combinations are obviously nonrandom, with two extreme patterns (examples comparing the Arabidopsis lineage against the other three lineages are shown in Fig. 3). The first is “convergent expansion,” where two or more genes were gained independently in different lineages. The other is “single-lineage expansion,” where gene gains have predominantly occurred in one lineage and are coupled with a loss in the other.
Figure 3.

Expansion patterns of OGs. A, Example topologies for two OGs and log ratios for each OG expansion pattern for the Arabidopsis-moss comparison. In the matrix on the right, the x and y axes indicate the numbers of Arabidopsis and moss genes in an OG, respectively. In each cell, a log ratio was generated between the observed number of OGs found to have a specific gene number combination (Obs) and the expected number of OGs (Exp; see “Materials and Methods”). This ratio serves as a measure of the relative enrichment of the number of OGs with a particular gene number combination (the color scale is shown under the phylogenies). Positive log ratios are shown in shades of red, and negative log ratios are shown in shades of blue. Also shown are log ratios for OG expansion patterns for Arabidopsis and rice (B) and Arabidopsis and poplar (C).
To determine whether either of the two expansion patterns is correlated with tandem or nontandem duplication, we classified genes into tandem and nontandem categories (see “Materials and Methods”). The ratios between the numbers of tandem and nontandem duplicated genes in OGs that expanded in either a convergent or single-lineage fashion were used to assess the significance of enrichment (Table II). Here, OGs are regarded as convergently expanded if the number of genes is more than two in both lineages. OGs are defined as expanded in a single-lineage fashion if the number of genes is two or more in one lineage and zero in the other. Here, we show the results where the tandem/nontandem genes are defined solely based on that of the Arabidopsis genome in comparison with the other three lineages. We found that tandem duplicates tend to be in OGs that experienced single-lineage expansion (χ2 test; P < 10−12). In contrast, genes duplicated via nontandem mechanisms are mostly found in OGs that underwent convergent expansion (χ2 test; P < 10−12). These findings are consistent regardless of which types or sets of OGs (A-P, A-R, or A-M; similarity- or tree-based) were examined. We also observed that in OGs experiencing single-lineage expansion, the ratio of tandem to nontandem duplicated genes is highest in the A-P pairs and decreases as the divergence time between the species pair increases (Table II). One potential explanation for this trend is that some of these tandem genes may look younger because of gene conversion (Gao and Innan, 2004). However, gene conversion tends to occur between highly similar duplicate genes, and the sequence divergence between tandem duplicates is mostly quite large (Rizzon et al., 2006). The second explanation is that tandem duplicates have a relatively faster turnover rate; that is, tandem duplicates were not retained as long as nontandem duplicates. This is consistent with our observation that the ratio of tandemly to nontandemly duplicated genes decreases more rapidly in single-lineage expanded than in convergently expanded OGs (Table II).
Table II.
Ratio of Arabidopsis genes in OGs that expanded via tandem duplication and nontandem mechanisms
| OG Type | Method for Defining OG | Expansion Pattern
|
Pc | |
|---|---|---|---|---|
| Convergenta | Single-Lineageb | |||
| A-M | Similarity | 0.17 (756/4,500) | 0.30 (848/2,918) | 2.2 × 10−23 |
| Tree | 0.16 (831/5,297) | 0.40 (1,443/3,566) | 9.4 × 10−88 | |
| A-R | Similarity | 0.31 (959/3,115) | 0.47 (644/1,375) | 3.1 × 10−12 |
| Tree | 0.27 (844/3,073) | 0.50 (1,631/3,294) | 2.3 × 10−33 | |
| A-P | Similarity | 0.29 (1,141/3,944) | 0.60 (741/1,234) | 7.2 × 10−38 |
| Tree | 0.26 (1,014/3,930) | 0.64 (1,578/2,452) | 1.0 × 10−83 | |
OGs showing convergent expansion are defined based on the presence of more than two genes in both lineages. In the parentheses, the numerator is the number of genes in OGs expanded via tandem duplication and the denominator is the number of genes in OGs expanded via nontandem mechanisms.
OGs showing single-lineage expansion are defined as having two or more genes in the Arabidopsis lineage and zero in the other. Numbers in parentheses are as described above.
The ratios of tandem to nontandem duplicates in OGs experiencing convergent and single-lineage expansion were compared using χ2 tests.
Most highly duplicated genes have been reported to reside in OGs that expanded in parallel in different lineages (Oliver et al., 2000; Aravind et al., 2001; Holub, 2001; Hughes and Friedman, 2003). In contrast, we found that expansion can occur in either a parallel (convergent) or a single-lineage fashion. It should be noted that earlier studies did not distinguish between tandem and nontandem duplication. Single-lineage expansion is expected for genes in families experiencing rapid birth-and-death evolution, where rapid gene turnover is fueled by repeated gene duplication and frequent gene “death” due to pseudogenization (Nei and Rooney, 2005). Rapid birth-and-death evolution has been reported in multiple gene families with a large number of tandem duplicates, such as plant Leu-rich repeat disease resistance genes and mammalian olfactory receptors (Parniske et al., 1997; Michelmore and Meyers, 1998; Rouquier et al., 1998; Young et al., 2002; Mondragon-Palomino and Gaut, 2005). Our finding that single-lineage expansion is mainly due to tandem duplication is consistent with the findings from these gene families.
We have also conducted analyses to identify functional categories enriched in OGs that expanded in a single-lineage fashion (Supplemental Fig. S4). A large number of these categories, including response to biotic stress and secondary metabolism, are similar to those identified as being overrepresented among tandem duplicates. Therefore, genes found in OGs that experienced single-lineage expansion are likely involved in adaptive evolution in a lineage-specific fashion and turn over rapidly. In an earlier study, it was shown that genes derived from small-scale (mostly tandem) duplication events tend to have low decay rates (Maere et al., 2005). Based on our findings, this apparently lower decay rate may not be interpreted as a higher longevity of these stress-responsive tandem duplicates. Instead, the expansion of stress-responsive tandem duplicates is potentially a consequence of high duplication rate accompanied by rapid losses, as demonstrated by studies of sequence variation in plants (Borevitz et al., 2007).
CONCLUSION
Plant genes in OGs that expanded via tandem duplication tend to be involved in responses to biotic stress and environmental stimuli based on GO categories. Furthermore, by examining stress microarray data sets, we found that tandem duplicates are enriched in stress-responsive genes. Therefore, genes influencing stress response have an elevated probability of retention following tandem duplication. Why do these stress-responsive tandem genes tend to be retained? An important feature of tandem genes is their high rate of duplication per generation. As a result, new tandem gene paralogs are continuously generated, likely providing a pool of highly dynamic targets for selection. In addition, tandem genes are highly variable within species. For example, sequence variation between Arabidopsis ecotypes is enriched in regions containing tandem duplicates (Borevitz and Nordborg, 2003; Clark et al., 2007), potentially due to elevated rates of recombination in regions with tandem duplicates (Zhang and Gaut, 2003). This high level of within-species variation among tandem genes further increases the number of targets (paralogs and alleles) that can be selected in ever-changing environments.
Interestingly, gene families that have been shown to be important for responding to biotic stresses, such as Leu-rich repeat disease resistance gene and receptor kinase, have a higher proportion of members in polymorphic regions than other genes families (Clark et al., 2007). These two gene families also have significant numbers of tandem members (Meyers et al., 2003; Shiu et al., 2004). In addition, biotic stress may further increase the variation in tandem genes, since recombination rate is elevated under pathogen attack (Lucht et al., 2002). Therefore, there is a strong correlation between tandemness and biotic stress that is corroborated by our studies of stress expression data. The selection pressure imposed by biotic agents is not only intense, due to, for example, the arms race between hosts and pathogens, but also relentless, since interactions with the environment only cease upon death. Because tandem genes represent a significantly larger pool of standing variation than can be generated by nontandem mechanisms, they have a higher probability of being represented among genes that meet the challenges of the biotic agents and are retained.
MATERIALS AND METHODS
Defining Gene Families and Tandem Duplicate Clusters
The amino acid sequences of four plant species (Arabidopsis [Arabidopsis thaliana], TAIR6; poplar [Populus trichocarpa], version 1.1; rice [Oryza sativa japonica], version 2; and the moss Physcomitrella patens, version 1.1) were obtained from The Arabidopsis Information Resource (www.arabidopsis.org), The Institute for Genomic Research (www.tigr.org), and the Joint Genome Institute (www.jgi.doe.gov). To define gene families among sequences from these four species, all-against-all similarity searches were conducted using BLAST with an E-value cutoff of 1e-5 (Altschul et al., 1997). Based on the transformed E-values (Shiu et al., 2005), we generated similarity clusters representing gene families with the Markov clustering program (http://micans.org/mcl/ [van Dongen, 2000]).
Tandem duplicated genes were defined as genes in any gene pair, T1 and T2, that (1) belong to the same gene family, (2) are located within 100 kb each other, and (3) are separated by zero, one or fewer, five or fewer, or 10 or fewer nonhomologous (not in the same gene family as T1 and T2) spacer genes. Therefore, there are four sets of tandem gene definitions. All analyses were conducted using all four sets, and we found that the results were consistent regardless of the criteria. Therefore, only the analysis results based on the 10 or fewer spacer gene criteria are reported.
Inference of OGs
We took two approaches to infer OGs between Arabidopsis and poplar, rice, and moss. In the first approach, protein sequences of members in each family were aligned with ClustalW (Thompson et al., 1994), and the alignments were used to generate neighbor-joining trees (Saitou and Nei, 1987) with the two-parameter substitution correction (Kimura, 1980). The phylogenetic trees were rooted at midpoints. OGs were identified by reconciling between family phylogenies and the species tree of these four plants with Notung (Chen et al., 2000). The OGs identified with the first approach are referred to as tree-based OGs (Supplemental Table S3).
In the second approach, we used a search algorithm similar to Inparanoid (Remm et al., 2001). We first conducted all-against-all BLAST searches between two species, A and B, in each gene family, and found reciprocally best matching pairs as the seeds for OGs. Given a reciprocal best match pair, A1 and B1, if any sequence, Ax (or By) from species A (or B) that has a smaller sequence distance to A1 (or B1) than the distance between A1 and B1, Ax (or By) was added to the OG containing A1 and B1 (seeds). This process was continued until all qualified sequences were assigned to seed OGs. At this point, many genes were not assigned to any OG because they belong to OGs with a potential gene loss in one lineage. To assign these types of genes to OGs, we used genes without an OG assignment (e.g. Ai) to search against the genes assigned to OGs to identify the best match (Bi) for B. If Bi was already assigned to any OGs, Ai was treated as the seed of an OG with lineage-specific loss. Any sequence Aj from A was added to the OG with Ai if the Ai-Aj distance was smaller than the Ai-Bi distance. The same procedure was repeated for sequences from B. The OGs inferred based on pair-wise similarity are called similarity-based OGs. Since the inference of similarity-based OGs depends on the starting point, we constructed 10 sets of similarity-based OGs for each species pair for subsequent analysis.
To evaluate the consistency of OGs constructed with the tree- and similarity-based approaches, we determined the degree of overlap between these two OG data sets. Since each gene is assigned to tree- and similarity-based OGs, the number of overlapping genes was counted between the tree- and similarity-based OGs containing the gene. The proportion of the overlapping genes was calculated for tree, and similarity-based OGs. The average proportion was calculated for all genes.
Statistical Tests for Determining Overrepresented and Underrepresented GO Categories and the Influence of Duplication Mechanisms on Degree of Expansion
GO assignments for Arabidopsis genes were obtained from The Arabidopsis Information Resource (http://www.arabidopsis.org/). Three top GO categories, cellular components, molecular functions, and biological processes, were analyzed as described earlier (Shiu et al., 2006). Among these GO categories, we obtained the numbers of Arabidopsis genes residing in expanded OGs and nonexpanded OGs. Expanded and nonexpanded OGs are defined as OGs having two or more genes and only one gene, respectively, in the Arabidopsis lineage. For analyses of similarity-based OGs, the numbers of genes in expanded OGs and nonexpanded OGs in a GO category were generated by averaging the numbers of genes among 10 runs. The expected numbers of genes in expanded OGs and in nonexpanded OGs were defined as the number of all Arabidopsis genes in expanded OGs and nonexpanded OGs, respectively. The expected values were then compared with the observed values with the χ2 test to determine whether the ratio of observed gene numbers in expanded OGs to those in nonexpanded OGs was significantly higher than the expected ratio.
For functional categories with overrepresented numbers of genes in expanded OGs, the observed ratio of tandem to nontandem duplicated genes in each category was compared with the expected ratio to determine whether the overrepresentation is due mainly to the contribution of tandem or nontandem duplications. The expected ratio was estimated from the number of all Arabidopsis genes duplicated via tandem and nontandem mechanisms in expanded OGs. To correct for multiple testing, the moderated P value (q) was estimated from raw χ2 test P values with Q-VALUE software (Storey and Tibshirani, 2003). The null hypothesis was rejected if q values were <0.05.
To determine if stress-responsive genes (based on gene expression data) in expanded OGs derived from tandem or nontandem duplication tend to have certain molecular functions, we used the GO molecular function assignments of stress response genes for the overrepresentation analysis. Genes with only IEA (Inferred from Electronic Annotation) and IEP (Inferred from Expression Pattern) evidence were excluded.
Statistical Tests to Identify Responsive Genes for Abiotic and Biotic Stress Conditions
Gene expression data under eight abiotic and eight biotic stress conditions were obtained from AtGenExpress (http://www.uni-tuebingen.de/plantphys/AFGN/atgenex.htm). The array intensities were processed using the Bioconductor (www.bioconductor.org) affy package in the R software environment (www.r-project.org). After background correction and quantile normalization, significantly up- and down-regulated genes under each stress condition were identified by comparing the hybridization intensities of arrays hybridized with treated samples against their corresponding control with LIMMA (Wettenhall and Smyth, 2004). Up- and down-regulated genes for each stress treatment were defined as genes with significantly higher and lower intensities (at 5% false discovery rate) in arrays hybridized with treated samples than those in controls in at least one time point of a stress condition. Nonresponsive genes were defined as genes that were not significantly up- or down-regulated. The numbers of up- and down-regulated genes are shown in Supplemental Table S2. The three time points for the genotoxic condition have an insufficient number of up- or down-regulated genes and were excluded from further analyses.
Determination of Overrepresented OGs in Different Lineages
In every OG, there are two lineages (I and J), each with i and j genes. To determine if an OG with a particular gene number combination (i, j) is overrepresented or underrepresented, we determined a log ratio by the following equation:
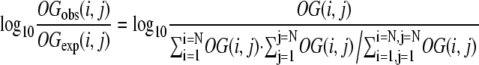 |
OGobs(i,j) is the observed number of OGs where the number of genes is i and j in lineages I and J, respectively. OGexp(i,j) is the expected number of OGs where the number of genes is i and j in lineages I and J, respectively. Since we have 10 sets of similarity-based OGs, the number OG(i,j) for similarity-based OGs is the average of OG(i,j) in 10 sets. The log ratio was independently estimated for tree- and similarity-based OGs, and the log ratio average was used to examine gene expansion patterns in two lineages.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. GO categories with overrepresented numbers of genes in different sets and types of OGs.
Supplemental Figure S2. GO categories with overrepresented numbers of genes in OGs expanded via tandem or nontandem mechanisms.
Supplemental Figure S3. Topology of GO categories with overrepresented numbers of genes in OGs expanded via tandem or nontandem mechanisms.
Supplemental Figure S4. Biological process categories overrepresented in tandem or nontandem duplicates in OGs showing single-lineage expansion.
Supplemental Table S1. Proportion of genes overlapped between OGs constructed via tree- and similarity-based methods.
Supplemental Table S2. Numbers of up- and down-regulated Arabidopsis genes in 16 stress conditions.
Supplemental Table S3. Genes in A-M, A-R, and A-P OGs.
Supplementary Material
Acknowledgments
We thank Takeshi Itoh and Takashi Makino for reading the manuscript and for discussion. We also thank The Arabidopsis Information Resource, The Institute of Genome Research, and the Joint Genome Institute for providing annotations and sequences and the Arabidopsis Functional Genomics Network for making the stress expression data sets available.
This work was supported by the Michigan State University Intramural Research Grant Program (grant no. 06–IRGP–875) and the National Science Foundation (grant nos. DBI–0638591 and MCB–00749634) to S.-H.S.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Shin-Han Shiu (shius@msu.edu).
The online version of this article contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 796–815 [DOI] [PubMed] [Google Scholar]
- Aravind L, Dixit VM, Koonin EV (2001) Apoptotic molecular machinery: vastly increased complexity in vertebrates revealed by genome comparisons. Science 291 1279–1284 [DOI] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16 1667–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz JO, Hazen SP, Michael TP, Morris GP, Baxter IR, Hu TT, Chen H, Werner JD, Nordborg M, Salt DE, et al (2007) Genome-wide patterns of single-feature polymorphism in Arabidopsis thaliana. Proc Natl Acad Sci USA 104 12057–12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz JO, Nordborg M (2003) The impact of genomics on the study of natural variation in Arabidopsis. Plant Physiol 132 718–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Vermunt JK, Roos DS (2007) Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS ONE 2 e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Durand D, Farach-Colton M (2000) NOTUNG: a program for dating gene duplications and optimizing gene family trees. J Comput Biol 7 429–447 [DOI] [PubMed] [Google Scholar]
- Clark RM, Schweikert G, Toomajian C, Ossowski S, Zeller G, Shinn P, Warthmann N, Hu TT, Fu G, Hinds DA, et al (2007) Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317 338–342 [DOI] [PubMed] [Google Scholar]
- Doebley J, Lukens L (1998) Transcriptional regulators and the evolution of plant form. Plant Cell 10 1075–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortna A, Kim Y, MacLaren E, Marshall K, Hahn G, Meltesen L, Brenton M, Hink R, Burgers S, Hernandez-Boussard T, et al (2004) Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol 2 E207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao LZ, Innan H (2004) Very low gene duplication rate in the yeast genome. Science 306 1367–1370 [DOI] [PubMed] [Google Scholar]
- Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296 92–100 [DOI] [PubMed] [Google Scholar]
- Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB (2001) Molecular evidence for the early colonization of land by fungi and plants. Science 293 1129–1133 [DOI] [PubMed] [Google Scholar]
- Holub EB (2001) The arms race is ancient history in Arabidopsis, the wildflower. Nat Rev Genet 2 516–527 [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R (2003) Parallel evolution by gene duplication in the genomes of two unicellular fungi. Genome Res 13 794–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K (2007) The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J 50 347–363 [DOI] [PubMed] [Google Scholar]
- Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16 111–120 [DOI] [PubMed] [Google Scholar]
- Kovalchuk I, Kovalchuk O, Kalck V, Boyko V, Filkowski J, Heinlein M, Hohn B (2003) Pathogen-induced systemic plant signal triggers DNA rearrangements. Nature 423 760–762 [DOI] [PubMed] [Google Scholar]
- Leister D (2004) Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet 20 116–122 [DOI] [PubMed] [Google Scholar]
- Lespinet O, Wolf YI, Koonin EV, Aravind L (2002) The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res 12 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockton S, Gaut BS (2005) Plant conserved non-coding sequences and paralogue evolution. Trends Genet 21 60–65 [DOI] [PubMed] [Google Scholar]
- Lucht JM, Mauch-Mani B, Steiner HY, Metraux JP, Ryals J, Hohn B (2002) Pathogen stress increases somatic recombination frequency in Arabidopsis. Nat Genet 30 311–314 [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290 1151–1155 [DOI] [PubMed] [Google Scholar]
- Lynch M, Force A (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, Van de Peer Y (2005) Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci USA 102 5454–5459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15 809–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8 1113–1130 [DOI] [PubMed] [Google Scholar]
- Mondragon-Palomino M, Gaut BS (2005) Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol Biol Evol 22 2444–2456 [DOI] [PubMed] [Google Scholar]
- Moore RC, Purugganan MD (2003) The early stages of duplicate gene evolution. Proc Natl Acad Sci USA 100 15682–15687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Purugganan MD (2005) The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol 8 122–128 [DOI] [PubMed] [Google Scholar]
- Nakazato T, Jung MK, Housworth EA, Rieseberg LH, Gastony GJ (2006) Genetic map-based analysis of genome structure in the homosporous fern Ceratopteris richardii. Genetics 173 1585–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39 121–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S (1970) Evolution of Gene Duplication. Springer-Verlag, New York
- Oliver MJ, Tuba Z, Mishler BD (2000) The evolution of vegetative desiccation tolerance in land plants. Plant Ecol 151 85–100 [Google Scholar]
- Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA, Harrison K, Wulff BB, Jones JD (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91 821–832 [DOI] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Chapman BA (2004) Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci USA 101 9903–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes J, Vandepoele K, Simillion C, Saeys Y, Van de Peer Y (2003) Investigating ancient duplication events in the Arabidopsis genome. J Struct Funct Genomics 3 117–129 [PubMed] [Google Scholar]
- Remm M, Storm CE, Sonnhammer EL (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol 314 1041–1052 [DOI] [PubMed] [Google Scholar]
- Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud PF, Lindquist EA, Kamisugi Y, et al (2008) The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319 64–69 [DOI] [PubMed] [Google Scholar]
- Rizzon C, Ponger L, Gaut BS (2006) Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput Biol 2 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostoks N, Borevitz JO, Hedley PE, Russell J, Mudie S, Morris J, Cardle L, Marshall DF, Waugh R (2005) Single-feature polymorphism discovery in the barley transcriptome. Genome Biol 6 R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquier S, Friedman C, Delettre C, van den Engh G, Blancher A, Crouau-Roy B, Trask BJ, Giorgi D (1998) A gene recently inactivated in human defines a new olfactory receptor family in mammals. Hum Mol Genet 7 1337–1345 [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406–425 [DOI] [PubMed] [Google Scholar]
- Seoighe C, Gehring C (2004) Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet 20 461–464 [DOI] [PubMed] [Google Scholar]
- Shiu SH, Bleecker AB (2001) Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc Natl Acad Sci USA 98 10763–10768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu SH, Byrnes JK, Pan R, Zhang P, Li WH (2006) Role of positive selection in the retention of duplicate genes in mammalian genomes. Proc Natl Acad Sci USA 103 2232–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu SH, Karlowski WM, Pan R, Tzeng YH, Mayer KF, Li WH (2004) Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 16 1220–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu SH, Shih MC, Li WH (2005) Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol 139 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simillion C, Vandepoele K, Van Montagu MC, Zabeau M, Van de Peer Y (2002) The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci USA 99 13627–13632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313 1596–1604 [DOI] [PubMed] [Google Scholar]
- van Dongen S (2000) Graph clustering by flow simulation. PhD thesis. University of Utrecht, Utrecht, The Netherlands
- Vision TJ, Brown DG, Tanksley SD (2000) The origins of genomic duplications in Arabidopsis. Science 290 2114–2117 [DOI] [PubMed] [Google Scholar]
- Walsh JB (1995) How often do duplicated genes evolve new functions? Genetics 139 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A (2007) Natural history and evolutionary principles of gene duplication in fungi. Nature 449 54–61 [DOI] [PubMed] [Google Scholar]
- Wettenhall JM, Smyth GK (2004) limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics 20 3705–3706 [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Gouy M, Yang YW, Sharp PM, Li WH (1989) Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data. Proc Natl Acad Sci USA 86 6201–6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Friedman C, Williams EM, Ross JA, Tonnes-Priddy L, Trask BJ (2002) Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum Mol Genet 11 535–546 [DOI] [PubMed] [Google Scholar]
- Zhang L, Gaut BS (2003) Does recombination shape the distribution and evolution of tandemly arrayed genes (TAGs) in the Arabidopsis thaliana genome? Genome Res 13 2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.