

Abstract
The enzyme thymidylate synthase (TS) catalyzes the reductive methylation of 2′-deoxyuridine 5′-monophosphate (dUMP) to 2′-deoxythymidine 5′-monophosphate. Using kinetic and x-ray crystallography experiments, we have examined the role of the highly conserved Tyr-261 in the catalytic mechanism of TS. While Tyr-261 is distant from the site of methyl transfer, mutants at this position show a marked decrease in enzymatic activity. Given that Tyr-261 forms a hydrogen bond with the dUMP 3′-O, we hypothesized that this interaction would be important for substrate binding, orientation, and specificity. Our results, surprisingly, show that Tyr-261 contributes little to these features of the mechanism of TS. However, the residue is part of the structural core of closed ternary complexes of TS, and conservation of the size and shape of the Tyr side chain is essential for maintaining wild-type values of kcat/Km. Moderate increases in Kms for both substrate and the cofactor upon mutation of Tyr-261 arise mainly from destabilization of the active conformation of a loop containing a dUMP-binding arginine. Besides binding dUMP, this loop has a key role in stabilizing the closed conformation of the enzyme and in shielding the active site from bulk solvent during catalysis. Changes to atomic vibrations in crystals of a ternary complex of E. coli Tyr261Trp are associated with a greater than 2000-fold drop in kcat/Km. These results underline the important contribution of dynamics to catalysis in TS.
The bisubstrate enzyme thymidylate synthase (TS)1 utilizes the cofactor CH2H4folate in catalyzing the reductive methylation of dUMP into dTMP, a nucleotide required for DNA synthesis. The indirect but essential role of TS in DNA synthesis makes the enzyme an important drug target and thus, an exhaustively studied system. The enzyme is an obligate dimer in which each active site has residues from both subunits. The combination of kinetic analysis and subsequent structural characterization of mutant enzymes that display altered catalytic efficiency has done much to elucidate the ordered Bi Bi kinetic mechanism of TS (Figure 1). A complete understanding of the mechanism of TS relies on defining the roles of conserved residues that participate in catalysis.
Figure 1.
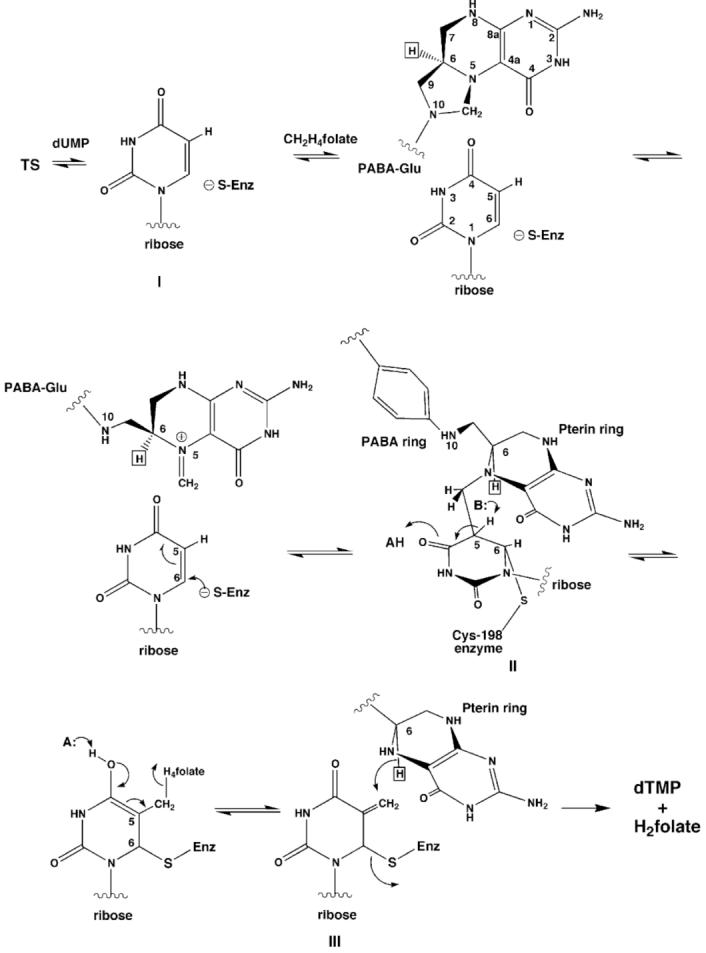
Schematic showing the minimum mechanism for TS catalysis.
Our study focuses on the highly conserved residue Tyr-261 that participates in substrate binding. In TS, Tyr-261 participates with His-259 in the only direct hydrogen bonding contacts between the ribose ring of dUMP and the enzyme (1). This residue is highly conserved: it is absent only in TS from Methanococcus jannaschii, a highly divergent TS with only 7% sequence identity to TS from Escherichia coli TS (EcTS) (2). In Lactobacillus casei TS (LcTS) almost all amino acid substitutions of Tyr-261 yield enzymes with less than 1% of wild-type activity; the only exception in an initial screen of 14 amino acid substitutions (all possible substitutions except Ala, Phe, Ile, Cys, and Lys) was methionine (3). We have employed x-ray crystallography and kinetic analysis to explore the role of Tyr-261 in substrate specificity, binding affinity and orientation. Further, we have solved and compared high-resolution crystal structures of wild-type EcTS and E. coli Y261(209)W2 in complexes with dUMP and a cofactor analog to assess the role Tyr 261 may have on cofactor binding and orientation and atomic thermal parameters. Our analysis shows that Tyr-261 is a linchpin of the closed enzyme and that altering its size and shape may subvert the protein dynamics required to drive catalysis.
EXPERIMENTAL PROCEDURES
Our approach to understanding the roles of conserved residues has been to mutate them to all possible alternatives using a synthetic LcTS gene that allows DNA cassettes containing mixtures of 32 codons to be constructed at any particular amino acid position (4). High-level expression of these variants in a TS-deficient strain of E. coli defines substitutions that complement the deficiency, and indicates interesting subsets of mutants for steady-state kinetic analysis (5). Crystal structures of the dUMP-bound LcTS mutants illustrate the structural basis for ligand binding effects. If a mutation affects kcat, the structure of an analog of the reaction intermediate preceding the rate-determining step is most informative. In this case, the analogous EcTS mutant is usually made, since EcTS analogs of reaction intermediates II and III (Figure 1) crystallize readily and diffract to higher resolution than the complexes from other TS species.
Construction, purification and kinetic analysis of mutants
Mutant LcTSs were constructed by cassette mutagenesis utilizing plasmids pSCTS9 containing a synthetic TS gene (4) as described previously (5). Mutants were expressed in E. coli χ2913recA (thy-), which lacks a gene for TS, and purified using automated sequential chromatography on phosphocellulose and hydroxylapatite (6). Site-directed mutagenesis of E. coli Thy A was done using the QuickChange site-directed mutagenesis kit (Stratagene, LaJolla, CA).
Steady-state kinetic parameters KmdUMP, KmCH2H4folate and kcat for dTMP formation were determined by monitoring the change in absorbance at 340 nm at 25° C that results from formation of H2folate, as described previously (7), using a Hewlett-Packard Model 8452A diode-array spectrophotometer. For determining the KmCH2H4folates for the LcTS wild-type and mutant proteins, the concentration of dUMP was fixed at a value between 100 and 300 mM as the concentration of CH2H4folate was varied. For measuring the KmCH2H4folate of EcTS Y261(209)W, a fixed dUMP concentration of 500 mM was used. Very high concentrations of cofactor (but not of dUMP) inhibit EcTS (8) and LcTS (unpublished); therefore we were careful not to use the regions of the Michaelis-Menten plots where the reaction rates began to decrease as a function of cofactor concentration in our calculations of kcat or KmCH2H4folate.
For calculating KmdUMPs, we fixed the concentrations of CH2H4folate at values that we estimated were saturating but not inhibitory. For wild-type LcTS this concentration was 150 mM, for the LcTS mutants, this value was 300 mM, and for E. coli Y261(209)W, we used a fixed concentration of 1mM. As a precaution we made sure the kcats derived from these Michaelis-Menten plots were the same as the kcats derived from the plots for the corresponding KmCH2H4folates, since we knew the data for the latter plots weren’t affected by substrate inhibition.
Steady-state kinetic parameters for methylation of UMP and ddUMP were also determined spectrophotometrically. UMP and ddUMP were very poor substrates for both wild-type LcTS and for the Y261 mutants. In order to measure initial reaction rates we needed to use fixed concentrations of CH2H4folate of 500mM or, in the case of KmUMP for Y261F, 1mM. We didn’t measure KmCH2H4folate for methylation of these substrates, so can’t be sure that the high fixed cofactor concentrations were not inhibitory. For methylation of ddUMP, kcats were only 2 to 3-fold less than for methylation of dUMP, thus substrate inhibition likely did not significantly affect the kinetic measurements. However, for methylation of UMP by Y261F, kcat was 70-fold less than for methylation of dUMP; substrate inhibition may have contributed in part to this impaired kcat. However, even if it were assumed that the kcats for Y261F methylation of UMP and dUMP were equal, the selectivity of Y261F for dUMP would still be almost as high as for wild-type LcTS (5.1 versus 5.7).
Dissociation constants for dUMP were determined by displacement of pyridoxal 5′-phosphate (PLP) from the LcTS-PLP complex as described (9).
Crystallization and X-ray diffraction
The LcTS variants were co-crystallized with dUMP at room temperature by vapor diffusion against 20mM phosphate buffer (pH 7.0) from protein solutions in the same buffer containing 15-20 mM ammonium sulfate, DTT, and 38mM dUMP. Hexagonal bipyramidal crystals isomorphous to the native binary complex crystal form grew to full size (0.5 × 0.5 × 0.75 mm) in approximately three days.
X-ray diffraction data for the LcTS binary complexes were collected using an RAXIS-II image plate detector and a Rigaku rotating anode generator operating at 50 kV and 300 mA, with λ=1.54178Å. Data were collected as a series of 1.5 or 2.0° oscillation frames of 20 and 25 minutes exposure, respectively. Crystals did not survive transfer to cryosolvents, so data were collected at room temperature. The crystals did not decay appreciably during data collection. Data were reduced using either the RAXIS-II software (10, 11) or Denzo/Scalepack from the HKL suite of programs (12) (Table 1).
Table 1.
Crystallographic Refinement Statistics for Tyr-261 mutants
| Statistic | Y261F | Y261M | Y261A | Y261W | nativea |
|---|---|---|---|---|---|
| Unit cell a(b)(Å) | 78.5 | 78.5 | 78.6 | 78.7 | 78.6 |
| c (Å) | 241.8 | 240.9 | 241.0 | 242.0 | 240.2 |
| Resln (Å) | 39.4-2.4 | 39.3-2.4 | 39.3-2.5 | 39.5-2.4 | 2.5 |
| Data cutoff (σ) | 0 | 0 | 0 | 0 | |
| High resln. Bin (Å) | 2.55-2.4 | 2.55-2.4 | 2.66-2.5 | 2.55-2.4 | |
| % Completeness | 91.5(87.5)b | 94.5(87.2) | 79.8(67.3) | 88.7(76.8) | |
| Avg. I/σ(I) | 17.4(3.1) | 16.7(3.4) | 11.5(2.2) | 11(2.3) | |
| # Reflections | 16639 | 17124 | 12873 | 16205 | |
| Rcryst (%)c | 17.4(21.5) | 20.3(35.1) | 18.3(33.6) | 18.6(29.4) | |
| Rfree (%) | 23.6(32.8) | 27(42.2) | 25.1(37.9) | 25.0(39.8) | |
| <B>all (Å2) | 43 | 36 | 45 | 39 | 30 |
| <B>core(Å2) | 29 | 18 | 31 | 22 | 21 |
| <B>dUMP (Å2) | 75 | 36 | 83 | 60 | 26 |
| <B>res.21-27 (Å2) | 78 | 52 | 69 | 83 | 45 |
| Rmsdbond (Å) | .022 | .029 | .026 | .026 | |
| Rmsdangle (°) | 2.2 | 2.6 | 2.5 | 2.4 |
Structure from Finer-Moore et al. (1)
Statistics for high-resolution bin in parentheses
Rmerge = Σ|I-<I>|/Σ|<I>|; negative intensities included as zero
EcTS Y261(209)W and wild-type EcTS were both co-crystallized with dUMP and CB3717 by vapor diffusion against 1.44M Na citrate, pH 7.39 (HEPES) from enzyme solutions containing 5.2mg/ml protein containing 17 mM phosphate buffer, 3.3 mM DTT, 3.3mM dUMP, 3.3 mM CB3717 and either 5% ethyl acetate or 40% n-propanol. Crystals were transferred to cryosolvent before data collection. Data from single crystals of the mutant and wild-type complexes were collected at -170° C on a QUANTUM image plate at beamline 8.3.1 at the Advanced Light Source, LBNL using a wavelength of 1.0Å. Data were processed and scaled using the HKL processing software (12) (Table 2).
Table 2.
Crystallographic Data and Refinement Statistics for EcTS structures
| Statistic | Y261(209)W | Wild-type EcTS |
|---|---|---|
| ligands | dUMP, CB3717 | dUMP, CB3717 |
| Space group | P63 | P63 |
| Unit Cell a(b), c (Å) | 125.8, 66.94 | 125.4, 66.6 |
| Resln. (Å) | 45.8 -1.3 | 45.6-1.3 |
| High resln. Bin (Å) | 1.32-1.3 | 1.33-1.3 |
| completeness (%) | 99.9 (99.9)a | 99.9 (100) |
| Rmerge(%)b | 8.0 (82.3) | 5.6 (60.6) |
| Avg. I/σ(I) | 25.1 (1.9) | 28.5 (2.7) |
| Data cutoff for refinement (σ) | 0 | 0 |
| reflections in refinement (#,%) | 146991, 99.9 | 141142, 96.6 |
| Rcryst | 15.1 (23.8) | 15.1 (22.0) |
| Rfree | 17.0 (24.4) | 16.6 (25.6) |
| <Biso>all (Å2) (prot. 1, prot. 2) | 13, 17 | 14, 18 |
| <Biso>core (Å2) (prot. 1, prot. 2) | 11, 15 | 12, 17 |
| <Biso> dUMP (Å2) | 12, 14 | 11, 13 |
| <Biso>CB3717 (Å2) | 16, 23 | 14, 18 |
| <Biso>res. 19-25 (Å2) | 28, 23 | 15, 15 |
| Rmsdbond (Å) | 0.01 | 0.01 |
| Rmsdangle (°) | 1.37 | 1.38 |
Statistics for high-resolution bin in parentheses
Rmerge = Σ|I-<I>|/Σ|<I>|; negative intensities included as zero
Structure solution and refinement
Structures of LcTS binary complexes were solved by the difference Fourier method (13) and refined using X-PLOR (14) or CNS (15) according to the following protocol: i) rigid-body refinement of the wild-type TS-dUMP structure (PDB code 1TDM) with dUMP and waters removed ii) manual rebuilding of the mutated side chains and building of the substrate into (Fo-Fc)αcalc density maps in O (16) and iii) several rounds of simulated annealing (17), positional, and atomic B-factor refinement alternated with manual rebuilding of out-of-density side chains and the addition of waters. Final cycles of refinement were done with REFMAC5 (18) using a maximum-likelihood target residual. (Table 1).
The complex of EcTS Y261(209)W with dUMP and CB3717 was solved by molecular replacement using EPMR (19), starting with the structure of wild-type EcTS·dUMP·CB3717 with ligands and waters removed (PDB accession code 2TSC). The structure of the isomorphous wild-type EcTS·dUMP·CB3717 crystal, grown under identical conditions as the Y261(209)W complex, was solved by difference Fourier methods, starting from the mutant ternary complex structure. The structures of both the wild-type and Y261(209)W ternary complexes were refined with CNS and REFMAC5 (18) using a maximum-likelihood target residual. Atomic anisotropic B-factors were refined during the final cycles (20) (Table 2).
Structure analysis
In order to compare structures of Y261(209) variants with their wild-type counterparts, Cαs in the structural cores of compared structures were overlapped using LSQMAN from the DEJAVU suite of programs. A previously described method (21), which uses difference-distance matrices to identify domains of conserved structure in two coordinate sets, was utilized to define the structural cores used for overlapping the structures. The rms errors in interatomic distances were estimated using an empirical approach that takes into account the resolution of the structure and the atomic B-factors of the atoms (22). Difference distance plots were generated using the DDMP software from the Center for Structural Biology at Yale University. Ligand-protein interactions were determined using LIGPLOT (23) and the program DISTANG from the CCP4 suite of programs.
Since B-factors in crystal structures are not only a function of inherent mobility of the atoms in the protein, but also depend on experimental conditions used for data collection, direct comparisons of B-factors between structures could be misleading. Therefore, when assessing changes in mobility resulting from a mutation, we computed relative B-factors by averaging the isotropic B-factors for atoms in the segment or residue of interest and dividing by the average B-factor of the buried helix in the core of the protein, as described previously (24) or by the overall average B-factor for the protein. Comparisons of the relative B-factors were only done between isomorphous structures whose data had been collected and processed the same way and to approximately the same resolution. Protein atoms in the E. coli TS ternary complex structures were tested for correlated motions by pairwise comparisons of the projections of their anisotropic displacement parameters along their interatomic vectors using the program ANISOANL from the CCP4 suite (25).
Figures 4, 5a and 6 were made using BOBSCRIPT (26, 27), Figures 3b a, 5b, 7e and 7f were prepared with the graphics program PyMOL (28), and Figure 3a was made with LIGPLOT (23). Figures 7a, b, c and d were produced with ANISOANL (33).
Figure 4.

Stereo plots of the overlapped structures of the dUMP complexes of wild-type LcTS (black), Y261F (dark gray), Y261M (medium gray), Y261W (light gray), and Y261A (white) in the region of the dUMP binding site. The left two panels comprise a divergent-eyes stereo pair and the right two panels comprise a crossed-eyes stereo pair.
Figure 5.

a. Stereo plot of 3σ level (Fo1-Fo2)αcalc density around the mutated residue Y261(209)W in protomer 1, where Fo1 comes from Y209W (shown in light bonds) and Fo2 is from the wild-type structure (shown in dark bonds). Positive density contours are shown with solid lines while negative density contours are shown with dashed lines. The left two panels comprise a divergent-eyes stereo pair and the right two panels comprise a crossed-eyes stereo pair. b. Density in the active site of Y209W protomer 1 from a 1.3Å (2Fo-Fc)αcalc, map contoured at 0.5 σ.
Figure 6.

Stereo plot of EcTS Y261(209)W·dUMP·CB3717 (light gray) in the region of the mutation, overlaid with wild-type EcTS·dUMP·CB3717 (dark gray). Hydrogen bonds are shown as dashed lines. The LcTS numbering convention is used. The left two panels comprise a divergent-eyes stereo pair and the right two panels comprise a crossed-eyes stereo pair.
Figure 3.

a. A diagram showing dUMP and CH2H4folate interactions with EcTS. Interactions are derived from the ternary complex EcTS·5-fluoro-dUMP·CH2H4folate (52) (PDB accession code 1TSN). Carbon atoms are black while heteroatoms are dark gray (oxygen, phosphorous), light gray (nitrogen) or white (sulfur). The LcTS numbering convention is used. LcTS residue numbers, m, can be converted to EcTS residue numbers, n, by n=m-2, for m less than or equal to 89, and n=m-52 for m greater than 89. b. Slice through the active site cavity of EcTS-dUMP-CB3717 rendered as cyan space-filling atoms for dUMP, purple space-filling atoms for protein residues from the same protomer as dUMP, and blue space-filling atoms for dUMP ligands from the second protomer [R178′ (126′) and R179′ (127′)]. Y261(209) is enclosed by D221(169) above, the phosphate-binding loop below, and on either side, the C-terminal residues (shown as stick bonds) and R178′ (126′). The cofactor analog, CB3717 and Trp-85(83), in the same layer of the protein as the C-terminus, are also plotted with stick bonds. The LcTS numbering convention is used in the figure.
Figure 7.

Grid plots showing degree of correlation in anisotropic displacement parameters between atoms identified by the (x, y) labels of the plot (EcTS numbering), for all atoms in one protomer of dimeric wild-type EcTS·dUMP·CB3717 (panel A) and Y261(209)W·dUMP·CB3717 (panel B). Correlation is indicated by similar values for the projections of the anisotropic displacement parameters along the interatomic vector (“deltas”- see text). The deltas within a small square have been averaged and the square is shaded according to the average delta value, with lighter shades indicating greater correlation. Blocks of light colored squares along the diagonal of a plot identify protein segments that potentially move as rigid bodies. Segments whose putative rigid body vibrations may be disrupted by the Y261(209) mutation are marked by red stars. Higher resolution plots of the lower left regions of (A) and (B) are shown in panels (C) and (D), respectively. These illustrate that in wild-type EcTS ternary complex, residues 22(20)-47(45) may vibrate as a rigid body, whereas in the Y261(209)W complex, resides19(17)-27(25) clearly vibrate independently of neighboring residues. The average degree of correlation of atomic B-factors (deltas) of a residue with those of its four nearest neighbors in the sequence (equivalent to the diagonal squares in panels A and B) is mapped onto a ribbon drawing of a wild-type EcTS protomer in (E) and a Y261(209)W protomer in (F), using the following color scheme: black, dark blue, purple, cyan, light blue and white, ranging in order from least to most correlated.
RESULTS
Substrate Specificity: The Y261η-O hydrogen bond as a specificity determinant
Besides the cluster of arginines and the conserved serine that form the phosphate-binding site for bound nucleotide in TS, only three other protein side chains form hydrogen bonds with dUMP. The invariant residue Asn-229, which provides the sole sidechain hydrogen bonds to the pyrimidine ring, is essential for discriminating against dCMP (29, 30). In order to test whether the Tyr-261 hydrogen bond to the 3′-O of dUMP similarly contributes to substrate specificity, we kinetically assayed wild-type and mutant enzymes with two alternate substrates, ddUMP and UMP, that differ from dUMP by the substitution pattern at the 3′ position of the ribose ring (Figure 2). These experiments served two purposes. First, the experiments could determine whether the Tyr-261 η-O hydrogen bond to the 3′-O of dUMP is a major factor in specificity for dUMP. Secondly, kcat/Km for ddUMP provides an estimate of the contribution of the hydroxyl group on the ribose ring of dUMP to the substrate binding step of the reaction that cannot be obtained by single mutants at Y261. The protein engineering experiment that would provide this same estimate would entail removing both of the hydrogen bonding side chains, His-259 and Tyr-261 (Figure 3a).
Figure 2.
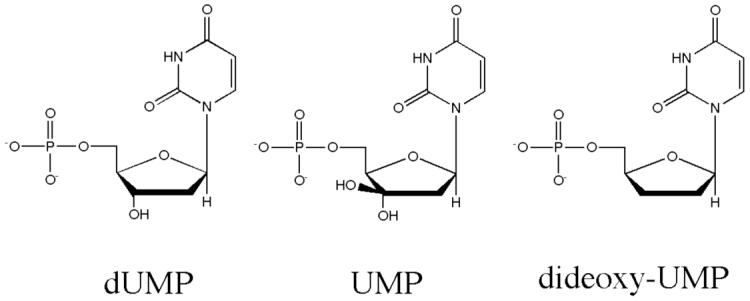
Chemical structures of dUMP, UMP and ddUMP.
In order to test whether Tyr-261 contributes to substrate specificity we calculated selectivity factors, ln[(kcat/KmdUMP)/( kcat/KmddUMP)] and ln[(kcat/KmdUMP)/( kcat/KmUMP)] for wild-type LcTS and for Y261F (Table 3). The selectivities for dUMP over ddUMP and UMP are the same or greater for Y261F as for the wild-type enzyme, showing that Tyr-261 η-O does not significantly enhance selectivity of LcTS for dUMP over either alternate substrate.
Table 3.
Kinetic constants for alternate substrates
| Substrate | Parameter | Wt TS | Y261F | Y261A |
|---|---|---|---|---|
| dUMP | Km, μM | 6+/- 2 | 6.6+/- 0.6 | 34+/- 6 |
| kcat, s-1 | 3.8+/- 0.2 | 1.7+/- 0.04 | 0.026+/- 0.001 | |
| UMP | Km, μM | 1165+/-136 | 1411+/- 101 | n.d.a |
| kcat, s-1 | 2.5+/- 0.1 | 0.022+/- .001 | n.d. | |
| Selectivityb | 5.7+/- 0.4 | 9.7+/- 0.1 | — | |
| ddUMP | Km, μM | 836+/- 143 | 1630+/- 216 | n.d. |
| kcat, s-1 | 1.17+/- 0.07 | 1.07+/- 0.06 | n.d. | |
| Selectivity | 6.1+/- 0.4 | 6.0+/- 0.2 | — |
n.d.= no detectable activity
Selectivity = ln[(kcat/KmdUMP)/( kcat/Kmx)], where x=UMP or ddUMP
Table 3 lists kinetic parameters (kcat, Km for substrate), for methylation of dUMP, ddUMP and UMP for wild-type LcTS, Y261F and Y261A. The Km for ddUMP is 139-fold higher than Km for dUMP in wild-type TS and kcat/Km is reduced more than 900-fold. As a consequence of the ordered Bi Bi mechanism used by TS, in which dUMP binds before cofactor, kcat/KmdUMP measured under saturating concentrations of cofactor is equal to the dUMP-binding rate, k1, and is related to the energy required for the rate-determining step in productive dUMP binding (31). Therefore, the free energy contribution of the dUMP 3′-hydroxyl group to the substrate binding step can be estimated by ΔΔGs = -RTln[(kcat/KmddUMP)/( kcat/KmdUMP)] (32) and is ∼4 Kcal/mol. Because of the redundancy of having two groups hydrogen bonding to the ribose 3′-hydroxyl, eliminating the Tyr-261 hydrogen bond by itself has a much smaller impact on ΔGs, <1 Kcal/mol (see below).
Enzyme kinetics: Tyr-261 mutations increase Km for both dUMP and CH2H4folate
In the initial assay of 14 Tyr-261 variants, only Met complemented TS-deficient E. coli, suggesting that the hydrophobicity and size of the residue at position 261 were important for function (5). To explore in greater detail the essential properties of Tyr-261 and their role in catalysis, we substituted Tyr-261 in LcTS with four hydrophobic residues that had side chains of various sizes, ranging from Ala (a single methyl group) to Trp (the bulkiest amino acid). The impact of mutagenesis on substrate binding was moderate and similar for each substitution (Table 4). KmdUMPs were the same within experimental error for wild-type LcTS, Y261F and Y261M and less than 10-fold higher for Y261A and Y261W. KmCH2H4folates increased by 4 to 13-fold. The effects of mutagenesis on kcat were more varied and correlated with the size of the substituted side chain. Phe, differing from wild-type residue Tyr by a hydroxyl group, had a kcat two-fold less than that of wild-type LcTS, Met was 15-fold less active, while the smallest substitution, Ala, showed a 146-fold decrease in kcat. Substitution of the larger residue Trp decreased kcat by 173-fold.
Table 4.
Kinetic constants for L. casei Tyr-261 mutants and E. coli Y209W
| Mutant | kcat, s-1 | KmdUMP, μM | KmCH2H4folate, μM | KmdUMP/KmCH2H4folate | KddUMP |
|---|---|---|---|---|---|
| WT LcTS | 3.8+/- 0.02 | 6+/- 2 | 17+/- 3 | 0.35 | 0.39+/- 0.05 |
| Y261F | 1.7+/- 0.04 | 6.6+/- 0.6 | 69+/- 13 | 0.08 | 0.44+/- 0.65 |
| Y261M | 0.26+/- 0.02 | 7+/- 2 | 68.7 | 0.10 | |
| Y261A | 0.026+/- .001 | 34+/- 6 | 221+/- 5 | 0.15 | |
| Y261W | 0.022+/- .001 | 78+/- 11 | 119.9 | 0.65 | |
| WT EcTSa | 8.8 | 4.1+/- 0.4 | 13.6+/- 0.2 | 0.29 | |
| Y209W | 0.05+/- 0.01 | 118+/- 40 | 172 +/- 26 | 0.68 |
Data from Reyes et al. (51).
Except for Y261W, the proportional increases in KmCH2H4folate were greater than those of KmdUMP, as seen by the wild-type and mutant ratios KmdUMP/KmCH2H4folate. This result is unusual for mutations of a dUMP-binding residue that does not also bind cofactor; such mutations generally have a significantly larger impact on KmdUMP than on KmCH2H4folate(33, 34). While Tyr-261 does not directly bind to the cofactor, mutations of this residue can indirectly affect KmCH2H4folate, presumably by changing the conformation or degree of ordering of dUMP or the neighboring residues that together form the cofactor-binding surface. In order to test this hypothesis we have determined the crystal structures of Y261A, Y261W, Y261M and Y261F in complexes with dUMP.
TS binds to its ligands in an ordered mechanism in which dUMP binds first to a preformed site. Subsequently, CH2H4folate binds and triggers a protein conformational change that helps open the cofactor imidazolidine ring, position the cofactor pterin ring on top of dUMP, and bring several residues into direct or water-mediated contact with the cofactor (35). Tyr-261 packs against Asp-221, which is a major cofactor-binding residue, a loop at the phosphate-binding site (the “phosphate-binding loop”), which participates in the cofactor-induced conformational change, and dUMP, which constitutes a major part of the cofactor-binding surface (Figure 3b). One or more of these groups are perturbed in all of the Tyr-261 mutant binary complexes, accounting for their higher KmCH2H4folates. The carboxyl group of Asp-221 is pre-oriented in wild-type TS-dUMP to accept a hydrogen bond from the cofactor pterin ring, whereas in Y261A-dUMP, Y261M-dUMP and Y261W-dUMP it is rotated 30-50° from the optimum orientation, and in Y261W-dUMP, the Asp-221 carboxyl group accepts a hydrogen bond from Trp-261 ε1-N. The average relative B-factor for dUMP (scaled by the average B-factor of the protein) is approximately twice that of wild-type TS-dUMP for the binary complexes of Y261A, Y261F and Y261W (Table 1) and the phosphate-binding loop is in a different conformation in these mutant complexes (35).
Substrate Orientation: Y261 determines dUMP localization, but not orientation in LcTS-dUMP binary complexes
The orientations of dUMP in the active sites of the four variants are very similar to that in wild-type TS (Figure 4). When the structural cores of the Y261M and Y261W complexes are aligned with that of LcTS-dUMP, dUMP is seen to occupy the same binding site in each complex within experimental error (∼+/-0.2Å), where the error is calculated as previously described (22). In the refined structures of both Y261A-dUMP and Y261F-dUMP, the planes of the dUMP pyrimidine rings are rotated away from the catalytic cysteines slightly, moving dUMP-O4 ∼0.9 +/-0.4Å away from the wild-type O4 position. However, the (2Fo-Fc)αcalc density at the dUMP sites, contoured at 1σ, is diffuse, and encompasses a narrow spectrum of dUMP conformations including that seen in wild-type TS-dUMP, so that dUMP spends at least some of the time in the wild-type binding site in the binary complexes of these two variants. Except for the absence of the Tyr-261 hydrogen bond to the ribose ring, the protein-dUMP interactions in the variant complexes are the same as in the wild-type structure (Figure 3a, Table 5). Thus, the hydrogen bond between Tyr-261 η-O and dUMP 3′-O contributes to localizing dUMP in the binary complex with TS and affects KmdUMP, but it is not essential for dUMP orientation.
Table 5.
Hydrogen bond distances (Å) between protein and ligands (dUMP, CB3717) in complexes of wild-type TS and the Tyr-261 variants Met, Phe, Ala, and Trp
| DUMP atom | Protein atoma | Wt Lc | M | F | A | W | Wt Ec | Ec W |
|---|---|---|---|---|---|---|---|---|
| O1P | R218Nη2 | 2.9 | 3.0 | 2.8 | 2.8 | 2.7 | 2.8 | 2.7 |
| R179Nε | 2.9 | 2.8 | 2.9 | 2.8 | 2.9 | 2.9 | 2.9 | |
| R179Nη2 | 3.0 | 2.9 | 3.1 | 2.9 | 2.9 | 2.8 | 2.9 | |
| O2P | S219Oγ | 2.8 | 3.1 | 2.7 | 3.0 | 3.0 | 2.7 | 2.7 |
| R218Nη1 | 2.8 | 2.7 | 3.1 | 3.1 | 2.8 | 2.8 | 2.8 | |
| R178Nε | 3.3 | 3.4 | 3.3 | 3.3 | 3.2 | 2.9 | 2.9 | |
| O3P | R23Nε | 3.0 | 2.6 | 2.8 | 3.4 | 2.7 | 2.8 | 2.7 |
| R178Nη2 | 3.0 | 3.1 | 3.3 | — | 3.3 | 2.9 | 2.9 | |
| Wat8 | 2.9 | 2.4 | 2.5 | 2.5 | — | 2.8 | 2.8 | |
| O5′ | R23Nη2 | 3.0 | 3.1 | 2.9 | 2.9 | 2.8 | 2.9 | 3.1 |
| R178Nη2 | 3.2 | 3.4 | 3.5 | 3.6 | 3.2 | 3.4 | 3.5 | |
| O3′ | H259Nε2 | 2. 9 | 2.6 | 2.7 | 3.1 | 2.7 | 2.7 | 2.8 |
| Y261Oη | 2.8 | — | — | — | — | 2.7 | — | |
| W261Nε1 | — | — | — | — | — | — | 3.2 | |
| O4 | N229Nδ2 | 2.9 | 2.9 | 2.5 | 2.5 | 2.6 | 3.0 | 3.0 |
| WAT1 | 3.2 | 3.1 | 3.1 | 3.7 | 3.2 | 3.0 | 3.0 | |
| H199Cδ2 | 3.0 | 2.8 | 3.3 | 3.3 | 3.2 | 3.2 | 3.2 | |
| O2 | D221N | 2.8 | 2.8 | 2.6 | 2.6 | 2.5 | 3.0 | 3.0 |
| N3 | N229Oδ1 | 3.1 | 2.9 | 2.9 | 3.0 | 2.9 | 3.0 | 3.0 |
| CB3717 | ||||||||
| N1 | WAT2 | 2.9 | 2.9 | |||||
| NA2 | A315O | 3.0 | 3.0 | |||||
| NA2 | WAT3 | 3.1 | 3.3 | |||||
| N3 | D221Oδ2 | 2.7 | 2.7 | |||||
LcTS numbering; LcTS residue numbers, m, can be converted to EcTS residue numbers, n, by n=m-2, for m less than or equal to 89, and n=m-52 for m greater than 89.
Tyr-261 mutations decrease dUMP affinity partly through destabilization of the active conformation of a key substrate-binding loop
The decreased localization of dUMP in the crystal structures of the Y261 mutants most likely reflects decreased binding affinity for dUMP, at least in the absence of cofactor. The KddUMP for the most conservative mutant, Y261F, is 11-fold higher than for wild-type LcTS. This corresponds to a decrease of 1.4 Kcal/mol in binding energy. This number is an upper limit to the contribution of the hydroxyl group of Tyr-261 to dUMP binding affinity since mutation of Tyr-261 alters the structure and mobility of the phosphate-binding loop, which contains Arg-23, itself a major dUMP contact residue. The phosphate-binding loop is highly mobile in apo-TS, and becomes progressively better ordered as dUMP then cofactor bind. The hydrogen bond between Tyr-261 η-O and dUMP 3′-O aids in this process by orienting the Tyr aromatic ring such that it provides a complementary surface for the ordered loop (Figure 3b). In the wild-type LcTS-dUMP complex, Tyr-261 δ1-C is 3.3Å from Thr-28 γ2-C, and Tyr-261 ε1-C is 3.2Å from Asp-22 δ1-O and 3.3Å from Thr-24 γ1-O. The latter two contacts may represent weak hydrogen bonds donated by the acidic ε1-C hydrogen. In Y261F, where the Phe-261 side chain is not constrained by a hydrogen bond, the aromatic ring is shifted approximately 0.6 Å from the wild-type position such that it can no longer make the close contacts with Asp-22, Thr-24 and Thr-28, and the phosphate-binding loop is more disordered and in a different conformation than in wild-type TS-dUMP.
Figure 4 illustrates the conformations for the phosphate-binding loop seen in the binary complexes of the four variants compared to the wild-type conformation. In the crystal structures of Y261F and Y261W, this loop has much higher B-factors, relative to both the protein as a whole and relative to the average B-factor of a helix in the protein core, than does the same loop in wild-type LcTS (Table 1) and the loop has broken (2Fo-Fc)αcalc density in the Y261F, Y261W, and Y261A structures. Nonetheless, Arg-23 maintains the wild-type orientation in these variants (Figure 4) and makes hydrogen bonds to the phosphate moiety of dUMP, although its B-factors are very high. In Y261W, the wild-type orientation of the Arg-23 side chain is one of at least two discrete conformations seen in the crystal structure.
In the structure of the Y261M-dUMP complex, a new ordered water near the location of the wild-type Tyr-261 hydroxyl group donates hydrogen bonds to Asp-22 δ1-O and M261 δ-S, and accepts a hydrogen bond from R178′ η2-NH from the second monomer. By so tethering the phosphate-binding loop to another phosphate binding arginine the new water may help to order the active site. In particular, electron density for all residues, including Arg-23 in the phosphate-binding loop, is continuous and well resolved, as it is for wild-type LcTS-dUMP, and the conformation of this loop more closely resembles that of wild-type LcTS than do the loops of the other Y261 mutants (Figure 4). In Y261M-dUMP the average atomic B-factor for dUMP is the same as the average B-factor for the protein, compared to wild-type LcTS where the average B-factor for dUMP is 0.86 that of the protein as a whole. In contrast, the average B-factor for dUMP is from 1.5 to 1.8 times higher than the average B-factor for the protein in Y261W, Y261F and Y261A. Thus for the binary complex structures we determined, the Y261 mutant whose phosphate-binding loop is least perturbed from the wild-type structure also has the most well-ordered dUMP. This result supports the hypothesis that a secondary effect of Y261 mutation, perturbation of the phosphate-binding loop, contributes to reduced binding affinity for dUMP.
Cofactor-protein contacts and dUMP and cofactor orientations in ternary complexes are unchanged by the Y261(209)W mutation
In the mechanism of TS, precise alignment of substrate and cofactor during first methyl, then hydride transfer is likely to be essential for catalytic rate. Since the steady-state kinetics and binary structures of the Tyr-261 variants suggested Tyr-261 might contribute to reactant orientation during catalysis, we needed to assess the affects of mutation on the structure of intermediate II (Fig. 1). Since the ternary complex of LcTS with substrate and co-factor or co-factor analogs is not routinely accessible via crystallography, we accomplished this through the study of the ternary complexes of E. coli TS with dUMP and CB3717. The E. coli TS enzyme readily yields ternary complex crystals of high quality, and the effects of mutation on the kinetic parameters for EcTS mirror those observed for LcTS. In order to determine the high-resolution structure of an analog of intermediate II in a catalytically impaired Tyr-261 mutant, we made the Y261(209)W variant of the homologous EcTS. For Y261(209)W, KmdUMP was 29-fold higher, KmCH2H4folate was 13-fold higher, and kcat was 176-fold lower than in wild-type EcTS, implicating identical roles for Y261(209) in structure and catalysis in EcTS and LcTS.
The Y261(209)W ·dUMP·CB3717 ternary complex does not crystallize under the same conditions we used previously to crystallize wild-type EcTS ternary complexes. However, through screening we discovered a set of conditions that grew crystals that diffracted to a better resolution (1.3Å) than has been reported for any other TS structure. In order to compare Y261(209)W·dUMP·CB3717 to wild-type EcTS·dUMP·CB3717, we crystallized the latter complex using the same conditions used to crystallize the Y261(209)W complex and determined its structure to 1.3Å resolution.
The structure of the Y261(209)W complex is remarkably similar to wild-type EcTS·dUMP·CB3717. The rmsd between Cαs after superposition of the two dimers is 0.22Å. Because both the wild-type and Y261(209)W complex structures are determined to high resolution and have low atomic B-factors it is possible to reliably identify small but real differences in their conformations. Significant differences in backbone structure, identified by reference to estimated random errors in coordinates as a function of B-factor (22), are limited to three segments. The changes include i) a shift of the mutated residue, W261(209), and the neighboring residues , 262(210) - 264(212) approximately 0.5+/- 0.1 Å towards the active site in protomer 2. ii) a shift of the phosphate-binding loop, residues 21(19) - 27(25), in both protomers approximately 1.0 +/- 0.2 Å out of the active site and iii) an adjustment to the loops at the edge of the dimer interface, near an unidentified ion or molecule whose shape and intensity could indicate a carbonate ion. These changes are clearly evident in a difference map calculated with coefficients (Fo1-Fo2)αcalc, where Fo1 is a structure factor amplitude from the wild-type complex crystal and Fo2 is the corresponding amplitude from the mutant complex crystal, and αcalc is the phase calculated from the Y209W structure (Figure 5a). These changes parallel changes seen in LcTS Y261W-dUMP versus wild-type LcTS-dUMP. All the structural shifts are small and do not affect interactions of the ligand-binding residues with dUMP or CB3717.
The crystals contain one dimer per asymmetric unit, with the substrate, dUMP, and cofactor analogue, CB3717, well ordered in both protomers (Figure 5b). After superimposing the Cαs of the Y209W·dUMP·CB3717 dimer on those of the wild-type EcTS·dUMP·CB3717 dimer, the substrates and cofactor analogues of the dimer overlap their positions in wild-type EcTS·dUMP·CB3717 within experimental error. In both protomers, there is a covalent bond between 6-C of dUMP and the active site sulfhydryl. The ligand-protein interactions are conserved (Table 5). The ε1-N of Trp-261(209) donates a hydrogen bond to 3′-O of the dUMP ribose ring that is analogous to the hydrogen bond between Tyr-261(209) η-O and dUMP 3′-O in wild-type complexes. In spite of the shift in the phosphate-binding loop, the hydrogen bonds Arg-23(21) makes with dUMP, CB3717 and the C-terminus are conserved. However, the hydrogen bond donated by Arg-178′ (126′) η1-N to Y261(209) η-O in wild-type complexes is not made in Y261(209)W because Trp does not contain a compensating hydrogen-bond acceptor atom (Figure 6). Since this wild-type TS hydrogen bond is made in ternary complexes but not in open conformations of the enzyme (apo-enzyme or TS-dUMP complexes), it provides stabilizing energy to the closed enzyme and would be expected to help drive the enzymatic reaction forward.
The phosphate-binding loops at both active sites of the dimer are more mobile in Y261(209)W than in wild-type EcTS. The isotropic B-factors of the phosphate-binding loops are 2.5 and 1.5 times higher than for the protein core helices in protomers 1 and 2, respectively, in the Y261(209)W complex, while in the wild-type complex dimer the B-factors of the loops are 1.2 and 0.9 times higher than those of the core helices in protomers 1 and 2, respectively.
Protein Vibrational States: The impact of Y261 mutants on correlated anisotropic B-factors
Changes in protein vibrational states were analyzed in greater detail by comparing the refined anisotropic B-factors of Y261(209)W and wild-type EcTS. Six anisotropic thermal parameters per atom specify the dimensions and orientation of an ellipsoidal electron distribution about each atom and indicate amplitudes and directions of atomic vibrations in the crystal. Anisotropic refinement is valid for a 1.3Å resolution data set provided refinement is restrained to effectively reduce the parameter to data ratio. We used a refinement procedure that restrained bond lengths, bond angles, chirality, planarity, torsion angles, steric clashes, and isotropic B-factor variation between neighboring atoms. We monitored the R-factor for a set of data excluded from refinement (Rfree) to guard against over-fitting the data. For the Y261(209)W·dUMP·CB3717 structure, Rfree decreased from 19.1% to 17.0% when anisotropic B-factors were refined. Rfree decreased from 18.4% to 16.6% when anisotropic B-factors of wild-type EcTS·dUMP·CB3717 were refined.
For both structures, anisotropy (the ratio of the smallest to largest dimension of the ellipsoid) was small in all regions of the protein: the average anisotropy for the atoms comprising a residue was usually in the range 0.6-0.9. As a measure of the correlation in movement of any two atoms, the differences in the projections of their anisotropic thermal parameters along their interatomic vector were calculated. The differences in the projections, referred to as “deltas”, should be zero if the atoms move as a rigid body; small deltas are necessary but not sufficient evidence of rigid body motion (36). Deltas for each pair of atoms within a subunit, weighted by the average isotropic B-factor of the pair, were binned according to sequence number (5 × 5 atom pairs per bin) averaged within the bins, and plotted on a two-dimensional grid using shading to indicated magnitude of the averaged delta value. Lighter shades of gray indicated smaller deltas. The plots for one subunit of wild-type EcTS and the corresponding subunit of Y261(209)W are shown in Figure 7. The patterns of light and dark squares are very similar for wild-type and mutant enzyme, showing that the B-factors did not vary randomly, but likely contained information about rigid body vibrations in the crystal that could be related to protein normal modes. The plots for the second subunits (not shown) were also similar to each other, but different in several respects from the first subunits, reflecting the well characterized asymmetry of TS dimers.
We inspected the diagonals of the plots for stretches of residues that might vibrate as rigid bodies. Several of these corresponded to protein segments that are known to shift as part of the conformational change triggered by CH2H4folate, consistent with the observation that conformational changes in proteins are often described by one or a few low frequency normal modes (37). The plots for wild-type and Y261(209)W were noticeably different in the same four regions of each subunit of the dimer. These regions, marked by red stars in Figure 7, corresponded to four possible rigid body segments in wild-type EcTS but not in Y261(209)W: residue segments 22(20)-47(45), 195(143)-204(152), 248(196)-262(210) and 302(250)-316(264). The average weighted deltas for atom pairs within each of the four segments in wild-type EcTS were 0.15, 0.17, 0.13, and 0.17, respectively, and for the corresponding segments in Y261(209)W they were 0.44, 0.31, 0.24, and 0.29. Thus, the anisotropic B-factors for the wild-type structure were more consistent with the segments vibrating as rigid bodies than were the B-factors of the Y261(209)W structure. The segments included a segment containing the mutated residue, Trp-261(209), and two segments that pack against this residue, namely, the phosphate-binding loop and C-terminus. The fourth segment was distant from the site of the mutation, on the opposite wall of the active site cavity, and contained the catalytic cysteine, Cys-198(146).
DISCUSSION
The invariant residue Tyr-261 donates one of only two hydrogen bonds to the ribosyl 3′-hydroxyl group of dUMP, therefore a logical role for this residue in catalysis is substrate binding and orientation. Surprisingly, we have found that substitutions at this site have far less effect on dUMP orientation than mutations of other dUMP-binding residues, such as Asn-229, which anchors the pyrimidine ring, or Arg-178, which correspondingly anchors the phosphate moiety (38, 39). This could be because the ribose lies in between the tethered uridine ring and phosphate moieties, and the tethers to the latter moieties are largely responsible for maintaining the productive substrate conformation (40). Furthermore unlike Asn-229, Tyr-261 does not appear to contribute to selectivity for dUMP over other potential nucleotide substrates.
The hydrogen bond between Tyr-261 and 3′-O dUMP probably contributes less than 1.4 Kcal to dUMP binding affinity, and less than 1 Kcal to the free energy, ΔGs, of productive dUMP binding during catalysis, based on the KddUMP and KmdUMP for the most conservative mutation Y261F. This is consistent with estimates of the strength of hydrogen bonds between uncharged groups in proteins (41). However when both hydrogen bonds between protein and the ribose moiety are eliminated, by using ddUMP, which has no 3′-hydroxyl, as substrate, the free energy of binding is ∼4 Kcal higher. This ΔΔGs is likely much bigger than the sum of the ΔΔGss for eliminating each hydrogen bond separately and suggests that at least one hydrogen bond to the ribosyl 3′-O is required to orient dUMP.
All mutations of Tyr-261 resulted in higher Kms for cofactor. These increases were only partly the result of substituting new side chains for Tyr-261 per se, rather, they correlated with a change in the ordering and conformation of the phosphate-binding loop, containing Arg-23, a critical binding determinant for both substrate and cofactor. The loop is fairly disordered in the apo-enzyme and its progressive ordering during dUMP then cofactor binding is part of the ligand binding mechanism. Tyr-261 stabilizes the conformation of the loop in two ways. First, the Tyr-261 side chain is in van der Waals contact with several residues in the loop (Figure 4b). Secondly, in closed conformations of the enzyme its hydroxyl group is connected via hydrogen bonds from Arg-178′ to the conserved residue Asp-22 in the phosphate-binding loop. Perturbations to the phosphate-binding loop were more pronounced in the mutant LcTS binary complexes and in apo-Y261(209)W (unpublished) than in E. coli Y209W·dUMP·CB3717. Thus we propose that the increases in KmCH2H4folate derive from the fact that the open (apo or dUMP-bound) conformations of the mutants are more disordered and further removed from the ternary complex structures than in wild-type TS. Increases in KmCH2H4folate were also related to greater disorder of dUMP in the dUMP binary complexes since dUMP forms part of the cofactor-binding surface.
We saw no evidence from the crystal structure of EcTS Y261(209)W·dUMP·CB3717, an analog of intermediate II in Figure 1, that misorientation of reactants was responsible for the 175-fold decrease in kcat for the Tyr to Trp mutation. Ligand orientations and protein-ligand interactions are the same in this complex as in the analogous wild-type complex. For other EcTS mutants with impaired kcats, crystal structures of ternary complexes with dUMP and CB3717 have been asymmetric, with significant ligand misorientation in one of the active sites (24, 42). These crystal structures have belonged to the same space group, had the same crystal packing interactions and almost identical cell constants as the Y261(209)W·dUMP·CB3717 structure. Therefore, crystal packing in the Y261(209)W complex crystals would not be expected to completely mask effects of the mutation on orientation of the reactants and it is likely that the structure of reaction intermediate II (Figure 1) is the same in Y261(209)W as in wild-type TS. However, we can’t rule out the possibility that the solution structure of Y261(209)W would show ligand misorientation not evident in the crystal structure.
On the other hand we observed significant changes in B-factors of the Y261(209)W complex crystal structure compared to the isomorphous structure of wild-type EcTS·dUMP·CB3717, which had been solved and refined to the same resolution using identical refinement methods. These differences included a doubling of the relative isotropic B-factors for the phosphate-binding loop in one protomer and a 1.5-fold increase in these B-factors in the other. Analysis of anisotropic B-factors, which indicate direction as well as amplitude of atomic motion, revealed protein segments whose anisotropic B-factors were correlated in a way that was consistent with rigid body vibration in the wild-type complex but not in the Y261(209)W complex. These segments included the site of the mutation and segments that pack against the mutated residue, and also a segment on the opposite side of the active site cavity, which contained the catalytic Cys. This result suggests that mutations of Y261(209) change protein dynamics not only in the immediate vicinity of the mutation, but also in regions of the protein close to the site of methyl and hydride transfer.
The changes in protein dynamics in that we deduce from the crystallographic B-factors could affect kcat in a number of ways. The kcats of the Y261(209) mutants may have decreased because of entropic effects. After the binding of CH2H4folate and the closure of the active site to form a covalent ternary complex, only small reorientations of substrate and cofactor occur as first the 5-N-methyl group and then the 6-C hydride are transferred from the cofactor to the substrate (43). It is critical that the enzyme maintain a closed conformation in order for the reactants to be properly aligned with each other and with groups on the protein (42). Tyr-261 has a key role in stabilizing the closed enzyme conformation since it serves as a hydrophobic core against which cofactor-binding segments pack, and it participates in a hydrogen bonding scheme that tethers the phosphate-binding loop to the rest of the protein. Thus mutation of Tyr-261 may decrease kcat by allowing a broader range of nonproductive conformational states, that is, increasing ground state entropy, at one or more steps in the enzyme reaction (44). Consistent with this hypothesis is the fact that mutations of Tyr-261 to smaller residues Phe, Met or Ala reduce kcats by amounts that are roughly proportional to the decrease in packing densities of the substituted side chains, and the decreases in specificity for these three variants (ΔΔGs = -RTln[(kcat/KmCH2H4folate)mut/(kcat/KmCH2H4folate)w.t.] = -RTln(k1mut/k1wt,), where k1 = rate of productive dUMP binding under saturating concentrations of CH2H4folate) are on the same order as the decreases in ΔG of unfolding for deletion mutants of residues in the hydrophobic core of a protein (∼1-4 kcal) (45) (46, 47). Arguing against this explanation is the fact that the active sites of the Y261(209)W structure are well ordered except for the phosphate-binding loop, which is far from the site of methyl and hydride transfer. The B-factors of the substrate and cofactor analogue are only slightly higher relative to those of the protein core in Y261(209)W than they are in the wild-type EcTS complex. We can’t rule out the possibility that the solution structure of the mutant complex would be more disordered.
The clearest difference between the E. coli Y261(209)W and wild-type complex structures is in the average B-factor of the phosphate-binding loop in Y261(209)W. Increased mobility of this loop was also observed in the crystal structures of the binary dUMP complexes of LcTS Y261 variants. Greater mobility of this loop could impair catalysis by admitting bulk solvent into the active site.
Finally, Tyr-261 mutations may disrupt protein motions that play a direct role in certain catalytic steps. Analysis of the anisotropic B-factors of Y261(209)W·dUMP·CB3717 clearly showed that the Y209W mutation alters correlated motions in TS. Protein dynamics enhances hydride transfer in EcTS (8) and dihydrofolate reductase (48). In dihydrofolate reductase, mutations distant from the active site, which had little effect on substrate binding, decreased the rate of hydride transfer by altering the dynamics of the enzyme (48, 49). Similarly, TS mutations such as Y261(209)W that are distant from the enzyme chemistry may decrease kcat by changing the dynamic motions contributing to hydride transfer, which is the rate determining step in the wild-type enzyme (8, 50).
ACKNOWLEDGEMENT
We thank Wayman Chan for producing E. coli Y209W and Lu Liu for his help with measuring the kinetics of the L. casei Tyr-261 mutants.
Footnotes
- TS
- thymidylate synthase
- LcTS
- Lactobacillus casei TS
- EcTS
- Escherichia coli TS
- dTMP
- 2′-deoxythimidine 5′-monophosphate
- dUMP
- 2′-deoxyuridine 5′-monophosphate
- UMP
- uridine 5′-monophosphate
- ddUMP
- 2′,3′-dideoxyuridine 5′-monophosphate
- FdUMP
- 5-fluoro-2′-deoxyuridine 5′-monophosphate
- CH2H4folate
- 5,10-methylenetetrahydrofolate
- H2folate
- 7,8-dihydrofolate
- CB3717
- 10-propargyl-5,8-dideazafolate
- DTT
- dithiothreitol
- Y261A (or M,F,W)
- Lactobacillus casei TS variant that has Ala (or Met, Phe, Trp) substituted for Tyr at residue 261
- Y261(209)W
- E. coli TS variant with Trp substituted for Tyr at residue 209 (261 in L. casei TS numbering)
- rmsd
- root mean square deviation
- (2Fo-Fc)αcalc map
- electron density map calculated with coefficients 2*|Fo|-|Fc| and phases derived from the coordinates of the structure
- ΔGs
- enzyme-substrate binding energy
- Cα
- alpha carbon
L. casei TS numbering is used in the text. In the discussion of the E. coli structures, the corresponding EcTS residue number is given in parentheses after the L. casei residue number. A “’” after a residue number signifies it contributes interactions across the dimer interface to the other protomer within the obligate dimer.
This work was supported by NIH grants CA 41323 to JFM and CA 63081 to RMS. Coordinates and structure factors for LcTS Y261A, Y261M, Y261F, Y261W, wild-type EcTS·dUMP·CB3717 and EcTS Y209W·dUMP·CB3717 have been deposited in the Protein Data Bank, with accession codes 2G89, 2G8A, 2G86, 2G8D, 2G8O, and 2G8M, respectively.
REFERENCES
- (1).Finer-Moore J, Fauman EB, Foster PG, Perry KM, Santi DV, Stroud RM. Refined structures of substrate-bound and phosphate-bound thymidylate synthase from Lactobacillus casei. J. Mol. Biol. 1993;232:1101–1116. doi: 10.1006/jmbi.1993.1463. [DOI] [PubMed] [Google Scholar]
- (2).Aurora R, Rose GD. Seeking an ancient enzyme in Methanococcus jannaschii using ORF, a program based on predicted secondary structure comparisons. Proc Natl Acad Sci U S A. 1998;95:2818–23. doi: 10.1073/pnas.95.6.2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Climie S, Ruiz-Perez L, Gonzalez-Pacanowska D, Prapunwattana P, Cho SW, Stroud R, Santi DV. Saturation site-directed mutagenesis of thymidylate synthase. J. Biol. Chem. 1990;265:18776–9. [PubMed] [Google Scholar]
- (4).Climie S, Santi DV. Chemical synthesis of the thymidylate synthase gene. Proc. Natl. Acad. Sci., USA. 1990;87:633–637. doi: 10.1073/pnas.87.2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Climie SC, Carreras CW, Santi DV. A Complete Replacement Set of Amino Acids at the C-Terminus of Thymidylate Synthase: Quantitative Structure-Activity Relationships of Mutants of an Enzyme. Biochemistry. 1992;31:6032–6038. doi: 10.1021/bi00141a011. [DOI] [PubMed] [Google Scholar]
- (6).Kealey JT, Santi DV. Purification Methods for Recombinant Lactobacillus Casei Thymidylate Synthase and Mutants: A General, Automated Procedure. Protein Expression and Purification. 1992;3:380–385. doi: 10.1016/s1046-5928(05)80039-7. [DOI] [PubMed] [Google Scholar]
- (7).Pogolotti AL, Jr., Danenberg PV, Santi DV. Kinetics and mechanism of interaction of 10-propargyl-5,8-dideazafolate with thymidylate synthase. J. Med. Chem. 1986;29:478–482. doi: 10.1021/jm00154a009. [DOI] [PubMed] [Google Scholar]
- (8).Agrawal N, Hong B, Mihai C, Kohen A. Vibrationally enhanced hydrogen tunneling in the Escherichia coli thymidylate synthase catalyzed reaction. Biochemistry. 2004;43:1998–2006. doi: 10.1021/bi036124g. [DOI] [PubMed] [Google Scholar]
- (9).Liu L, Santi DV. Exclusion of 2′-Deoxycytidine 5′-Monophosphate by Asparagine 229 of Tymidylate Synthase. Biochemistry. 1993;32:9263–9267. doi: 10.1021/bi00087a001. [DOI] [PubMed] [Google Scholar]
- (10).Higashi T. Auto-indexing of oscillation images. J. Appl. Crystallogr. 1990;23:253–257. [Google Scholar]
- (11).Sato M, Yamamoto M, Imada K, Katsube Y, Tanaka N, Higashi T. A high-speed data-collection system for large-unit-cell crystals using an imaging plate as a detector. J. Appl. Crystallogr. 1992;25:348–357. [Google Scholar]
- (12).Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- (13).Chambers JL, Stroud RM. Difference Fourier refinement of the structure of diptrypsin at 1.5 A with a minicomputer technique. Acta Crystallogr. Sect. B. 1977;B33:1824–1837. [Google Scholar]
- (14).Brunger AT. X-PLOR Version 3.1. A System for X-ray Crystallography and NMR. Yale University Press; New Haven: 1992. [Google Scholar]
- (15).Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- (16).Jones T, Zou J-Y, Cowan S, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. sect. A. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- (17).Brunger AT. Crystallographic refinement by simulated annealing. Application to a 2.8 A resolution structure of aspartate aminotransferase. J. Mol. Biol. 1988;203:803–816. doi: 10.1016/0022-2836(88)90211-2. [DOI] [PubMed] [Google Scholar]
- (18).Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- (19).Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55:484–91. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- (20).Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 1):247–55. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- (21).Montfort WR, Weichsel A. Thymidylate synthase: structure, inhibition, and strained conformations during catalysis. Pharmacol Ther. 1997;76:29–43. doi: 10.1016/s0163-7258(97)00099-5. [DOI] [PubMed] [Google Scholar]
- (22).Stroud RM, Fauman EB. Significance of structural changes in proteins: expected errors in refined protein structures. Protein Sci. 1995;4:2392–2404. doi: 10.1002/pro.5560041118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wallace AC, Laskowski RA, Thornton JM. Ligplot - a program to generate schematic diagrams of protein ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- (24).Sage CR, Rutenber EE, Stout TJ, Stroud RM. An essential role for water in an enzyme reaction mechanism: the crystal structure of the thymidylate synthase mutant E58Q. Biochemistry. 1996;35:16270–16281. doi: 10.1021/bi961269r. [DOI] [PubMed] [Google Scholar]
- (25).Collaborative Computational Project, Number 4 Acta Cryst. Sect. D. 1994;D50:760–763. [Google Scholar]
- (26).Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- (27).Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graphics. 1997;15:133–138. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- (28).Delano WL. Delano Scientific; San Carlos, CA: 2002. [Google Scholar]
- (29).Liu L, Santi DV. Mutation of asparagine 229 to aspartate in thymidylate synthase converts the enzyme to a deoxycytidylate methylase. Biochemistry. 1992;31:5100–4. doi: 10.1021/bi00137a002. [DOI] [PubMed] [Google Scholar]
- (30).Hardy LW, Nalivaika E. Asn177 in Escherichia coli thymidylate synthase is a major determinant of pyrimidine specificity. Proc. Natl. Acad. Sci., U S A. 1992;89:9725–9. doi: 10.1073/pnas.89.20.9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Santi DV, Pinter K, Kealey J, Davisson VJ. Site-directed mutagenesis of arginine 179 of thymidylate synthase. A nonessential substrate-binding residue. J. Biol. Chem. 1990;265:6770–5. [PubMed] [Google Scholar]
- (32).Wilkinson AJ, Fersht AR, Blow DM, Winter G. Site-directed mutagenesis as a probe of enzyme structure and catalysis: tyrosyl-tRNA synthetase cysteine-35 to glycine-35 mutation. Biochemistry. 1983;22:3581–6. doi: 10.1021/bi00284a007. [DOI] [PubMed] [Google Scholar]
- (33).Liu L, Santi DV. Asparagine 229 in thymidylate synthase contributes to, but is not essential for, catalysis. Proc. Natl. Acad. Sci., USA. 1993;90:8604–8608. doi: 10.1073/pnas.90.18.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kawase S, Cho SW, Rozelle J, Stroud RM, Finer-Moore J, Santi DV. Replacement set mutagenesis of the four phosphate-binding arginine residues of thymidylate synthase. Protein Eng. 2000;13:557–63. doi: 10.1093/protein/13.8.557. [DOI] [PubMed] [Google Scholar]
- (35).Stroud RM, Finer-Moore JS. Conformational Dynamics along an Enzymatic Reaction Pathway: Thymidylate Synthase, “the Movie”. Biochemistry. 2003;42:239–47. doi: 10.1021/bi020598i. [DOI] [PubMed] [Google Scholar]
- (36).Rosenfield RE, Trueblood KN, Dunitz JD. Rigid-body postulate. Acta Crystallogr. Sect. A. 1978;A34:828–829. [Google Scholar]
- (37).Krebs WG, Alexandrov V, Wilson CA, Echols N, Yu H, Gerstein M. Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic. Proteins. 2002;48:682–95. doi: 10.1002/prot.10168. [DOI] [PubMed] [Google Scholar]
- (38).Finer-Moore J, Liu L, Birdsall DL, Brem R, Apfeld J, Santi DV, Stroud RM. Contributions of orientation and hydrogen bonding to catalysis in Asn229 mutants of thymidylate synthase. J. Mol. Biol. 1998;276:113–129. doi: 10.1006/jmbi.1997.1495. [DOI] [PubMed] [Google Scholar]
- (39).Morse RJ, Kawase S, Santi DV, Finer-Moore J, Stroud RM. Energetic contributions of four arginines to phosphate-binding in thymidylate synthase are more than additive and depend on optimization of “effective charge balance”. Biochemistry. 2000;39:1011–20. doi: 10.1021/bi9918590. [DOI] [PubMed] [Google Scholar]
- (40).Stout TJ, Sage CR, Stroud RM. Ligand binding: modular components for enzyme specificity. Structure. 1998;6:839–848. doi: 10.1016/S0969-2126(98)00086-0. [DOI] [PubMed] [Google Scholar]
- (41).Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–8. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- (42).Birdsall DL, Finer-Moore J, Stroud RM. The only active mutant of thymidylate synthase D169, a residue far from the site of methyl transfer, demonstrates the exquisite nature of enzyme specificity. Protein Eng. 2003;16:229–40. doi: 10.1093/proeng/gzg020. [DOI] [PubMed] [Google Scholar]
- (43).Fritz TA, Liu L, Finer-Moore JS, Stroud RM. Tryptophan 80 and Leucine 143 are critical for the hydride transfer step of thymidylate synthase by controlling active site access. Biochemistry. 2002;41:7021–9. doi: 10.1021/bi012108c. [DOI] [PubMed] [Google Scholar]
- (44).Cannon WR, Singleton SF, Benkovic SJ. A perspective on biological catalysis. Nat Struct Biol. 1996;3:821–33. doi: 10.1038/nsb1096-821. [DOI] [PubMed] [Google Scholar]
- (45).Serrano L, Kellis JT, Jr., Cann P, Matouschek A, Fersht AR. The folding of an enzyme. II. Substructure of barnase and the contribution of different interactions to protein stability. J Mol Biol. 1992;224:783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- (46).Otzen DE, Rheinnecker M, Fersht AR. Structural factors contributing to the hydrophobic effect: the partly exposed hydrophobic minicore in chymotrypsin inhibitor 2. Biochemistry. 1995;34:13051–8. doi: 10.1021/bi00040a016. [DOI] [PubMed] [Google Scholar]
- (47).Xu J, Baase WA, Baldwin E, Matthews BW. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 1998;7:158–77. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rajagopalan PT, Lutz S, Benkovic SJ. Coupling interactions of distal residues enhance dihydrofolate reductase catalysis: mutational effects on hydride transfer rates. Biochemistry. 2002;41:12618–28. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- (49).Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36:15792–800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- (50).Spencer HT, Villafranca JE, Appleman JR. Kinetic scheme for thymidylate synthase from Escherichia coli: determination from measurements of ligand binding, primary and secondary isotope effects, and pre-steady-state catalysis. Biochemistry. 1997;36:4212–4222. doi: 10.1021/bi961794q. [DOI] [PubMed] [Google Scholar]
- (51).Reyes CL, Sage CR, Rutenber EE, Nissen R, Finer-Moore JS, Stroud RM. Inactivity of N229A thymidylate synthase is due to water-mediated effects, isolating a late stage in methyl transfer. J. Mol. Biol. 1998;284:699–712. doi: 10.1006/jmbi.1998.2205. [DOI] [PubMed] [Google Scholar]
- (52).Hyatt DC, Maley F, Montfort WR. Use of strain in a stereospecific catalytic mechanism: crystal structures of Escherichia coli thymidylate synthase bound to FdUMP and methylenetetrahydrofolate. Biochemistry. 1997;36:4585–4594. doi: 10.1021/bi962936j. [DOI] [PubMed] [Google Scholar]