

Abstract
MicroRNAs (miRNAs), ∼21 nt RNAs that mediate post-transcriptional regulation of mRNAs in animals and plants, are a diverse class of regulatory genes whose specific biological functions are largely unknown. Here, we detail a protocol to design and introduce into cultured Drosophila and human cells sequence-specific antisense oligonucleotides (ASOs) that block the function of individual miRNAs. Coupled with recent studies that catalog the miRNAs expressed in diverse cultured cells, our method offers a rapid (< 1 week) approach to validate miRNA targets and to study the cellular functions of individual human and Drosophila miRNAs. ASO-based inactivation of miRNAs is faster and simpler than comparable genetic or “sponge” based approaches, for which extensive recombinant DNA manipulation is required. We present our ASO design principles and an optimized transfection protocol in which transfection efficiency of Drosophila Schneider 2 cells can approach 100%. Our 3′-cholesterol modified ASOs have enhanced potency, allowing miRNA inhibition for at least 7 days from a single transfection.
Keywords: microRNA, miRNA, Antisense Oligonucleotide, ASO, Antagomir, Drosophila, Tissue culture, Schneider 2, S2, HeLa, NTera 2, NT2
Introduction
MicroRNAs (miRNAs) are an ancient class of ∼21 nt small silencing RNAs that mediate post-transcriptional regulation of mRNAs in animals and plants. When bound to partially complementary sequences in their target mRNAs, animal miRNAs tune gene expression by repressing translation or accelerating mRNA decay 1-5. This regulation is critical for diverse biological processes, including stem cell maintenance; musculoskeletal, circulatory, and nervous system development; insulin secretion; and oncogenic transformation6-12. More than 1000 miRNA genes have now been identified in animals, of which a large fraction are conserved between vertebrates and invertebrates. However, few specific functions have been described for individual miRNAs.
To accelerate the study of miRNA function and mechanism, we developed a method to disrupt individual miRNAs using antisense oligonucleotides (ASOs) in human tissue culture cells13 and adapted it for use in Drosophila S2 cells14. ASOs bind complementary miRNAs and selectively block silencing in cell extracts, in cultured cells, and in vivo in worms, flies, mice, and primates13,15-22. Studying miRNA function using classical genetic approaches is challenging, as miRNAs often form multi-gene families with common mRNAs targets, reside in introns of protein coding genes, or derive from polycistronic non-coding transcripts23-29. ASOs can be designed to inactivate a specific miRNA and its paralogs knowing only the sequences of the mature miRNAs. Recent studies cataloguing miRNA sequences and expression profiles in flies, worms, mice, humans, and cultured cells provide a road map for the use of ASOs to study miRNA targets and the biological pathways they regulate30-35. Here, we describe the principles used in our laboratory to design ASO miRNA inhibitors and provide protocols for the efficient delivery of ASO inhibitors into cultured Drosophila Schneider 2 (S2) and human cells.
miRNAs and Their Targets
miRNAs are transcribed as hairpin precursors, then sequentially processed by the RNase III enzymes, Drosha and Dicer, to yield double-stranded intermediates bearing 2 nt, 3′ overhanging ends36-39. These imperfectly paired duplexes are then assembled into long-lived, cytoplasmic protein-RNA complexes called RISCs (RNA-Induced Silencing Complexes) that mediate RNA silencing. Every RISC contains a single-stranded small RNA guide bound to a member of the Argonaute family of proteins40,41. The miRNA and Argonaute protein act together to bind and silence target mRNAs (Fig. 1). Perfectly complementary targets are efficiently silenced by the endonucleolytic cleavage activity of some Argonaute proteins41-43, but the vast majority of predicted targets in animals are only partially paired31,44-49 and likely cannot be cleaved50. Instead, they bind RISC using the “seed” of the miRNA, nucleotides 2-7, and are translationally repressed and/or degraded by a pathway distinct from the endonucleolytic activity of RISC48,51,52.
Figure 1.

Model for Antisense Oligonucleotide Disruption of miRNA Silencing. Single-stranded, mature miRNAs bound to an Argonaute protein can cleave perfectly complementary mRNA targets. Imperfectly paired mRNAs are translationally repressed or degraded. ASOs bind miRNAs in RISC, thereby preventing miRNA from binding its target mRNA. In some cases, ASOs may secondarily promote miRNA degradation by an unknown mechanism.
The remarkably small number of nucleotides required for miRNA-directed target repression suggests that each miRNA may regulate hundreds of mRNA species31,44-49. Experimentally validating computationally predicted miRNA targets and proving the biological significance of each mRNA candidate remains a daunting challenge. Selective inactivation of miRNAs with ASOs has already helped accelerate this task.
Selection of Antisense Oligonucleotide Chemistry
miRNA activity has been blocked effectively using ASOs containing several distinct nucleic acid modifications. In general, an effective ASO is (1) resistant to non-specific cellular ribonucleases, (2) resistant to miRNA-directed cleavage by RISC, and (3) binds miRNAs in RISC with high affinity, effectively out-competing binding to target mRNAs. We designed ASO inhibitors containing exclusively 2′-O-methyl (2′-O-Me) ribose sugars (Fig. 2). 2′-O-Me oligonucleotides are resistant to cleavage by both RISC and other cellular ribonucleases13,18,53. Moreover, 2′-O-methyl-modified RNA:RNA hybrids are more thermodynamically stable than either RNA:RNA or DNA:RNA duplexes53,54. Other base modifications with enhanced hybridization stability have also been used successfully to inhibit miRNA function, including ASOs combining 2′-deoxy and Locked Nucleic Acid (LNA) nucleotides55-58, 2′-O-methyl and LNA59, all 2′-O-methoxyethyl (2′-O-MOE) ASOs, and ASOs incorporating pyrimidines bearing 2′-O-fluoro modifications19,20. (2′-O-MOE-modified oligonucleotides are not available commercially.) Nuclease-resistant phosphorothioate backbone linkages, in combination with ribose modifications, have also been employed in cultured cells, in vivo in mice and non-human primates16,19,20,22,60 (Fig. 2). miRNA inhibition by peptide nucleic acid oligonucleotides has been reported for cultured cells59.
Figure 2.

Chemical Structures of Antisense Oligonucleotides Used to Block miRNA Function. Anti-miR-277 is represented to illustrate the design principles used in different studies. All ASOs contain core sequences that are perfectly complementary to mature miR-277. A number of flanking sequence, backbone, base, and terminal modifications have been incorporated in the indicated studies. Chemical structures correspond to color coded sugar, backbone, and terminal modifications. The linkers used for 3′-cholesterol conjugation by Dharmacon and Alnylam (not commercially available) are also illustrated.
A 3′ terminal cholesterol group appears to aid delivery of ASOs to cells. Originally used to enhance delivery of ASOs targeting mRNAs61-63, cholesterol modification has been adapted to deliver both small interfering RNAs and miRNA-blocking ASOs to the liver and other tissues in mice16,64. We find that 3′ conjugation of cholesterol to ASOs makes them ∼8-fold more potent when transfected into S2 cells14 (Fig. S3). While cholesterol conjugation likely aids ASO delivery into cells, it may have properties that further enhance ASO activity, such as improved intracellular escape from liposomes, relocalization of the targeted miRNAs, or enhancement of ASO stability. Recent studies in mice support such ideas60.
Does Length Really Matter?
To make the ASO more “target-like,” we added 5 extra nucleotides to each of its ends, for a total of 31 nucleotides13. 21 nt 2′-O-Me ASOs have also been used in cultured human and Drosophila cells and in fly embryos17,18,20. Esau and coworkers reported no significant difference between 21 nt and longer ASOs in HeLa cells, but Hutvágner et al. found that 21-mer ASOs were measurably less potent. Similarly, Berger and co-workers observed only partial inhibition of miR-2 in Drosophila S2 cell reporter assays using a 21-mer ASO65. Vermeulen and coworkers found that increasing ASO length by adding as many as 16 nt to each side of the miRNA-complementary core increased ASO potency as much as 10-fold66.
Consistent with these observations, Ameres and colleagues recently described a sequence-independent, single-stranded RNA-binding activity associated with human RISC67. Non-sequence-specific binding by RISC of ASO sequence flanking the miRNA-complementary core may account for the enhanced potency of ASOs longer than 21 nt. These additional ASO sequences might also protect the core from cellular exonucleases. Krutzfeldt suggests this possibility to explain a “tendency” toward improved disruption of miR-122 in vivo when they extend the ASO by only 1 base on each end60. Interestingly, Vermeulen observed that addition of double-stranded, 8 base-pair hairpins to the ends of the ASO increased its potency more than the addition of 16 single-stranded nucleotides66. Double-stranded ends may protect the ASO from exonucleolytic destruction or the terminal hairpins may participate in coaxial stacking interactions with the ASO:miRNA duplex, increasing its thermodynamic stability68. Supporting the idea that ASOs must have high thermodynamic stability, Elmen and colleagues recently showed that full complementarity of an ASO is not required if a very high affinity 16-mer LNA targeting the 5′ end of the miRNA is used22,58. In our experience, 21 nt LNA-substituted ASOs and 31-mer 2′-O-Me ASOs performed similarly in S2 cells (PDZ and MDH, unpublished). While the higher affinity of LNAs may permit design of shorter effective ASO inhibitors, we believe that the bulk of published data suggest that the addition of single- or double-stranded sequences flanking the 21 ASO nucleotides complementary to the targeted miRNA potentiates miRNA inhibition by 2′-O-Me ASOs.
The Mechanism of miRNA Inhibition
The mechanism by which ASOs block miRNA function remains controversial. Initial studies suggested that ASOs block miRNA function by binding mature miRNAs in RISC. 31-mer 2′-O-Me ASOs act as stoichiometric inhibitors in vitro and in HeLa cells and bind miRNAs in RISC in vivo in C. elegans13. Unexpectedly, ASO-targeted miRNAs could not be detected by northern hybridization in mice that were injected with “antagomirs”, cholesterol modified, 2′-O-Me ASOs with terminal phosphorothioate modifications, (Fig. 2)16,60. Similarly, Esau and coworkers found that fully modified 2′-O-methoxyethyl phosphorothioate ASOs targeting the same miRNA, miR-122, induced the apparent destruction of the miRNA. Potentially, the phosphorothioate modification used in these studies triggered miRNA degradation. Chan and colleagues also report that 2′-O-methyl or LNA/DNA ASOs bearing phosphodiester backbones reduced apparent miRNA levels, but these authors suggest that standard northern hybridization probes may be unable to disrupt the high affinity of miRNA:ASO duplexes, even under denaturing conditions55.
If high affinity ASO:miRNA duplexes block access of standard DNA oligonucleotide northern probes, higher affinity LNA probes coupled with an increased hybridization temperature might enhance detection of a miRNA that is tightly bound to an ASO rather than degraded. Indeed, we found that an LNA probe and higher hybridization temperature enhanced detection of synthetic let-7 incubated with ∼1000 fold excess antagomir (Fig S1A). Similarly, endogenous miR-277 was undetectable by northern blot in miR-277 LNA ASO treated Drosophila S2 cells if a DNA probe was used, but miR-277 was detected with an LNA probe and higher hybridization temperature. Interestingly, miR-277 2′-O-Me ASOs had no significant effect on miR-277 levels using DNA or LNA probes. These data suggest that in Drosophila cells ASOs do not promote miRNA degradation and that in some cases, ASOs can interfere with miRNA detection by northern blot with DNA probes (see supplementary text).
Alternatives to ASOs for Studying miRNA Function
A number of alternative approaches are available to study the loss of function of specific miRNA genes.
Gene targeting using homologous recombination is possible in mice and flies69,70, and has revealed roles for individual miRNAs in the mouse immune system71 and heart9, and in the development of fly muscle8 and sensory organs72. While roles for miRNAs in development had been suggested by the analysis of fly embryos injected with 2′-O-Me ASOs17 or developing zebrafish injected with pre-miRNA-complementary “morpholinos” (∼25 nt oligonucleotides containing 6-carbon morpholine rings and a phophoramadite backbone instead of ribose sugars and a phosphodiester backbone)73, ASOs have yet to be utilized in developing mice. Thus, targeted deletion is currently the only way to study loss of miRNA function in mouse development.
Gene targeting is also possible in human and mouse tissue culture cells74. Only a single study to date has used this approach75, likely because gene targeting in somatic cells is technically challenging and laborious, requiring construction of a targeting vector, recombination selection schemes that can vary widely in efficiency depending on the gene to be targeted, and screening to verify genotype. Moreover, the results obtained by somatic gene targeting closely mirror those from parallel experiments using ASOs (see Anticipated Results for more discussion).
A “knock-out” phenotype in an intact animal is, of course, the most convincing proof of the biological function of a miRNA. Often, however, no observable phenotype results from miRNA loss of function. In a recent study in which deletions were reported for 83% of known miRNA genes in C. elegans, loss of most individual miRNA genes caused no obvious phenotype76. Functional redundancy likely explains this result, as combining deletions of several let-7 family members that alone had no phenotype caused distinct phenotypes25. In mice, deletion of the miR-17∼92 cluster produced dramatic defects in heart, lung, and B-cell development, accompanied by inappropriate apoptosis, but deletion of two other paralogs (miR-106a∼363 or miR-106b∼25) produced no obvious phenotype77. Still, it is unclear if one or all of the 6 miRNAs in the cluster contribute to the observed phenotype. Highlighting the utility of ASOs as an adjunct for rapid functional studies of individual miRNAs, Matsubara and colleagues had already found that lung cancer cell lines expressing the miR-17∼92 cluster undergo apoptosis when miR-17 or miR-20a are inhibited with ASOs78. Thus, while gene deletion remains the biological gold standard, its technical difficulty paired with the complex organization of miRNA genes makes it a high risk, time consuming approach.
“miRNA sponges” offer a new alternative to inhibit miRNA function that may be superior for inhibiting whole miRNA families. Sponges are highly expressed transgenes bearing multiple, bulged (i.e., non-cleavable) miRNA-binding sites complementary to a miRNA of interest. These abundant RNAs compete with endogenous targets, and thus “soak up” RISC79. miRNA sponges have been transfected into tissue culture cells and appear to function as well as ASOs. Ebert and colleagues observed that sponges can inhibit miRNAs whose only shared sequence is their seed. This should be considered when targeting a single miRNA that is part of a family—sponges may not distinguish among family members, while ASOs appear to selectively silence family members that differ by fewer than two bases80. In theory miRNA sponges could be used to make transgenic animals, but to date they have only been tested in cultured cells. Limitations of sponges, compared to ASOs, include 1) the time needed to design, build, and test the recombinant sponge vectors, 2) the reliance on plasmid DNA transfection, which is less efficient than oligonucleotide transfection, and 3) their limited selectivity for a single member of a miRNA family.
Transfected siRNAs directed against pre-miRNA loop regions have been reported to decrease miRNA abundance in human tissue culture cells81,82. This method should be used with caution as it appears to be comparatively inefficient, reducing the level of a mature miRNA ∼80% at best, with the remaining 20% free to interact with targets. A number of issues likely account for the relative inefficiency of siRNA-directed depletion of miRNAs, including the secondary structure of pre-miRNAs which likely limits access to RISC. Secondary structure surrounding target sites is a well documented anti-determinant for RNAi67,83. An siRNA targeting a pre-miRNA must also be able to cleave the pre-miRNA before Dicer converts it to a mature miRNA. To date, no evidence suggests that RISC has a kinetic advantage over Dicer. Additionally, miRNA depletion by pre-miRNA-directed siRNAs is limited by mature miRNA turnover. In contrast, ASOs directly inhibit mature miRNAs in RISC. Because RISC-bound miRNAs may be quite stable, this is an important consideration.
Advantages and Limitations of miRNA inhibition by ASOs
In principle, transfected ASOs can be used to study the loss-of-function phenotype for any miRNA expressed in cultured cells by measuring growth rate, induction of apoptosis, or changes in mRNA or protein abundance. Derepression of miRNA-regulated genes in the presence of an ASO is especially convincing evidence for a proposed miRNA:target interaction. In contrast to target validation approaches that use cloned 3′ untranslated region (UTR) reporters and over-express miRNAs, ASO approaches can demonstrate that an endogenous miRNA interacts with an endogenous target mRNA.
To study miRNA function in human somatic cells, where genetic knockouts are difficult at best, ASO transfection is an essential tool. Studies using cell lines are, of course, limited by the repertoire of miRNAs expressed and by the cellular processes that can be recapitulated in immortalized or transformed cells. To study the role of miRNAs in complex developmental or physiological processes involving the interaction of multiple cell types, the use of model organisms in which miRNAs can be inactivated in vivo with ASOs or by targeted deletions (see above) may be preferable. However, even when studying miRNA function in vivo, validation of miRNA:target regulation in cell lines has the advantage that all the cells studied are essentially identical.
The main technical limitations to the use of ASOs are their delivery, duration of action, and specificity. Delivery and duration of action can be assessed using miRNA sensors, genes engineered to place a reporter, such as green fluorescent protein (GFP) or luciferase, under the control of the miRNA of interest (see Experimental Design and Anticipated Results). Using the protocol described below, transfection is efficient (>90%) and miRNA inhibition is long lasting (>7 days). ASOs likely do not discriminate among miRNAs that differ by a single nucleotide. This can be a limitation or an advantage. Because many miRNAs are members of highly related families, i.e., they contain identical seeds, a single ASO likely blocks the function of more than one miRNA in a family. However, a single ASO may not strongly inhibit miRNAs whose only common sequence is the seed19,79. For such applications, co-transfection of multiple ASOs targeting various isoforms is possible75. In this respect, genetic approaches are superior for studying individual miRNA family members, whereas miRNA sponges or multiple ASOs are appropriate for studying miRNA families whose members only contain a common seed sequence; single ASO studies may simplify the study of nearly identical miRNA paralogs.
Combinations of ASOs targeting unrelated miRNAs have also been used to disrupt more than one miRNA in the same transfected cells, obviating the need to make and combine multiple genetic knockouts. Because combinatorial control of targets by miRNAs may be common84, this approach may prove particularly important for uncovering networks of miRNAs that act together. Vermeulen and co-workers showed that co-transfection of a six-ASO mixture can effectively de-repress reporters for each individual miRNA66. Functional studies also suggest that co-transfection of ASOs is effective. For instance, Pedersen and colleagues asked if five interferon-β induced miRNAs with seed matches to Hepatitis C genes had anti-viral effects; indeed simultaneous cotransfection of all five ASOs, but not controls, significantly enhanced Hepatitis C RNA production85.
Experimental Design
Control experiments are required to ensure that miRNAs are indeed inactivated by the transfected ASOs. All experiments should employ a miRNA-specific ASO and a mismatched or unrelated control ASO. Suitable controls include
Antisense sequence of a non-homologous gene from another species,
A randomly scrambled version of the experimental ASO,
A sequence derived from the experimental ASO incorporating purine:purine mismatches in at least 4 evenly spaced positions spanning the sequence of the miRNA. In theory, an ideal control would be a single mismatch in the miRNA seed region, but the enhanced binding affinity of 2′-O-Me:RNA may compensate for a single seed mismatch. 4 mismatches spanning the miRNA sequence should be sufficient to disrupt binding.
After transfecting cells with these ASOs, miRNA inhibition can be assessed by measuring the abundance of a protein encoded by (1) a validated miRNA target, relative to a control gene, or (2) a reporter protein that is regulated by one or more miRNA-binding site(s) in the 3′ UTR of its mRNA. If miRNA mediated repression is blocked by the ASO, expression of target genes will increase compared to control genes. miRNA reporter or “sensor” constructs typically place one or two perfectly complementary miRNA sites in the 3′ UTR of reporter gene. In mammalian cells, we use a dual luciferase reporter system in which Photinus pyralis luciferase containing a perfectly paired 3′ UTR miRNA site is cotransfected with an unregulated Renilla Reniformis luciferase control gene (or vice versa). The relative expression of these two enzymes can be easily quantified by measuring luminescence activity13 (Box 4, Fig. 3).
BOX 4. Assessing miRNA Inhibition with the Dual Luciferase Assay.
The dual luciferase system offers a relatively simple approach to assess miRNA inhibition. The psiCheck2 vector system from Promega is a commercially available vector which encodes both Photinus pyralis and Renilla Reniformis luciferase genes on a single plasmid with a multiple cloning site in the 3′ UTR of Renilla luciferase for insertion of synthetic oligonucleotides encoding the miRNA target sites (or other cloned regulatory sequences, such as target 3′UTRs). We have not tested this system in S2 cells, but its use has been previously reported28. Here we briefly list the steps necessary to adapt this reporter system for miRNA sensing and assessment of miRNA inhibition in cultured cells. Additional information is available from the manufacturer (Dual luciferase Assay Manual: www.promega.com/tbs/tm040/tm040.pdf ):
Design a miRNA Target Site Insert.Design a set of two oligonucleotides corresponding to two tandem copies of the miRNA of interest and its reverse complement. Add appropriate overhanging bases corresponding to the restriction enzyme(s) used to digest the psiCheck vector such that the mature miRNA’s reverse complement is in the 5′ to 3′ orientation (See manufacturer’s information for restriction map; www.promega.com/tbs/tb329/tb329.pdf). XhoI and NotI sites are convenient for directional cloning of inserts (Fig. 3). [AU: for formatting reasons, we cannot have figures within a Box; please format this as a separate figure and refer to from the Box and an appropriate place in the main text.]
Clone the miRNA Site Insert Into psiCheck 2 Vector. Digest psiCheck 2 vector, ligate miRNA site insert, transform into E. coli, and sequence the insert using a custom sequencing primer to confirm correct orientation of insert (Vector sequence is available in GenBank, accession number AY535007).
Transfect Mammalian or S2 Cells with ASO as described in the main Procedure. Prepare mammalian or S2 cells for 6 triplicate transfections (18 wells in a 24 well plate). Transfect 6 wells of cells with miRNA directed ASO and 6 wells with control ASO as described in procedure Step 9A (S2 cells) or step 9B (mammalian cells). The remaining 6 wells will be used as a no transfection control.
Transfect Mammalian or S2 Cells with psiCheck 2 Vector. 24 hours later (mammalian cells) or 48 hour later (S2 cells) perform a second transfection with psiCheck2 (control) or psiCheck2+miR (miRNA sensor). Each vector should be transfected into 3 wells of miRNA ASO treated cells, 3 wells of control ASO treated cells, and 3 wells of untreated cells. Lipofectamine 2000 (Invitrogen) can be used for mammalian cell transfection according to the manufacturers instructions (Promega recommends using 0.1 μg vector/ well of a 24 well plate). siLentFect can be used for transfection of psiCheck2 and psiCheck2+miR vectors into S2 cells as described above, with the following alterations: step 9A(iv) - DNA mix: 0.1 μg vector + Schneider media to 25 μl final volume , step 9A(v) - Lipid Mix: 2 μl siLentFect + 23 μl Schneider media.
Perform Luciferase Assay. 24 hours (mammalian cells) or 48 hours (S2 cells) after transfection of psiCheck vectors, cells can be assayed for luciferase activity. Briefly, cells are washed with PBS, lysed in passive lysis buffer, luciferin reagents are added, and samples are read in a luminometer. Note: Adherent cells can be grown, washed, and lysed in the same plate. S2 cells must be pelleted in Eppendorf tubes (1000 × g for 2 min) each time the media is changed. Details for the dual luciferase assay reagents and protocol are provided by Promega (see above).
Assessing miRNA Inhibition. The relative levels of Renilla luciferase should reveal first, whether the vector was regulated by the endogenous miRNA, and second, whether the ASO blocked this regulation. In control ASO treated cells, the endogenous miRNA should only regulate psiCheck2+miR, giving it lower Renilla luciferase levels than psiCheck2. However, when the miRNA is inhibited in miRNA ASO treated cells, Renilla luciferase levels should be similar in all samples. If they are not, miRNA silencing was not fully blocked. Potential problems are described below (see Troubleshooting).
TIMING
Step i: 10 minutes
Step ii: 3-5 days
Step iii: 3-5 days
Step iv: 24-48 hours
Step v: ∼2 hours
Step vi: 30 minutes
Figure 3.

Design of miRNA Sensor Reporter Target Sites. Two synthetic oligos are illustrated that contain, 1) appropriate “sticky” ends for cloning into XhoI and NotI sites in the psiCheck 2 vector, and 2) tandem miRNA sites with perfect complementarity to a miRNA (miR-277 is shown here).
In Drosophila S2 cells, we typically use stably integrated GFP sensor reporters and derive clonal cell lines14. Changes in GFP expression are measured by flow cytometry (FACS). An advantage of this approach is that transfection efficiency can be assessed by observing the fraction of cells that have increased GFP levels after ASO transfection (Fig. 5).
Figure 5.
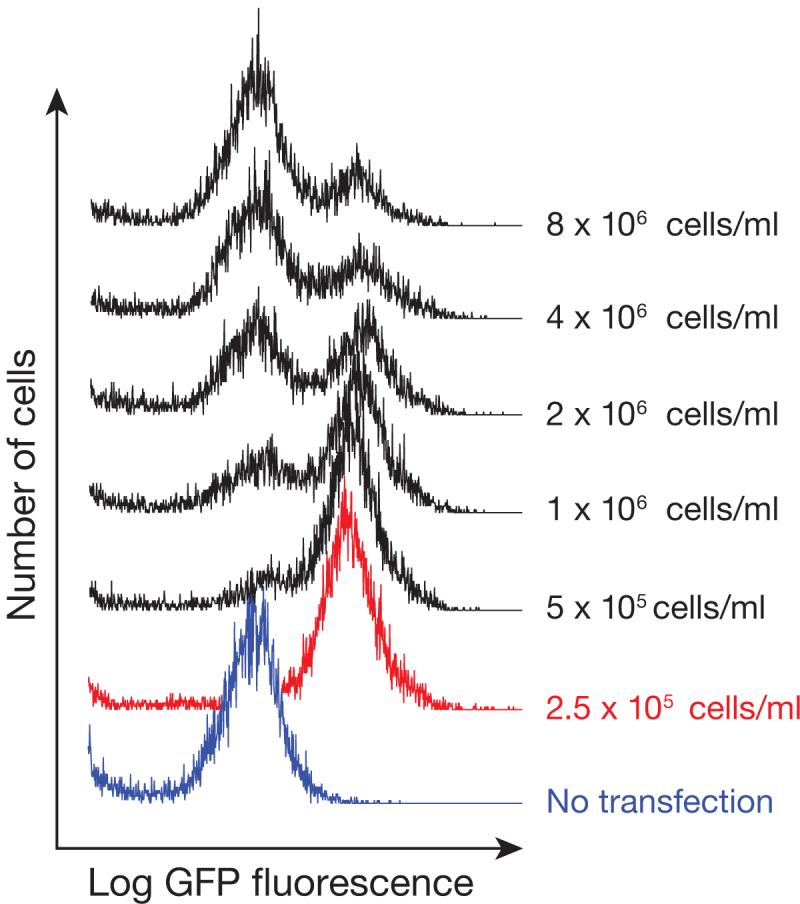
Efficient Transfection Requires Optimal Cell Density. S2 cells were transfected at 20 nM with 33 nt 2′-O-Me 3′-cholesterol modified miR-277 ASO, using Dharmafect 4 at the indicated cell density.
After miRNA inhibition is established, any number of functional assays can be performed in the absence of reporters. Examples are discussed in Anticipated Results.
Gain-of-function studies can also be conducted to complement ASO loss-of-function studies. The finding that introduction of a miRNA into cultured cells has the opposite effect of ASO inhibition on an mRNA provides strong support for a regulatory relationship between the miRNA and the mRNA target. Importantly, it also reduces the likelihood that a cellular phenotype observed with an ASO is due to a non-specific effect. miRNAs can be introduced using a pri-miRNA expression plasmid14,86-88, an siRNA whose sequence corresponds to a miRNA3,89, or a synthetic pre-miRNA90. For instance, we used a pri-miR-277 expression vector to augment miR-277 levels and thus increase silencing of GFP reporters responsive to this miRNA14. Pedersen and colleagues complemented their ASO loss-of-function studies (see above), with a gain-of-function study in which they transfected a mix of 5 miRNA-like siRNAs into cultured cells and found that Hepatitis C virus replication was reduced, even in the absence of IFN-β induction85.
MATERIALS
REAGENTS
Fetal Bovine Serum, Heat Inactivated (Invitrogen, 10082-139)
Dharmafect 4 Transfection Reagent (Dharmacon, T-2004)
siLentFect (Bio-Rad, 170-3360)
Lipofectamine 2000 (Invitrogen, 11668-027)
Dulbecco’s Phosphate Buffered Saline, no Calcium or Magnesium (Invitrogen, 14190)
Antisense oligonucleotides (Dharmacon, Sigma, or many other oligo synthesis vendors)
Trypan Blue (Invitrogen, 15250-061)
Sterile deionized H2O
Drosophila Schneider 2 (S2) cells (Invitrogen, R690-07)
Schneider’s Drosophila Medium (Invitrogen, 11720-034)
HeLa cells (ATCC, CCL-2.2) or NTera2 cells (ATCC, CRL-1973)
Dulbecco’s Modified Eagle Media (Invitrogen, 11965)
Trypsin-EDTA Solution (Invitrogen, 12605)
psiCheck 2 Vector (Promega, C8021)
EQUIPMENT
Sterile 24-well Dishes (Beckton Dickinson, 35 3047)
Sterile 6-well Dishes (Beckton Dickinson, 35 3046)
Sterile 10 cm Plate (Nunc, 172958)
Laminar Flow Hood (class II)
Stereomicroscope with 50× magnification and a bright field setting
Hemocytometer
Sterilized standard 100-1000 μl, 20-200 μl, 1-20 μl, and 0.1-10 μl pipette tips
Micropipettors, P-1000, 200, 20, and 10 (Gilson)
Serological Pipets: 5 ml, 10 ml, and 20 ml
Pipet-aid (Becton Dickinson, 357565)
Sterile Plasticware: 1.5 ml Eppendorf tubes (Eppendorf, 22 36-411-1), 15 ml conical tubes (Beckton Dickinson, 35 2097)
Centrifuges for 1.5 ml and 15 ml tubes
Tissue Culture Incubator at 25°C (without CO2; NuAire, 8700)
Tissue Culture Incubator at 37°C with 5% CO2 (NuAire, 8700)
BD FACScan Flow Cytometer (Becton Dickinson) or equivalent
Veritas Microplate Luminometer (Tuner Biosystems, 9100-000) or equivalent
REAGENT SETUP
Antisense Oligonucleotide Stock Solution
Dilute antisense oligo to 100 μM with sterile dH2O. To be certain the concentration is correct, we check the stock concentration by measuring its absorbance at 260 nM using a spectrophotometer. Extinction coefficients and molecular weights are usually provided with the oligo. 2′-O-Me oligonucleotides do not require deprotection. For pipetting accuracy it may also be necessary to make a 10 μM stock, especially when transfecting a single well or very small wells with an ASO. ASO stock solutions can be stored short term (<1 month) at -20°C and long term at -80°C.
S2 + FBS Media
Combine Schneider’s media with 1/10 volume of fetal bovine serum. Warm to room temperature (∼23°C) before use. Store at 4°C.
S2 Media
Schneider’s Drosophila Medium (no serum) used for transfection mixes Warm to room temperature before use. Store at 4°C.
DMEM + FBS Media
Combine DMEM with 1/10 volume of fetal bovine serum. Warm to 37°C before use. Store media at 4°C.
DMEM
DMEM (no serum) used for transfection should be warmed to room temperature.
Trypsin-EDTA
Pre-warm Trypsin-EDTA solution to 37°C.
PBS
Pre-warm Dulbecco’s Phosphate Buffered Saline solution to 37°C.
PROCEDURE
Design of Antisense Oligonucleotides to Disrupt miRNA Function: Timing — ∼1 hour
Retrieve the sequence of the miRNA(s) of interest from miRBase (http://microrna.sanger.ac.uk/sequences/)91.
Record the reverse complement of the miR strand sequence.
Add 5 arbitrary bases to both the 5′and 3′ ends of the antisense miR sequence. Our inhibitors have used the sequence 5′-UCUUA—antisense miRNA—ACCUU-3′.
Check the full length sequence for potential secondary structure using mFold: http://mfold.bioinfo.rpi.edu/cgi-bin/rna-form1-2.3.cgi92. We use the default settings.
If the flanking sequences are predicted to be involved in formation of strong secondary structure elements (ΔG> user defined cutoff value), alter the flanking sequence base composition and repeat step 4.
Perform a BLAST search using the full length oligonucleotide. If fortuitous stable base pairing to an mRNA is predicted involving the flanking sequences and 13 or more bases within the targeting sequence, repeat step 3-6.
Design a control oligo (see Experimental Design for a discussion of appropriate controls). Flanking sequences can be identical to those in the experimental ASO. All control oligos should also be checked for strong secondary structure or unintended complementarity to mRNAs as detailed in Steps 4-6. Strong secondary structure elements in any part of the sequence should be minimized by altering that part of the sequence.
Order 2′-O-Me-modified oligonucleotides (Dharmacon, Sigma, or many other oligo synthesis vendors). We find that 3′ conjugation of cholesterol enhances the potency of miRNA inhibition in Drosophila S2 cells (Fig. S2A). We order custom cholesterol conjugated oligonucleotides from Dharmacon using the custom RNA module(www.dharmacon.com/rna/rna.aspx).
Cell culture, transfection and analysis of inhibition
9. If using Drosophila S2 cells to study miRNA function, follow option A. If using mammalian cells, follow option B; in our lab, this strategy has been used successfully to transfect both HeLa and NTera2 cells. These transfection protocols can be adapted for RNAi using siRNA or dsRNA by altering only a few steps, as outlined in Box 1.
BOX 1. Transfection of siRNA and dsRNA for RNAi.
The main Procedure can be adapted for RNAi using siRNA and dsRNA with just a few alterations, as detailed below. In our experience, transfection of dsRNA into Drosophila S2 cells is far more effective than soaking102 and dsRNA transfection is more effective than siRNA transfection in S2 cells (MDH unpublished).
For Drosophila S2 cells:
Step 9A(iv): Instead of ASO, use 10 pmol siRNA or 1 μg dsRNA per well of a 24 well plate (scaled linearly for other volumes).
Step 9A(ix): Allow S2 cells to grow for 5 days, and then check protein or mRNA abundance. In our experience the best knock-down is achieved if a second transfection is performed at day 5. The cells are analyzed on day 10.
For mammalian cells
Step 9B(v): Instead of ASO, use 10 pmol siRNA per well of a 24 well plate (scaled linearly for other volumes).
Step 9B(x): For mammalian cells, reduction of protein from the gene targeted by the siRNA can usually be achieved after 3-4 days. A second transfection can be performed if knockdown is insufficient, e.g. if the protein half-life is long.
TIMING
siRNA or dsRNA Transfection of Drosophila S2 Cells
Step 9A(iv): 5 min
Step 9A(ix): 5-10 days
siRNA Transfection of Mammalian cells
Step 9B(iv): 5 min
Step 9B(ix): 3-8 days
TROUBLESHOOTING
Option A: Culture and Transfection of S2 Cells: Timing - Up to 14 days
-
Grow S2 cells in S2-FBS Media in a 25°C incubator to a density of 8-10 × 106 cells/ml with at least 90% viability; cell number and viability should be checked using Trypan blue and a hemocytometer (see Box 2). If starting from a frozen stock, up to two weeks of growth may be necessary to reach >90% viability. Cells can be propagated in one well of a 6 well plate with 2 ml total volume per well.
CRITICAL STEP: Having S2 cells at high density before transfection is important because at lower densities S2 cells form clumps that may limit transfection efficiency. As the cells reach high density these clumps disperse.
-
In a laminar flow hood, dilute cells from a density of 8-10 × 106 cells/ml to a density of 2.75 × 105 cells/ml with S2 + FBS media. Cells can be removed from the 6-well plate and diluted in a 50 ml conical tube, and then dispensed into the desired dishes.
CRITICAL STEP: Diluting the cells to this density is very important. Transfection of too few cells will result in cell death. Transfection of too many cells will cause uptake of the ASO to be inefficient (see Fig. 5).
For a 24-well plate add 450 μl of diluted cells per well (i.e., 90% of the 500 μl final volume per well) and place the plate in the 25°C incubator. For other types of plates or dishes, all the transfection mixes can be scaled linearly. See Table 1.
Vortex the ASO stock. In a new 1.5 ml Eppendorf tube mix 1.25 μl of a 10 μM ASO stock solution with 23.75 μl of Schneider media per well of a 24 well plate. Gently mix by tapping the side of the tube. If transfecting multiple wells with the same ASO, make 5% extra to avoid running short due to pipetting errors (1.31 μl 10 μM ASO/well X number of wells, and 25 μl S2 Media/well X number of wells). CRITICAL STEP: Cholesterol-modified ASOs can pool near the walls of the tube. It is important to mix both the ASO stock and the S2 Media-ASO mixture.
In a new 1.5 ml Eppendorf tube, mix 1 μl Dharmafect 4 transfection reagent with 24 μl of S2 media per well of a 24 well plate. If transfecting multiple wells with the same ASO, add 5% extra to compensate for pipetting errors.
Combine the mixtures from step 9A(iv) and 9A(v) by adding the Dharmafect 4 mixture to the tube containing the ASO. Gently mix by tapping the side of the tube.
Incubate transfection mixture(s) at room temperature for 20 minutes.
-
Remove cells from the incubator and add 50 uL of the ASO-Dharmafect 4 transfection mix (10% total volume) to each well of the diluted cells. Try to distribute drops evenly over the wells and mix by gentle agitation after the mix is added.
CRITICAL STEP: Move the plate back and forth in straight lines rather than circles to evenly distribute cells and transfection mix. Circular motions cause cells to pool in the middle, which may reduce transfection efficiency.
Allow cells to grow for 1-8 days depending on experimental design. For example, if validating the effectiveness of miRNA inhibition with a dual luciferase miRNA sensor system (Box 4), transfect sensor/control reporters at 48 h with siLentfect (Bio-Rad); perform dual luciferase assay at 72 h. Alternatively, if checking miRNA inhibition with a stable GFP cell line, FACS can be performed at 72 h. Because S2 cells are semi-adherent, GFP reporter cells can be transferred to test tubes and assayed directly in the FACS machine according to the manufacturer’s instructions. Because Dharmafect 4 is not very toxic to S2 cells, media does not need to be changed until cells reach a density of 1 × 107 cells/ml. The effects of miRNA inhibition on the expression of regulated mRNAs and proteins can be seen as early as 24 h and persist beyond 8 days (Fig. S3C).
BOX 2. Analysis of cell number and viability.
To dislodge S2 cells, pipette media across the bottom of the well or plate several times. S2 cells are semi-adherent and form loose clumps at low density. As the cells approach the correct density they become less adherent and the clumps disperse.
Remove 50 μl of freshly dislodged cells and mix with 50 μl Trypan blue solution in a 1.5 ml Eppendorf tube. Wait 3 min.
Add ∼20 μl of the cell suspension to the hemocytometer slide.
Count the number of live cells (clear) and dead cells (blue) contained in the largest box in the field. Count two other complete fields and calculate the average.
Multiply by 2 × 104 to determine cells/ml. If the cell number is too great to count (>200 cells in the field), dilute the cells 1:5 in PBS, then proceed from step ii, and multiply by 1×105 to determine cells/ml.
When propagating S2 cultures, cells can be diluted to ∼2.5 × 105 cells/ml and split each time they reach 8-10 × 106 cells/ml. Growth between these densities typically takes ∼5 days.
TIMING
Steps i-vi: 5-10 minutes
Table 1.
Cell, ASO, and Lipid Volumes for Transfection of Cultured Drosophila Cells
| Volume of cells (2.75 × 105/ml) | ASO mix | Lipid mix | |||
|---|---|---|---|---|---|
| ASO (10 μM) | Schneider media | Dharmafect 4 | Schneider media | ||
| 24 well plate | 450 μl / well | 1.25 μl | 23.75 μl | 1 μl | 24 μl |
| 6 well plate | 1.8 ml / well | 5 μl | 95 μl | 4 μl | 94 μl |
| 10 cm Dish | 9 ml / dish | 25 μl | 475 μl | 20 μl | 480 μl |
Option B: Culture and Transfection of HeLa or NTera2 (NT2) Cells: Timing — 72 h
Grow cells to confluence in 10 ml DMEM + FBS in a 10 cm dish in a 37°C incubator with 5% CO2.
24 h before transfection, treat cells with Trypsin-EDTA and split cells (Box 3) into a 6-well plate so that they will be 30-40% confluent at the time of transfection (∼2 × 105 cells/well in 2 ml DMEM + FBS in one well of a 6-well plate). For other types of plates or dishes, cell and transfection mixes can be scaled linearly. See Table 2 for volumes.
-
Check that cells are ∼30-40% confluent.
CRITICAL STEP: Transfection of too few cells can be toxic. Transfection of too many cells is inefficient. Be sure your cells are at the correct density before transfection.
Remove media and replace with 1.8 ml of fresh pre-warmed DMEM + FBS.
Vortex 10 μM ASO stock solution. For each well of a 6-well plate, to be transfected, add 5 μl of a 10 μM ASO stock solution to 95 μl of DMEM in a sterile 1.5 ml Eppendorf tube. Gently mix by tapping the side of the tube. If transfecting multiple wells with the same ASO, add 5% extra to compensate for pipetting errors.
For each well of a 6-well plate, add 4 μl Dharmafect 4 to 96 μl of DMEM in a sterile 1.5 ml Eppendorf tube. If transfecting multiple wells with the same ASO, add 5% extra to compensate for pipetting errors. Gently mix by tapping the side of the tube.
Combine mixtures from step 9B(v) and 9B(vi) by adding the Dharmafect 4 mixture to the tube containing the ASO. Gently mix by tapping the side of the tube.
Incubate the transfection mixture(s) at room temperature for 20 min.
-
Remove the cells from incubator and add 200 μl of the ASO/Dharmafect 4 mix to each well of the 6-well plate. Try to distribute drops evenly over the dish or plate and mix by gently agitating after the mix is added.
CRITICAL STEP: Move the plate back and forth in straight lines rather than circles to evenly distribute cells and transfection mix.
Allow cells to grow for 24-48 h depending on experimental design; for example, if validating the effectiveness of miRNA inhibition with a dual luciferase miRNA sensor (Box 4), transfect the sensor/control reporters at 24 h with Lipofectamine 2000 (Invitrogen) according to the manufacturer. Perform the dual luciferase assay at 48 h. With Dharmafect 4, the media does not need to be changed until cells reach confluence (∼48 h). Effects of miRNA inhibition on target mRNA and protein levels can be seen as early as 24 h post-transfection and are still present after 48 h. We have not defined the persistence of the miRNA inhibition in mammalian cells.
BOX 3. Splitting Adherent Mammalian Cells.
To split cells using Trypsin-EDTA, pre-warm Trypsin-EDTA solution, PBS, and DMEM-FBS media to 37°C (45min).
Remove media and rinse cells twice with 10 ml pre-warmed PBS.
Add 1 ml Trypsin-EDTA solution evenly cover the cells and place cells in the 37°C , 5% CO2 incubator for ∼5 min.
Briefly check to see that cells have been liberated from the dish and each other using a 50× bright field microscope. If not, place cells back in the 37°C, 5% CO2incubator for several additional minutes and re-check them.
Add 9 ml pre-warmed DMEM-FBS to cells to inactivate the Trypsin.
Count cells using a hemocytometer and dilute to a density of 1 × 105cells/ml with pre-warmed DMEM-FBS in a 15 ml conical.
Add 2 ml of cells for each well of a 6-well plate.
TIMING
Steps i-vii: 10-15 minutes
Table 2.
Cell, ASO, and Lipid Volumes for Transfection of Cultured Mammalian Cells
| Volume fresh DMEM+10% FBS | ASO mix | Lipid mix | |||
|---|---|---|---|---|---|
| ASO (10 μM) | DMEM | Dharmafect 4 | DMEM | ||
| 24 well plate | 450 μl / well | 1.25 μl | 23.75 μl | 1 μl | 24 μl |
| 6 well plate | 1.8 ml / well | 5 μl | 95 μl | 4 μl | 94 μl |
| 10 cm Dish | 9 ml / dish | 25 μl | 475 μl | 20 μl | 480 μl |
TIMING
ASO Transfection of Drosophila S2 Cells
Step 1: 1-14 days
Steps 2-8: approximately 1 hour
Step 9A: 1-8 days
ASO Transfection of Mammalian cells
Step 1: 1-4 days
Steps 2: approximately 30 min, then wait 24 h
Step 3-8: approximately 1 h
Step 9B: 1-3 days
TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
Table 3.
TROUBLESHOOTING
| PROBLEM | POSSIBLE REASONS | SOLUTION |
|---|---|---|
| ASO does not derepress reporter. |
ASO forms strong secondary structure. |
Redesign flanking sequences. |
| ASO transfection was not efficient. |
Co-transfect plasmid and ASO. If derepression is now observed, ASO transfection was inefficient. Repeat ASO transfection or optimize transfection. |
|
| Endogenous miRNA is not abundant enough to repress the reporter. |
Check miRNA level by northern blot. Try co- transfection with a “frayed” (guide nucleotide p1 mismatched) siRNA corresponding to the miRNA strand104 to be sure it is not a problem with the reporter. A pri- miRNA expression vector could also be used. |
|
| Incorrect reporter target site. |
If the reporter cannot be repressed, check the miRNA target site in reporter sequence. |
|
| Cholesterol modified ASO was not mixed. |
Vortex ASO stock before use and mix ASO-S2 cell mix by tapping the tube. |
|
| Cells die after ASO transfection. |
Too much transfection reagent or ASO used. |
Possible if cell death is seen in miRNA-specific ASO and control ASO transfected samples. Repeat with correct amount of transfection reagent or ASO or replace media after 12-24 h. |
| Cell density was too low. | Repeat with correct cell density. |
|
| miRNA inhibited is required for cell survival. |
Possible if miRNA ASO kills cells, but control ASO does not. Determine mechanism of cell death. Perhaps the miRNA is anti-apoptotic. Gain of function studies may also be informative (e.g. does overexpression protect cells from apoptosis?). |
|
ANTICIPATED RESULTS
Reporter Assays
In a typical experiment, a reporter mRNA is used to detect the activity of a specific endogenous miRNA and an ASO is used to inhibit that miRNA. In our experience, inhibiting a miRNA with an ASO typically produces a 3-5 fold increase in reporter protein expression when the reporter mRNA contains perfect target sites that can be cleaved by RISC (Fig. 4). miR-277, a miRNA that is expressed in Drosophila S2 cells, does not regulate a GFP reporter lacking target sites. Control and miR-277 ASOs did not change the levels of the non-targeted reporter, ruling out any non-specific effects of the ASOs on the reporter. When two miR-277-binding sites were placed in the 3′ UTR of the reporter mRNA, the miR-277-complementary ASO inhibited miR-277 regulation, increasing expression of the GFP reporter 4-5-fold ; the control ASO had no effect. miR-277 directs repression of a reporter with two, fully complementary sites (4-5 fold increase) to a greater extent than of a reporter bearing four, imperfectly paired miR-277-binding sites (∼30% increase). For this reason, we recommend perfectly matched reporters for testing ASOs. miR-277 inhibition by our ASOs was nearly complete, because the degree of derepression was similar to that observed when Ago2 and Ago1, the core RISC proteins that guide miRNA silencing, were depleted by dsRNA-triggered RNAi14.
Figure 4.
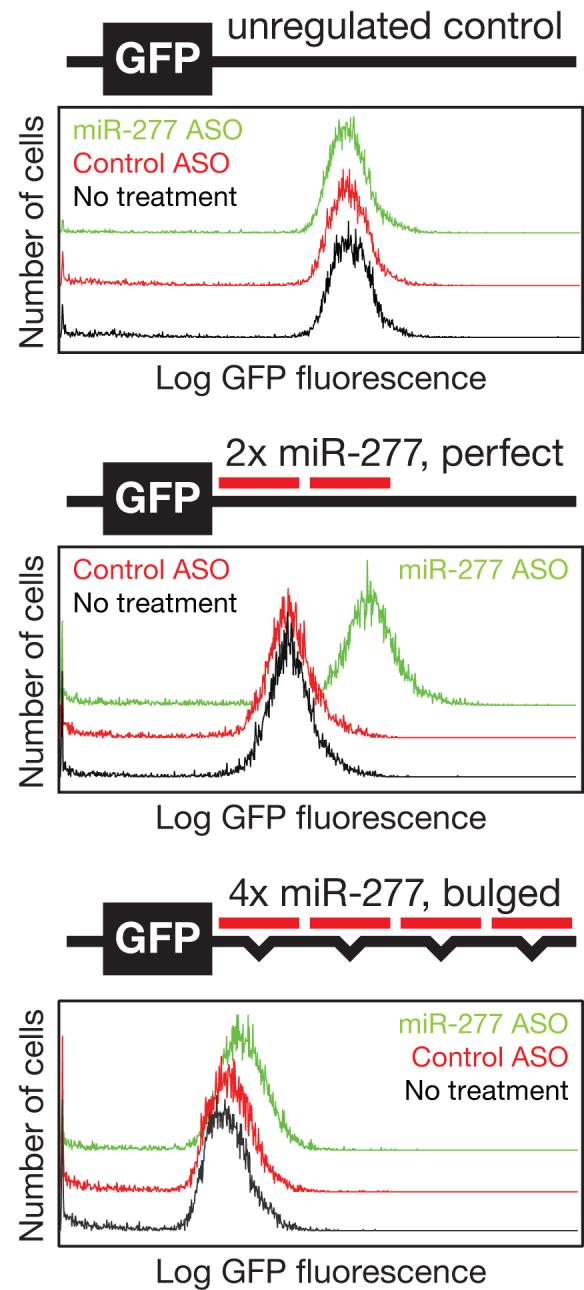
A Sensitive Reporter System For miR-277 Silencing. Clonal Drosophila S2 cell lines bearing stably integrated GFP transgenes containing no sites, two perfect or four bulged miR-277-complementary sites in the 3′ UTR were transfected with control or miR-277-complementary ASOs (33 nt 2′-O-Me bearing a 3′ cholesterol modification). Five days after transfection, GFP was measured by FACS.
Using our optimized protocol we see a uniform shift in the FACS curve when miR-277 is inhibited indicating that transfection was very efficient (>90%) (Fig. 4 and 5). If transfection is incomplete, two peaks will be observed: one corresponding to cells that received the ASO and the other from those that did not. This is seen if cells are transfected at too high a density (Fig. 5). Unlike stable GFP cell lines in which fluorescence is measured in every cell, it is more difficult to assess transfection efficiency using the luciferase assay, in which an average from all cells is obtained. In the sequential transfection procedure detailed in Box 4, if ASO treatment derepresses the Renilla luciferase reporter to a level near that of the non-targeted reporter, ASO transfection must have been efficient (i.e., miRNA was inhibited by ASO in all cells that received the reporter). However, if miRNA repression cannot be inhibited by an ASO, low transfection efficiency may be an explanation. Low potency may also be an explanation. This can be tested by co-transfecting the ASO with the reporter instead of sequential transfection; all transfected cells should receive both the reporter and the ASO. If incomplete derepression is still seen, potency may be the problem rather than transfection efficiency. A dose response curve can be performed to ensure that maximal derepression is being achieved (sample dose response curve, Fig. S3A).
Endogenous Target Validation
Having established that an ASO disrupts the function of an endogenous miRNA, it is now possible to study the cellular consequences of miRNA loss of function. One common application of ASOs is miRNA target validation—testing if computationally predicted target mRNAs93 are, in fact, regulated by a given miRNA in cultured cells. For instance, the Bartel and Dutta groups studied the oncogene HMGA2, which contains seven predicted target sites for the miRNA let-794,95. HMGA2 protein and mRNA increase 4-6 fold in HeLa cells treated with a let-7 ASO, but not a control. Conversely, transfected let-7 siRNAs cause a ∼3 fold reduction in HMGA2 protein. With so many sites, HMGA2 probably represents an extreme case for regulation by a single miRNA (Lee and coworkers note that the HMGA2 mRNA changed more than any other mRNA following Dicer or Drosha depletion by RNAi).
Regulation of targets by miRNAs can be quite modest, depending on miRNA and target abundance; two-fold regulation or less is not uncommon. While mRNA abundance is often affected by miRNA regulation, some targets appear to be regulated solely by translational repression52,96,97. Thus the absence of a change in mRNA abundance of a putative target is not always meaningful, especially if regulation at the mRNA level has not been previously demonstrated. When possible, measurements of the protein regulated is preferred, as changes in mRNA stability or translation should both affect protein levels. When direct measurement of target protein levels is not feasible, protein level regulation by a miRNA can be assessed using luciferase (Box 4) or GFP reporter assays in which the putative target 3′ UTR is cloned into the reporter mRNA. In this case, ASO treatment is predicted to increase reporter levels; conversely, increasing miRNA levels should reduce reporter levels.
When specific target seed sequences are suspected, analysis of 3′ UTRs with mutant seeds can reveal the importance of a specific site for regulation. Such mutational analysis is especially convincing, as it shows miRNA:target complementarity is required for regulation in the context of a potentially quite large 3′ UTR. However, the approach assumes that 1) the reporter target expression level is physiologically relevant, and 2) the target and the miRNA are in fact expressed in the same cells. Whenever possible it is preferable to examine endogenous target mRNA and/or protein levels. Reporter assays are best used to augment analysis and allow more focused studies of 3′ UTR sequence motifs.
Proving that a miRNA regulates a given target may implicate it in a particular biological pathway, giving a hint at its function. However, with each miRNA having hundreds of putative targets, proving a biological function for any one interaction may be difficult: an individual miRNAs’ critical function may be modest regulation of hundreds of genes or more substantial regulation of just a few.
Cellular Assays
Functional analysis of loss of miRNA function in cultured cells can be performed using any number of cellular assays. Because a growing body of evidence implicates miRNAs in cancer development, maintenance, and metastasis, several studies have now employed standard assays for proliferation, apoptosis, and metastasis. Using MTT assays and propidium iodide staining coupled with FACS analysis, Matsubara and colleagues found that blocking miR-20a and miR-17-5p with ASOs reduced cell viability and increased the proportion of sub G1 cells. They also used a TUNEL assay to show increased apoptosis when these same miRNAs are inhibited with ASOs78. Similarly, Bommer and colleagues used propidium iodide staining and FACS to show that miR-34 inhibition results in increased viability of colon cancer cells. In parallel, these authors demonstrated that ES cells genetically deleted of all three miR-34 isoforms had essentially the same phenotype as pooled ASO inhibition of miR-34: increased cell viability and increased concentration of anti-apoptotic Bcl2 protein75.
Ma and colleagues found that miR-10b expression enhances metastasis; invasive breast cancer cells failed to migrate as far when treated with miR-10b ASO98. ASOs have also been used in xenograft cancer models to demonstrate that some miRNAs affect metastatic potential and in vivo growth of tumors. While Tavazoie and colleagues found that miR-335 limits breast cancer metastasis (ASO inhibition increases metastasis of xenografted cell lines)99, Corsten and colleagues saw that transplanted gliomas treated with miR-21-specific ASO were sensitized to a chemotherapeutic agent100.
Several studies use ASOs in conjunction with in vitro differentiation systems to show the importance of individual miRNAs in cellular differentiation. Esau and colleagues showed that miR-143 expression increases during cultured adipocyte differentiation and that this differentiation is inhibited by a miR-143 ASO21. Similarly, Naguibneva and colleagues showed that miR-181 expression is induced upon myoblast terminal differentiation and that an ASO prevents this differentiation by blocking repression of the miR-181 target mRNA, Hox-A1196.
Finally, multiple studies using ASOs suggest roles for endogenous miRNAs in viral defense or replication. Lecellier and colleagues found that ASO inhibition of miR-32, a miRNA with potential target sites in primate foamy cell virus genes, permits enhanced production of viral RNA in human tissue culture cells56. Similarly, inhibition of IFN-β induced miRNAs permits Hepatitis C viral production85. However, ASO inhibition of liver specific miR-122 in cultured hepatocytes cripples Hepatitis C replication, suggesting its requirement in the Hepatitis C viral life cycle85,101. These ASO studies suggest that combinatorial expression of pro- or anti-viral miRNAs may affect tissue tropism of some viruses.
In sum, miRNA function can be efficiently disrupted in cultured mammalian and Drosophila S2 cells. The approaches detailed here should be readily adaptable to study miRNA function in any cell line.
Supplementary Material
ACKNOWLEDGMENTS
We thank Klaus Förstemann for constructing GFP reporters and stable S2 cell lines. This work was supported in part by grants from the National Institutes of Health to P.D.Z. (GM62862 and GM65236) and an NRSA MD/PhD pre-doctoral Fellowship from the National Institute on Aging to MDH (F30AG030283).
Footnotes
COMPETING INTERESTS STATEMENTS
The authors declare competing financial interests (see the HTML version of this article for details).
REFERENCES
- 1.Bagga S. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–563. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 2.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin- 4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 3.Lim LP. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 4.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin- 4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–62. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 5.Olsen PH, Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999;216:671–80. doi: 10.1006/dbio.1999.9523. [DOI] [PubMed] [Google Scholar]
- 6.Förstemann K. Normal microRNA maturation and germ-line stem cell maintenance requires Loquacious, a double-stranded RNA-binding domain protein. PLoS Biol. 2005;3:e236. doi: 10.1371/journal.pbio.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatfield SD. Stem cell division is regulated by the microRNA pathway. Nature. 2005;435:974–978. doi: 10.1038/nature03816. [DOI] [PubMed] [Google Scholar]
- 8.Sokol NS, Ambros V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 2005;19:2343–2354. doi: 10.1101/gad.1356105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Giraldez AJ. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–838. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- 11.Poy MN. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 12.He L. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutvágner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Förstemann K, Horwich MD, Wee L, Tomari Y, Zamore PD. Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell. 2007;130:287–297. doi: 10.1016/j.cell.2007.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boutla A, Delidakis C, Tabler M. Developmental defects by antisense-mediated inactivation of micro-RNAs 2 and 13 in Drosophila and the identification of putative target genes. Nucleic Acids Res. 2003;31:4973–4980. doi: 10.1093/nar/gkg707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krutzfeldt J. Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 17.Leaman D. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell. 2005;121:1097–1108. doi: 10.1016/j.cell.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 18.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–2304. doi: 10.1093/nar/gkl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esau C. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Esau C. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 22.Elmen J. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 23.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 24.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of Novel Genes Coding for Small Expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 25.Abbott AL. The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell. 2005;9:403–414. doi: 10.1016/j.devcel.2005.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aravin AA. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5:337–350. doi: 10.1016/s1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- 31.Ruby JG. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 32.Berezikov E. Diversity of microRNAs in human and chimpanzee brain. Nat Genet. 2006;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- 33.Gaur A. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–2468. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 34.Blower PE. MicroRNA expression profiles for the NCI-60 cancer cell panel. Mol Cancer Ther. 2007;6:1483–1491. doi: 10.1158/1535-7163.MCT-07-0009. [DOI] [PubMed] [Google Scholar]
- 35.Landgraf P. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 37.Hutvágner G. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 38.Grishok A. Genes and Mechanisms Related to RNA Interference Regulate Expression of the Small Temporal RNAs that Control C. elegans Developmental Timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 39.Ketting RF. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 41.Hutvágner G, Zamore PD. A microRNA in a Multiple-Turnover RNAi Enzyme Complex. Science. 2002;297:2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 42.Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- 43.Davis E. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol. 2005;15:743–749. doi: 10.1016/j.cub.2005.02.060. [DOI] [PubMed] [Google Scholar]
- 44.Grun D, Wang YL, Langenberger D, Gunsalus KC, Rajewsky N. microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput Biol. 2005;1:e13. doi: 10.1371/journal.pcbi.0010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krek A. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 46.Rajewsky N, Socci ND. Computational identification of microRNA targets. Dev Biol. 2004;267:529–535. doi: 10.1016/j.ydbio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 48.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 49.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haley B, Zamore PD. Kinetic analysis of the RNAi enzyme complex. Nat Struct Mol Biol. 2004;11:599–606. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- 51.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aleman LM, Doench J, Sharp PA. Comparison of siRNA-induced off-target RNA and protein effects. RNA. 2007;13:385–395. doi: 10.1261/rna.352507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inoue H. Synthesis and hybridization studies on two complementary nona(2′-O-methyl)ribonucleotides. Nucleic Acids Res. 1987;15:6131–6148. doi: 10.1093/nar/15.15.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsourkas A, Behlke MA, Bao G. Hybridization of 2′-O-methyl and 2′- deoxy molecular beacons to RNA and DNA targets. Nucleic Acids Res. 2002;30:5168–5174. [PMC free article] [PubMed] [Google Scholar]
- 55.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 56.Lecellier CH. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 57.Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–141. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 58.Elmen J. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fabani MM, Gait MJ. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krutzfeldt J. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–2892. doi: 10.1093/nar/gkm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desjardins J. Cholesteryl-conjugated phosphorothioate oligodeoxynucleotides modulate CYP2B1 expression in vivo. J Drug Target. 1995;2:477–485. doi: 10.3109/10611869509015917. [DOI] [PubMed] [Google Scholar]
- 62.Krieg AM. Modification of antisense phosphodiester oligodeoxynucleotides by a 5′ cholesteryl moiety increases cellular association and improves efficacy. Proc Natl Acad Sci U S A. 1993;90:1048–1052. doi: 10.1073/pnas.90.3.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Letsinger RL, Zhang GR, Sun DK, Ikeuchi T, Sarin PS. Cholesteryl-conjugated oligonucleotides: synthesis, properties, and activity as inhibitors of replication of human immunodeficiency virus in cell culture. Proc Natl Acad Sci U S A. 1989;86:6553–6556. doi: 10.1073/pnas.86.17.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soutschek J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 65.Berger EM, Dubrovsky EB, Appleby L, Dubrovskaya V. Inhibition of micro-RNA-induced RNA silencing by 2′-O-methyl oligonucleotides in Drosophila S2 cells. In Vitro Cell Dev Biol Anim. 2005;41:12–18. doi: 10.1290/040902.1. [DOI] [PubMed] [Google Scholar]
- 66.Vermeulen A. Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA. 2007;13:723–730. doi: 10.1261/rna.448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130:101–112. doi: 10.1016/j.cell.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 68.Walter AE. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc Natl Acad Sci U S A. 1994;91:9218–9222. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Thomas KR, Capecchi MR. Introduction of homologous DNA sequences into mammalian cells induces mutations in the cognate gene. Nature. 1986;324:34–38. doi: 10.1038/324034a0. [DOI] [PubMed] [Google Scholar]
- 70.Rong YS, Golic KG. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–2018. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- 71.Rodriguez A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Wang F, Lee JA, Gao FB. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes Dev. 2006;20:2793–2805. doi: 10.1101/gad.1466306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kloosterman WP, Lagendijk AK, Ketting RF, Moulton JD, Plasterk RH. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biol. 2007;5:e203. doi: 10.1371/journal.pbio.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AA, Seidman JG. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bommer GT. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 76.Miska EA. Most Caenorhabditis elegans microRNAs Are Individually Not Essential for Development or Viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ventura A. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsubara H. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007;26:6099–6105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 79.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 81.Lee YS, Kim HK, Chung S, Kim KS, Dutta A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem. 2005;280:16635–16641. doi: 10.1074/jbc.M412247200. [DOI] [PubMed] [Google Scholar]
- 82.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 83.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 84.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 85.Pedersen IM. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zeng Y, Wagner EJ, Cullen BR. Both natural and designed microRNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- 87.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 88.Voorhoeve PM. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 89.Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34:1646–1652. doi: 10.1093/nar/gkl068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 94.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007 doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let- 7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Naguibneva I. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol. 2006;8:278–284. doi: 10.1038/ncb1373. [DOI] [PubMed] [Google Scholar]
- 97.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 98.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 99.Tavazoie SF. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Corsten MF. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 101.Jopling CL, Yi M, Lancaster AM, MLemon S, Sarnow P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 102.Clemens JC. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci U S A. 2000;97:6499–6503. doi: 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Valoczi A. Sensitive and specific detection of microRNAs by northern blot analysis using LNA-modified oligonucleotide probes. Nucleic Acids Res. 2004;32:e175. doi: 10.1093/nar/gnh171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schwarz DS. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.