

Abstract
The interface of a protein molecule that is involved in binding another protein, DNA or RNA has been characterized in terms of the number of unique secondary structural segments (SSSs), made up of stretches of helix, strand and non-regular (NR) regions. On average 10-11 segments define the protein interface in protein-protein (PP) and protein-DNA (PD) complexes, while the number is higher (14) for protein-RNA (PR) complexes. While the length of helical segments in PP interaction increases with the interface area, this is not the case in PD and PR complexes. The propensities of residues to occur in the three types of secondary structural elements (SSEs) in the interface relative to the corresponding elements in the protein tertiary structures have been calculated. Arg, Lys, Asn, Tyr, His and Gln are preferred residues in PR complexes; in addition, Ser and Thr are also favoured in PD interfaces.
Keywords: protein-protein interactions, protein-DNA interactions, protein-RNA interactions, binding interface, protein secondary structure
Abbreviations
PP - protein-protein, PD - protein-DNA, PR - protein-RNA, SSE - secondary structural element, SSS - secondary structural segment
Background
Characterization of protein-protein (PP), protein-DNA (PD) and protein-RNA (PR) interactions is essential for understanding the mechanisms of biological processes on a molecular level. Interactions are highly specific and any distortion may be deleterious to the cellular function. Various experimental techniques have been employed to identify the interactions [1], with X-ray crystallography and NMR spectroscopy providing the most detailed view. The atomic coordinates of the complexes stored in the Protein Data Bank (PDB) [2] have been analyzed to derive information on the physicochemical features of the interface formed between the two components. PP interactions [3-5] have attracted the maximum attention. These can vary in strength – some are obligatory (permanent), as can be seen in the formation of the quaternary structures, while others are non-obligatory, in which the individual protomers exist independently in the stable form [6], but the time scale of interaction can vary widely from ~10-3 to 103s (transient to stable complexes, exemplified by electron transfer in redox proteins and antigen-antibody complexes, respectively). Studies in PD interactions have aimed at unravelling of the sequence specificity of nucleotide recognition [7-11]. In comparison PR interactions have been relatively fewer in number as data have been scarce till only recently [12-14]. Most of the complexes contain double-stranded DNA and the RNA is usually single stranded, though in a few cases depending on the sequence and length, it may fold into stem-loop structures including double helical segments. Akin to the non-obligatory PP complexes, PD and PR complexes are mostly transient, forming only when the protein encounters the nucleic acid, and exhibit a wide range of stability and lifetimes. With increase in our understanding of protein structure and interactions attempts are now geared towards synthetic biology for designing receptors for proteins and nucleic acids [15]. In this connection it is important to know what types of secondary structures are used in the interface and the residue usage vis-à-vis the rest of the protein structure. In this article these features are derived for PD and PR interfaces and compared to those observed in PP complexes [16].
Methodology
The list of 128 protein-DNA complexes used has been given in [11]. A search of PDB [2] (August, 2007) yielded 381 hits for the query “protein-RNA complex”. The list of entries was culled using PISCES [17], such that the maximum percentage identity was 25% and the resolution not worse than 3.0 Å. The minimum chain length for the protein part was kept at 40 and for RNA, at least 3 bases. For this non-redundant dataset of 50 protein-RNA complexes, the information on the biologically relevant assembly was obtained from the Nucleic Acid Database (NDB) [18] (since many PDB files have coordinates only for the crystallographic asymmetric unit, which may just contain a part of the whole molecule).
The protein secondary structural elements (SSEs) were assigned using DSSP [19]. Only three types of SSEs were considered. All helices (with DSSP notations ‘H’ and ‘G’) were included irrespective of their type, ‘E’ and ‘B’ constituted strands; turns (‘T’ and ‘S’) and the unclassified residues (with assignment ‘ ’) together formed the nonregular (NR) region. Based on the presence of interface residues in distinct SSEs along the chain, the interface can be split into secondary structural segments (SSS) - a segment is specified by the span between the two extreme locations of the interface residues on that SSE (with or without intervening non-interface residues) [16]. The propensity (Pi)SSE of a residue i to occur in a given secondary structural element (SSE) was calculated by the following formula (1) under supplementary material.
Results and discussion
Basic RNA-binding module and the interface area
Among the 50 RNA-binding proteins (Table 1, supplementary material) many are multimeric, each having distinct recognition sites which are structurally equivalent. Any one of them can be assumed to be the basic unit that gets repeated. We define this basic unit as one RNA-binding module (akin to what we have done for protein-DNA complexes [11]). The basic RNA-binding module that has been constructed can be repeated (by the application of simple symmetry operators) to generate the complete biological assembly. Thus for a homodimeric molecule (such as 1ooa), only one subunit interacting with the RNA was considered. In some other cases with more than two identical protein-RNA units (as in 2gic, where five identical protein chains bind symmetrically to five individual RNA strands), only one protein chain complexed with one RNA was considered. A considerable number (5) of the complexes in the dataset are coat proteins or nucleocapsids of viruses and bacteriophages. Basically, these are huge complexes (eg., 2fz2) formed by the application of a number of symmetry operators to a simple protein-RNA unit. For such complexes too, one subunit of the protein with one strand of the RNA was considered. 42 of the 50 complexes had the protein monomer binding to single-stranded RNA, and the rest to double-stranded RNA.
The interface area is given by the sum of the accessible surface area of the two isolated components minus that of the complex. This is the area that gets buried between the two components, which usually contribute almost equally [4,5]. The average interface area in PR complexes is comparable to that observed in PD and PP complexes (Table 2 under supplementary material), though there is a larger variation around the mean. This is expected as the length of RNA located in the interface varies considerably (range: 3 to 37) among the different structures.
Secondary structural segments in the interface
Data presented in Table 2 (see supplementary material) indicate that there is not much distinction between the numbers of SSSs present in PP and PD interfaces, even when the value is normalized for a fixed size (1000 Å2) of the interface. However, both these numbers are higher for PR interfaces. When the three SSSs (helix, strand and NR) are considered individually, the numbers are comparatively higher for PR than those in PD and PP interfaces. In contrast, the average lengths of the SSSs remain more or less the same in the three categories.
Variation of SSS length with interface size
The majority of the PP complexes have an interface with an area of 1600±400 Å2 that has been termed as the standard size [4]. The variation of the segment lengths as a function of the interface size has also been addressed [16]. It was found that the average length of helix is doubled from ~4 when the area increases ten-fold from 500 Å2; however, such changes were not observed for strand and NR segments. In comparison, in PD complexes (Figure 1a), a variation in the length of helical segments is not seen (the last bin is based on just single interface and is not considered) and a rather uniform length of 5.1-5.8 residues is observed, corresponding to ~1.5 turn of an α-helix interacting with the major groove of DNA. Interfaces have been classified as helical when the number of helical residues in the interface is more than 40% [16]. Considering strand and NR segments, in contrast to PP complexes, there are changes in PD complexes - the strand length decreases by about two fold and that of NR increases to the same extent. In PR complexes (Figure 1b), the length distribution, irrespective of segment type, is fairly uniform over the range of 500 to 3000 Å2; but below 500 Å2, the helical segments that are part of the interface contribute only two residues.
Figure 1.
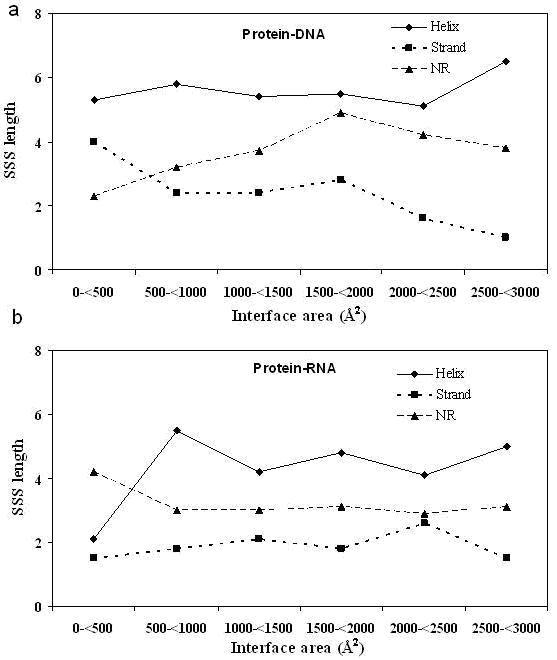
Variation in the average length of the three types of SSSs with the interface area contributed by the protein for (a) protein-DNA and (b) protein-RNA complexes. The protein interfaces have been grouped into bins of size 500 Å2 and the average in each bin is plotted (the last bin in PD complexes has only one data point).
Secondary structure preferences of interface residues
Calculation of the propensities of residues to occur in a SSE in the PP interface relative to the same element in the overall protein tertiary structure indicated that Arg and the aromatic residues are observed more in all interface SSEs [16]. In PD complexes (Figure 2a), propensities > 1.5 are observed for the basic residues (Arg, Lys and His). Residues with hydroxyl groups (especially Ser and Thr) also have higher values. Residues with amide side-chains, Asn in particular, is found more in PD interfaces. Of the aromatic residues, Phe is less abundant. Gly, which is underrepresented in interface SSEs in PP complexes, is found more in PD complexes. It may be noted that unlike the PP complexes, the hydrophobic residues are unfavoured in PD interfaces, which are more polar in nature [7,8,11]. In PR complexes the highest propensities are observed in Arg, Asn and Lys (Figure 2b). Tyr, His and Gln are also preferred, but Ser and Thr, unlike in PD complexes, are not as favoured. Of interest is the fact that Asp, which is known to have a high propensity to be located in strands that form β-sheet across PP interfaces [16], is also highly represented in strands in PR interfaces. This is in sharp contrast to the protein-DNA interfaces where Asp is poorly represented. Met located in NR segments is also preferred in PR complexes.
Figure 2.
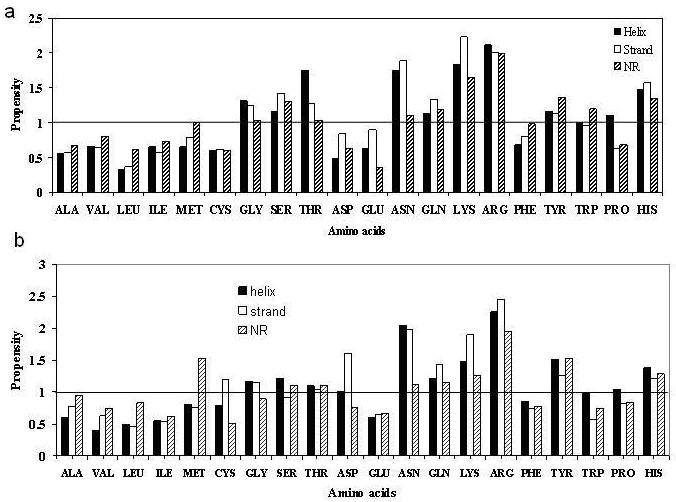
Propensities of residues to occur in a particular SSE in the interface relative to the same SSE in the tertiary structure in (a) protein-DNA and (b) protein-RNA complexes. A propensity of 1.0 indicates that the frequency of observing a particular residue in a given SSE in the interface is the same as that of the corresponding frequency for the entire tertiary structure; a value >1 (or <1) indicates higher (or lower) occurrence at the interface.
Conclusion
A non-redundant dataset of PR complexes has been created. This and a similar dataset of PD complexes [11] have been analyzed in terms of SSSs that constitute the protein part of the interface. PP complexes can bury a larger surface area by the use of longer helical segments [16]. However, in PR complexes the SSS length is rather invariant (Figure 1), but their number tends to increase regularly with the interface size (Figure 3). At any given size of the interface, the number of segments in PR complexes is usually more than that in PP complexes. In PD complexes the helices are of uniform length, but the strands get shorter with the concomitant increase in the length of NR, as the size of the interface increases. Relative to the tertiary structure the interface SSEs are depleted in Ala, Val, Ile and Leu in all types of complexes - interestingly these are the residues that have high α-helix or β-sheet propensities [20]. Compared to PP interfaces, aromatic residues are less favoured at the binding sites of nucleic acids. Preferred residues in PR complexes are Arg, Lys, Asn, Tyr, His and Gln; PD interfaces are also enriched in Ser and Thr. One feature that is common to both PP and PR interfaces is the presence of Asp in interface strands. The residue usage in a SSE in the interface relative to that in the overall tertiary structure would be useful in the design of structural motifs capable of interacting with another protein or nucleic acid.
Figure 3.
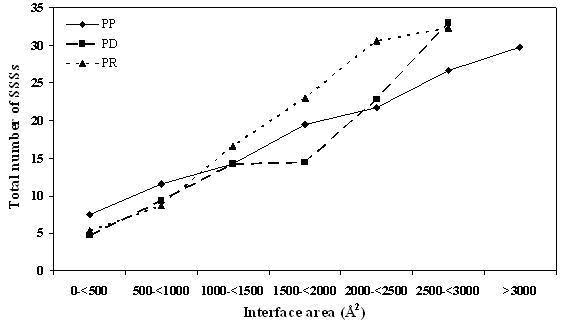
Variation in the average number of SSSs with the interface area (the details are in Figure 1 legend).
Supplementary material
References
- 1.Shoemaker BA, Panchenko AR. Plos Comp Biol. 2007;3:337. doi: 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman HM, et al. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, Thornton JM. Proc Natl Acad Sci USA. 1996;93:13. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo Conte L. J Mol Biol. 1999;285:2177. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 5.Chakrabarti P, Janin J. Proteins. 2002;47:334. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 6.Nooren M, Thornton JM. EMBO J. 2003;22:3486. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nadassy K, et al. Biochemistry. 1999;38:1999. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 8.Jones S, et al. J Mol Biol. 1999;287:877. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 9.Luscombe NM, et al. Genome Biol. 2000;1:001.1. doi: 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarai A, Kono H. Annu Rev Biophys Biomol Struct. 2005;34:379. doi: 10.1146/annurev.biophys.34.040204.144537. [DOI] [PubMed] [Google Scholar]
- 11.Biswas S, et al. Proteins. 2008 [Google Scholar]
- 12.Jones S, et al. Nucleic Acids Res. 2001;29:943. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treger M, Westhof E. J Mol Recogn. 2001;14:199. doi: 10.1002/jmr.534. [DOI] [PubMed] [Google Scholar]
- 14.Ellis JJ, et al. Nuc Acids Res. 2007;66:903. [Google Scholar]
- 15.Endy D. Nature. 2005;438:449. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 16.Guharoy M, Chakrabarti P. Bioinformatics. 2007;23:1909. doi: 10.1093/bioinformatics/btm274. [DOI] [PubMed] [Google Scholar]
- 17.Wang G, Dunbrack RL. Bioinformatics. 2003;19:1589. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 18.Berman HM, et al. Biophys J. 1992;63:751. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabsch W, Sander C. Biopolymers. 1983;22:2577. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 20.Chakrabarti P, Pal D. Prog Biophys Mol Biol. 2001;76:1. doi: 10.1016/s0079-6107(01)00005-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.