

Abstract
The usage by enzymes of specific binding pathways for gaseous substrates or products is debated. The crystal structure of the redox enzyme cholesterol oxidase, solved at sub-Ångstrom resolution, revealed a hydrophobic tunnel that may serve as a binding pathway for oxygen and hydrogen peroxide. This tunnel is formed by a cascade of conformational rearrangements and connects the active site with the exterior surface of the protein. To understand the relationship between this tunnel and gas binding and release, three mutant enzymes were constructed to block the tunnel or its putative gate. Mutation of the proposed gating residue Asn485 to Asp or the tunnel residues Phe359 or Gly347 to Trp or Asn, reduces the catalytic efficiency of oxidation. The KmO2 increases from 300 ± 35 μM for the wild-type enzyme to 617 ± 15 μM for the F359W mutant. The kcat for the F359W mutant catalyzed reaction decreases 13-fold relative to the wild-type catalyzed reaction. The N485D and G347N mutants could not be saturated with oxygen. Hydride transfer from the sterol to the flavin prosthetic group is no longer rate limiting for these tunnel mutants. The steady-state kinetics of both wild-type and tunnel-mutant enzymes are consistent with formation of a ternary complex of steroid and oxygen during catalysis. Furthermore, kinetic cooperativity with respect to molecular oxygen is observed with the tunnel mutants, but not with the wild-type enzyme. A rate-limiting conformational change for binding and release of oxygen and hydrogen peroxide, respectively are consistent with the cooperative kinetics. In the atomic resolution structure of F359W, the indole ring of the tryptophan completely fills the tunnel and is only observed in a single conformation. The size of the indole is proposed to limit conformational rearrangement of residue 359 that leads to tunnel opening in the wild-type enzyme. Overall, these results substantiate the functional importance of the tunnel for substrate binding and product release.
Keywords: GMC oxidoreductase, channel, hydrophobic tunnel, gaseous substrate, kinetically cooperative
Enzymes often catalyze reactions in which one or more of the substrates is a gas, for example, small diatomic molecules like oxygen or hydrogen. Due to the small, non-polar nature of these molecules, the requirements for their specific binding have long been debated. It is possible that these gasses may reach the active sites of enzymes by passive diffusion through proteins as they undergo “breathing” vibrations. The passive diffusion of small gasses could occur along many different trajectories through a protein. However, there are many atomic resolution structures of enzymes and proteins in which possible binding cavities, and even tunnels have been observed. In some cases, these tunnels have been saturated with xenon, an efficient diffractor of X-rays to demonstrate that a gas may diffuse through a channel or tunnel (1-4). Here, we present functional evidence that the observed hydrophobic tunnel in type I cholesterol oxidase is required for efficient gas binding and release.
Enzymes that channel an intermediate between two active sites are the best characterized examples of functional tunnels (5). These tunnels sequester a gaseous or reactive intermediate that is generated at one active site, and direct its diffusion to a second active site. For example, tryptophan synthase channels indole generated in one subunit to the adjacent subunit where it is converted to tryptophan (6). The amidotransferase family, of which carbamoyl phosphate synthetase is an exemplary member, channels ammonia produced from substrate, e.g. glutamine, from one active site to a second where the ammonia is utilized in a second reaction. In the case of carbamoyl phosphate synthetases, carbamate is subsequently channeled to a third active site (7-9). 4-Hydroxy 2-ketovalerate aldolase/aldehyde dehydrogenase (acylating) channels a toxic acetaldehyde intermediate between two distant active sites located 25 Å from each other as a means of protecting the cell from the adverse effects of the intermediate before it is converted to acetyl coenzyme-A (10). In these examples, blocking diffusion of an intermediate into bulk solution and the ensuing improvement in catalytic efficiency is the primary mechanistic advantage that results from tunneling of metabolites. In the reaction catalyzed by carbon monoxide dehydrogenase/acetyl coenzyme A synthase A, a channel between subunits is proposed to allow transport of CO or CO2 to the active site and between active sites (11). Mutation of amino acids to block this channel appear to block CO and CO2 access to the active site (12). These enzymes are primarily multimeric and require a tunnel or channel to pass the intermediate from one subunit to the next.
Enzymes that utilize a gas as a substrate, as opposed to as an intermediate, have different mechanistic requirements. In these cases, the substrate is already present in the bulk solution and must reach the active site. Binding gaseous substrate through a tunnel allows temporal and regiocontrol of its reaction with a second substrate, thus conferring a mechanistic advantage. For example, soybean lipoxygenase generates an alkyl radical intermediate. If activated oxygen is not aimed at a specific carbon, many regioisomers could result. A channel has been proposed to act as a reservoir for O2 that directs it through the protein to react predominantly at C-13 of the linoleyl radical (13, 14). Mutation of Ile553 to Phe in this channel reduces oxygen access (kcat/KmO2) to the radical intermediate without altering the rate of other steps in the catalytic cycle. Similarly, a related channel has been shown to function identically in the rabbit lipoxygenase, although other tunnels may function less efficiently when the main tunnel is blocked by mutagenesis (15). The cycloxygenase system is believed to behave similarly with oxygen channeling responsible for stereochemical control of peroxy radical formation and channeling of oxygen to form the required regioisomer (16). However, in this system reversible kinetic partitioning of the undesired peroxy radical intermediates also provides regiocontrol (17). In a well-studied family, the copper amine oxidases, the oxygen binding properties have been examined extensively by potential of mean force calculations and crystallography (18, 19). In this case, several distinct oxygen-binding pathways were identified. Based on mutagenesis studies, it was concluded that multiple pathways are utilized to different extents analogous to the observations in the rabbit lipoxygenase system (15). The classic example of a protein binding oxygen is that of heme proteins myoglobin and hemoglobin (20). Although, these proteins are not enzymes, several low affinity-binding sites for oxygen (or CO) have been observed by crystallographic and spectroscopic methods (1, 21-23).
In other cases, tunnels have been observed in enzymes, but their role as gas access routes has not been functionally demonstrated. For example, 3-hydroxybenzoate hydroxylase has a hydrophobic tunnel that leads to a discrete binding pocket that is proposed to be the oxygen-binding site (24). The structure of L-amino acid oxidase in the presence of L-phenylalanine reveals a Y-shaped channel that may bind the substrate oxygen and release the product hydrogen peroxide (25). A hydrophobic tunnel proposed to bind oxygen and hydrogen peroxide is also present in the type II cholesterol oxidase (26). Hydrophobic channels that could bind hydrogen gas were observed in the xenon-saturated structure of Ni-Fe hydrogenase (27).
The atomic structure of type I cholesterol oxidase has been solved to 0.95 Å resolution (28). At this resolution, the two conformations of approximately 80 amino acid residues are resolved. In one conformation, several amino acids are oriented such that a narrow, hydrophobic tunnel extending from the outside surface of the enzyme to the buried active site is formed. In the alternate conformation, the tunnel is closed (Figure 1A, 1B). The orientation and size of this tunnel suggests that it provides a passageway for oxygen access to the active site and hydrogen peroxide release to the aqueous solvent. Phe359, Val189, Val124 and Gly347 frame the entrance to the tunnel. At the other end of the tunnel, Asn485 and Met122 form a gate between the tunnel and the isoalloxazine ring of the flavin. The tunnel is only long enough to house two oxygen molecules (Figure 1B).
Figure 1.
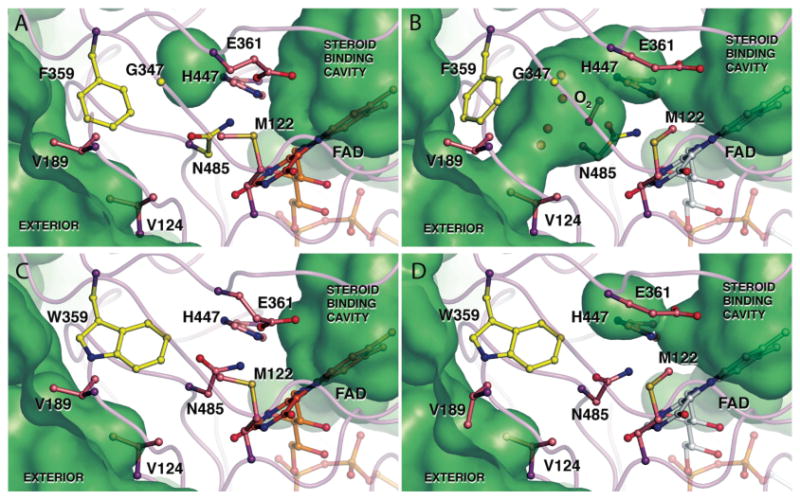
Atomic resolution structure of (A, B) wild-type (1MXT) and (C, D) F359W (####) cholesterol oxidases with key residues in the proposed oxygen tunnel shown in their closed (A, C) and open (B, D) conformations. The solvent accessible surface calculated using the probe radius of 1.4 Å is shown in green. The hydrophobic tunnel between the flavin and the exterior is present when the wild-type tunnel residues are in their open conformation (B) but blocked in the wild-type closed conformation (A). The open conformation includes a dioxygen species which was modeled in the tunnel with a fixed occupancy of 0.25 (37). However, the density may also represent two alternate water conformations. Inclusion in the model of either a dioxygen species or two alternate water molecules requires the adjacent active site and tunnel residues to be present in conformer B. Tryptophan 359 obstructs the tunnel in both conformations (C, D). However, the other tunnel residues, e.g., Glu361, Met122 or Val124, can adopt the closed (C) or open (D) conformation. Images were created using Pymol (41).
Cholesterol oxidases are bacterial, monomeric flavoenzymes that catalyze the oxidation of cholesterol to cholest-5-en-3-one. The flavin dinucleotide cofactor is simultaneously reduced. The reduced flavin is in turn oxidized by oxygen with concomitant formation of hydrogen peroxide. The cholest-5-en-3-one that is formed is isomerized to cholest-4-en-3-one in the same active site. Cholest-5-en-3-one is susceptible to radical oxygenation and most likely is isomerized in situ to the more stable cholest-4-en-3-one to prevent this side reaction (29).
An amide-π interaction between the side chain of Asn485 in cholesterol oxidase and the pyrimidine ring of the FAD stabilizes the reduced flavin (30). When the substrate is oxidized and the FAD cofactor is reduced, the side chain of Asn485 rotates toward the cofactor and a series of conformational changes involving other tunnel residues are initiated. These conformational changes result in the tunnel open conformation, suggesting that the conformation of Asn485, and, by implication, the oxidation state of the flavin regulate access of oxygen to the active site from this tunnel.
Despite the structural observation of a tunnel in this enzyme, no functional evidence that there is a specific binding pathway, e.g., a tunnel, has been forthcoming. To test the functional importance of the tunnel, we investigated the effects of mutating tunnel residues, Phe359, Asn485 and Gly347. The kinetics of the reactions catalyzed by mutant cholesterol oxidases were interpreted with respect to the Phe 359 mutant enzyme and wild-type atomic structures.
Materials and Methods
General materials and methods
Water for assays and chromatography was distilled followed by passage through an 18 MΩ filtration system. A Shimadzu UV2501 PC Spectrophotometer was used for kinetic assays and acquisition of UV spectra. Fluorescence measurements were made with a Spex Fluorolog 3-21 spectrofluorometer. The buffers used were A: 50 mM sodium phosphate, pH 7.0; B: buffer A + 0.025% triton X-100 (w/v); C: buffer B + 0.01% BSA (w/v); D (10X HEPES): 1.37 M NaCl, 0.06 M D-(+) glucose, 0.05 M KCl, 0.007 M Na2HPO4·7H2O, 0.2 M HEPES, pH 7.1.
Construction of pCO303 and other mutant expression plasmids
A 214 base oligonucleotide was made by PCR using 2 primers. Primer 1 contained the designated mutation site and a MluI site (5′-CggCATCgACgCgTgggACAACAgCgACTCCTCggTCTgggCggAgATCgCCCCCATgCC-3′). Primer 2 corresponded to the anticoding strand and contained a HindIII site (5′-gCTCACAATTCCACACAA-3′). With these 2 primers and using XhoI-restricted pCO202 (30) as a template, 35 cycles of PCR were performed with iProof polymerase (Bio-Rad Laboratories, Hercules, CA) at an annealing temperature of 72 °C. The PCR fragment was digested with MluI and HindIII, purified, and subcloned into pCO200(31) that had been similarly digested to yield the F359W ChoA mutant expression plasmid pCO303. The N485D and G347N mutants were constructed in a similar fashion to yield plasmids pCO300 and pCO302 with the following primers and restriction sites: pCO300, 5′-ggCTggggCCCCAACggTAACATCATgACCgCCCgggC CAACCACATgTggAACCCCACCggCgCCC-3′ (StuI). and 5′-gCTCACAAgCTTACgACgCCgTgACgTCCTgCTTgATgA TgCgCTCgACgTTCCgCTCggCCAgCgCCgTgATggTCACgAACgggAggACgCCgACgg-3′ (HindIII) pCO302, 5′-ggAACCCCACCggCgCCCACCAgTCCTCCATCCCCgCCCTCAATATCgACgCgTgggAC-3′ (SgrAI) and 5′-GCTCACAATTCCACACAA-3′ (HindIII). Sequencing of these clones was conducted at the Stony Brook University sequencing facility using an ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Protein purification of wild-type and mutant cholesterol oxidases
Wild-type and mutant cholesterol oxidases were purified as previously described (30). Fractions were assayed for content and purity by SDS-PAGE. Fractions containing pure cholesterol oxidase (>98%) were combined and ultrafiltered (YM30 membrane) into buffer A for storage.
Steady-state kinetic assays of wild-type and mutant cholesterol oxidases
Solutions of cholesterol oxidase were prepared in buffer C. Stock solutions of steroids were prepared by dissolving the appropriate sterol in propan-2-ol. The initial velocities of wild-type and mutant cholesterol oxidases were measured in one of two ways. (1) The formation of conjugated enone was followed as a function of time at 240 nm (ε240 =12,100 M-1 cm-1). The detergent micelles were allowed to form at 37 °C for 10 min, cholesterol was added, and the solution was equilibrated for 10 min. The reaction was initiated by adding cholesterol oxidase (wild type or mutant). (2) The rate of H2O2 formation was determined using a horseradish peroxidase coupled assay. The reaction was followed by excitation at 325 nm and monitoring the fluorescence emission at 415 nm (slits = 1.5 nm) to quantitate the rate of formation of H2O2. The standard assay conditions were the same as the UV A240 assay with the addition of 1.0 mM p-hydroxyphenylacetic acid and 10 U of horseradish peroxidase.
Steady-state kinetics for two varied substrates were measured using the UV A240 assay except that the assay solution (buffer C + cholesterol) was equilibrated with mixtures of N2 and O2 to vary the concentration of oxygen. Different ratios of oxygen and nitrogen were generated using a two-gas mixer (Aalborg Corporation, Orangeburg, NY). The assay solution was evacuated under high vacuum and purged with the appropriate gas mixture every 10 min over a period of 2 h. The oxygen concentration in the assay buffer was determined using an oxygen meter (YSI model 53, Yellow Springs Instruments, Yellow Springs, Ohio).
Independent sets of initial velocity data were globally fit to the following equations using Grafit software (Version 4.0.10). Primary isotope effects were determined by fitting all the possibilities and selecting the best fit (DV, DV/K, or DV and DV/K).
Fixed second substrate Michaelis-Menten equation
| (1) |
DV and DV/K kinetic isotope effect equation
| (2) |
Ternary complex Michaelis-Menten equation
| (3) |
Kinetic cooperativity steady-state rate equation
| (4) |
Secondary equations for two-substrate, ternary complex reaction
| (5) |
| (6) |
where KmO2 and KmChol are the Michaelis constants for oxygen and cholesterol at saturating concentrations of cholesterol and oxygen, respectively, KiO2 is the dissociation constant for enzyme and oxygen, Vm is the maximum velocity. Vmapp and Kmapp are the apparent rate constants for a varied substrate S at a fixed concentration of the second substrate, fi is the fraction of heavy isotope present, KIE is the kinetic isotope effect, and h is the Hill or cooperativity coefficient.
Reduction potential measurements
Potentiometric titrations were performed to determine the midpoint potential of wild type and mutant cholesterol oxidases in a spectroelectrochemical cell using the method of Stankovich (32, 33). Methyl viologen (100 μM) was used as the mediator dye to transfer electrons from the electrode to the protein (30 μM) and the redox indicator dye (2-10 μM). For wild-type cholesterol oxidase, indigo disulfonate, 2-amino-1,4-napthoquinone, and cresyl violet were used as redox indicator dyes. For N485D, riboflavin and phenosafranin were used, and for F359W indigo disulfonate and cresyl violet were used as indicator dyes. Before each titration, the enzyme solutions in buffer A underwent 12 cycles of vacuum and Ar purging over 2 h. Then the electrodes (working, auxiliary and reference electrodes) were inserted into the spectroelectrochemical cell under positive Ar pressure and the titration was initiated by adding electrons from a potentiostat (Model 800B, CH Instruments, Austin, TX). After each addition of electrons, the system was allowed to equilibrate at 25 °C for 30 min, then a visible spectrum and the system potential were recorded. The redox indicator dyes were titrated under the same conditions. The enzyme titration data were corrected for the absorbance of the oxidized and reduced dye at each potential to obtain the spectra of enzyme alone. The potentials were measured with a Ag/AgCl electrode and corrected to the standard hydrogen electrode. The concentrations of all species of the enzyme were calculated from the spectra and the molar absorptivities, and were plotted versus the equilibrium potential of the system using the Nernst equation (7),
| (7) |
where Ecell is the measured cell potential, E°′enzyme is the potential of the enzyme, [ox] and [red] are the concentrations of oxidized and reduced enzyme species, respectively.
Crystallization, data collection, and data processing
The atomic resolution crystal structure of the WT cholesterol oxidase (1MXT) was reported previously (28, 34). Crystals of the F359W mutant enzyme were obtained by hanging drop vapor diffusion method in 15% polyethylene glycol (PEG) 8000, 75 mM MnSO4 and 100 mM sodium cacodylate, pH 5.2. Growth of mutant enzyme crystals was stimulated by microseeding from crystals of the WT enzyme. Crystals suitable for data collection grew within two weeks.
X-ray diffraction data were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 9-2 in two sweeps. The first set was collected to the maximum resolution attainable, which in this case was 0.95 Å, while the second set was collected to the resolution of ∼1.5 Å to minimize the number of overloaded reflections at lower resolutions. The two datasets were integrated individually, then scaled together using the d*Trek suite of software (35). Final data collection statistics are shown in Table 4.
Table 4.
Crystallographic data and refinement statistics for the F359W cholesterol oxidase structure.
| Data collection | |
|---|---|
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 51.28, 72.87, 62.95 |
| α, β, γ (°) | 90.0, 105.11, 90.0 |
| Resolution (Å) | 46.7 − 0.95 (0.98 − 0.95) |
| No. reflections (total) | 1,141,795 |
| No. reflections (unique) | 270,322 |
| Rmerge | 0.047 (0.48) |
| Mean I / σ(I) | 11.2 (1.9) |
| Completeness (%) | 96.3 (67.6) |
| Redundancy | 4.22 (2.56) |
|
| |
| Refinement | |
|
| |
| Rwork / Rfree | 14.0 / 17.5 |
| Number of atoms | |
| Protein | 4714 |
| Water | 695 |
| Ligand / ion | 63 |
| Isotropic B-factors | |
| Protein (mainchain) | 12.11 |
| Protein (sidechain) | 15.52 |
| FAD | 8.4 |
| RMSD | |
| Bond lengths (Å) | 0.013 |
| Bond angles (Å) | 0.030 |
| Planarity (Å3) | 0.084 |
| Alternate conformations | 101 |
The structure of the F359W mutant enzyme was solved by difference Fourier techniques, using a starting model adapted from the 0.95 Å structure of the wild-type enzyme (PDB ID: 1MXT). This model was modified as follows: all anisotropic B-factor parameters were removed, side chains for Asn485, Met122, Val124, Val191, Phe359, Leu377, His447 and Asp459 were changed to alanines. Finally, the FAD cofactor, and all the solvent molecules (including water, glycerol and SO4) were removed. The starting model was subjected to a round of rigid body refinement using the program Refmac5 (36) after which the FAD molecule, many of the truncated sidechains and many of the best-coordinated water molecules became visible as difference electron density features and were modeled. Subsequently, several cycles of restrained refinement using isotropic temperature factors were carried out in Refmac5, followed by manual modeling of features emerging in improved electron density maps using the program Coot (37).
Thereafter, refinement was carried out using the program SHELXH (38) as described previously (28, 34). Briefly, anisotropic temperature factor parameters and alternate sidechain conformations were gradually introduced at this time, resulting in a dramatic overall improvement of R-values. Later in the refinement process, hydrogen atoms were modeled using the “riding model”, where the hydrogen atom's position is fixed relative to the atom to which it is bonded. The final stage of refinement involved minor adjustments, mostly involving water molecules with partial occupancy. The structure was validated using the programs SFCHECK (39) and PROCHECK (39) from the CCP4 suite of software (40). The final refinement statistics are reported in Table 4. All structure figures were generated using the program PyMOL (41). The structure of the F359W mutant cholesterol oxidase has been deposited to the Protein Data Bank with the accession number ####.
Results
Construction and purification of WT and mutant cholesterol oxidases
The mutant cholesterol oxidase genes were constructed, and the corresponding proteins were heterologously expressed in E. coli and purified as previously described for other mutants and wild-type enzyme (30). The isolated proteins were determined to be greater than 99% pure by SDS-PAGE analysis and UV/vis spectroscopy.
Kinetic characterization of tunnel mutants
Using cholesterol as a substrate at a fixed O2 concentration of 256 μM (buffer saturated with air), steady-state kinetic assays were used to initially characterize the wild-type and mutant cholesterol oxidases. Individual mutation of the tunnel residues reduced kcatapp for substrate conversion 30- to 575-fold and had little effect on the KmCholapp (Table 1).
Table 1.
Assessment of mutation impact on oxidation, isomerization and steroid binding by measurement of apparent steady-state Michaelis-Menten rate constants for cholesterol turnover.a
| Substrate turnoverb | Substrate turnoverb | Intermediate turnoverc | ||||
|---|---|---|---|---|---|---|
| H2O2 detection | Cholest-4-en-3-one detection | Cholest-4-en-3-one detection | ||||
| Enzyme | kcatapp (s-1) | KmCholapp (μM) | kcatapp (s-1) | KmCholapp (μM) | kcatapp (s-1) | Km5EOapp (μM) |
| WT | 47 ± 4 | 2.7 ± 0.3 | 42 ± 1 | 2.7 ± 0.3 | 61 ± 4 | 7 ± 1 |
| F359W | 1.3 ± 0.1 | 4 ± 1 | 0.86 ± 0.05 | 2.7 ± 0.8 | 23 ± 1 | 7 ± 1 |
| G347N | 0.85 ± 0.04 | 5 ± 1 | 1.0 ± 0.1 | 1.5 ± 0.6 | 12.0 ± 0.5 | 5.0 ± 0.8 |
| N485D | 35 ± 4.4 × 10-3 | 6.2 ± 2.2 | 73 ± 5 × 10-3 | 7 ± 1 | 10 ± 1 | 7.2 ± 0.9 |
Initial velocity data were fit to the Michaelis-Menten equation for a single substrate, equation (1). Values reported are the mean of three independent measurements, and errors are the standard deviation of the mean.
Cholesterol was used as a substrate at an ambient O2 concentration of 256 μM.
Cholest-5-en-3-one was used as a substrate at an ambient O2 concentration of 256 μM.
Cholesterol oxidase catalyzes the oxidation of cholesterol to cholest-5-en-3-one followed by its isomerization to cholest-4-en-3-one. Two steady-state kinetic assays were used to elucidate the individual effect of the mutations on these two chemical steps. One assay follows the formation of product cholest-4-en-3-one, and the second assay follows the formation of product H2O2. If the isomerization reaction is slowed or cholest-5-en-3-one binding is weakened by mutation, the intermediate cholest-5-en-3-one is released before it is isomerized (29), and different rates will be measured by the two assays. Comparison of the rate constants from the two assays shows that the intermediate is not released by any of the mutants. Direct measurement of the rate of isomerization using the intermediate cholest-5-en-3-one as a substrate confirmed that isomerization is not perturbed more than 6-fold in kcatapp by mutation (Table 1) and the Kmapp's for steroid are unaffected. Thus, reduced rates of steroid binding and release, or a reduced rate of isomerization are not responsible for the 40 to 600–fold rate reductions caused by these mutations.
These data suggested mutation of the tunnel residues primarily affected the oxidation chemistry catalyzed by cholesterol oxidase. In the wild-type catalyzed reaction, a primary kinetic isotope effect on hydride transfer from cholesterol to FADH- is observed on both kcatapp and (kcat/KmChol)app, requiring that oxidation of cholesterol is at least partially rate limiting at both sub-saturating and saturating cholesterol concentrations. The primary kinetic isotope effects on hydride transfer are greatly reduced in the cholesterol oxidation reactions catalyzed by all of the mutants except N485D (Table 2). The effects of asparagine 485 mutation on catalytic efficiency are pleiotropic (42, 43). The majority of the rate reduction upon asparagine mutation is due to lessening the oxidation power of the flavin cofactor (30, 42). The midpoint potential of N485D is -211 ± 6 mV and is reduced 80 mV with respect to wild-type enzyme. In contrast, to N485D, the midpoint potential of F359W is only reduced 20 mV to -153 ± 4 mV. The combination of reduced catalytic activity in the redox step, reduced sensitivity to deuterium substitution, and marginally reduced rates of isomerization suggests that either oxygen binding or FADH- oxidation has become rate limiting in the reactions catalyzed by the F359W and G347N mutants.
Table 2.
Assessment of mutation impact on the kinetic isotope effect for hydride transfer from cholesterol to FAD by measurement of apparent steady-state Michaelis-Menten rate constants for cholesterol turnover.a
| Enzyme | DV & DV/K |
|---|---|
| WTb | 2.2 ± 0.1 |
| F359W | 1.2 ± 0.2 |
| G347N | 1.7 ± 0.2 |
| N485D | 2.5 ± 0.2 |
Initial velocity data were measured for cholesterol or [3-2H]-cholesterol as a substrate at a fixed O2 concentration of 256 μM and fit to equation (2) for the same isotope effect on kcatapp (V) and (kcat/Km)app (V/K). Values reported are the mean of three independent measurements, and errors are the standard deviation of the mean.
Taken from Ye et al. (30).
Further steady-state assays were performed to identify the source of the rate decrease. The oxygen concentration was varied in addition to varying the cholesterol concentration and the formation of product cholest-4-en-3-one was followed. The dependence of wild-type rate on varied oxygen concentrations fit a classical hyperbolic saturation curve (Figure 2A), as did the dependence of rate on varied cholesterol. The combined oxygen and cholesterol data fit a model in which a ternary complex of steroid, oxygen and enzyme is formed (Figure 2B). The data were inconsistent with a ping-pong model in which cholest-4-en-3-one, the first product, is released before binding of O2, the second substrate.
Figure 2.

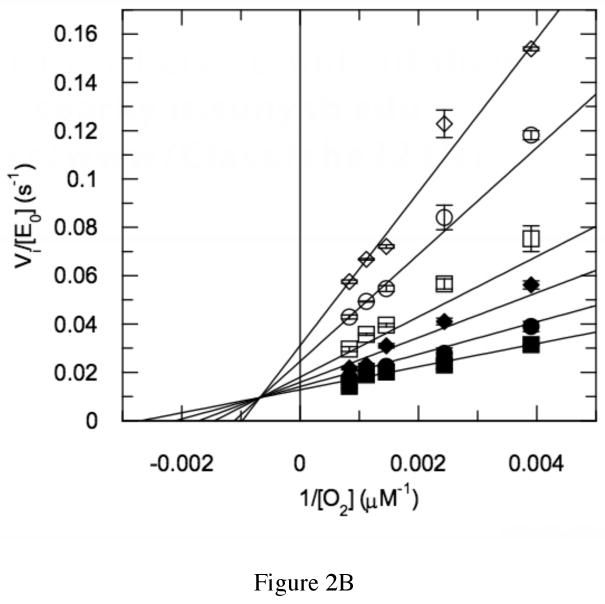
Steady-state kinetic profile of wild-type cholesterol oxidase. Initial velocities for wild-type cholesterol oxidase were measured over a range of O2 concentrations with varied cholesterol concentrations. The data were globally fit to equation (3) for a sequential ternary mechanism or they were fit to equation (4) for a cooperative mechanism at each individual cholesterol concentration. For the fit to equation (4), the Hill coefficient, h, was 1.0 at all cholesterol concentrations and the fit shown is for equation (3). The data shown are the average of two independent experiments, and the errors are the standard deviation of measurement. (A) Michaelis-Menten plots for νi/[Eo] versus [O2] at varied cholesterol concentrations: ◇, 30 μM; ○, 15 μM; □, 9 μM; ◆, 6 μM; ●, 3 μM; and ■, 2 μM cholesterol. (B) Double-reciprocal plot of the data in panel (A). The intersecting line pattern is consistent with the formation of a ternary complex between the enzyme, oxygen and steroid.
The rate dependencies on oxygen concentration of the mutant-catalyzed reactions were strikingly different than the wild-type catalyzed reaction. The N485D catalyzed reaction was linearly dependent on oxygen concentration within the experimentally accessible range of oxygen concentration (Figure 3A). In this concentration range, the reaction catalyzed by N485D is second order, i.e., dependent on both oxygen and enzyme concentration, suggesting that the Km for oxygen has been increased to a concentration above the solubility limit of oxygen in aqueous buffer. The F359W and G347N catalyzed reactions were sigmoidally dependent on oxygen concentration and were fit to the Hill equation for cooperativity (eq. (4), Figures 3B and 3C, Table 3). The Hill coefficient for the reaction catalyzed by F359W increased with increasing concentrations of cholesterol concentration and the maximal cooperativity coefficient was 1.8 ± 0.2 (Table 3). Although, the reaction catalyzed by G347N appeared cooperative, the data could not be fit reliably because the Km for O2 is close to the aqueous solubility limit of oxygen. Therefore these data were not analyzed further. Reanalysis of the data for the wild-type catalyzed reaction utilizing the Hill equation confirmed that there was no apparent oxygen cooperativity in the wild-type catalyzed reaction; the Hill coefficients at all cholesterol concentrations were 1, that is, the curves were hyperbolic, not sigmoidal (Figure 2A). The KmO2 is increased 2-fold to 617 μM upon F359W mutation and kcat is decreased 13-fold relative to wild type.
Figure 3.



Steady-state kinetic profiles of mutant cholesterol oxidases. Initial velocities for mutant cholesterol oxidases were measured over a range of O2 concentrations with varied cholesterol concentrations and the data fit to equation (4) for a cooperative mechanism at each individual cholesterol concentration: ◇, 30 μM; ○, 15 μM; □, 9 μM; ◆, 6 μM; ●, 3 μM; and ■, 2 μM cholesterol. (A) N485D; (B) F359W; (C) G347N. The data shown are the average of two independent experiments, and the errors are the standard deviation of measurement.
Table 3.
Steady-state rate constants for wild-type and F359W cholesterol oxidases.
| Enzyme | KiO2 (μM) | KmO2 (μM) | KmChol (μM) | kcat(s-1) | hmax | (kcat/KmO2)wt/(kcat/KmO2)mut | (kcat/KmChol)wt/(kcat/KmChol)mut |
|---|---|---|---|---|---|---|---|
| WTa | 1383 ± 120 | 300 ± 35 | 4.0 ± 0.5 | 96 ± 6 | 1.0c | 1 | 1 |
| F359Wb | 1381 ± 1649 | 617 ± 15 | 2.1 ± 2.5 | 7.4 ± 1 | 2.8c | 27 | 6.8 |
A compulsory order ternary-complex mechanism as described in equation (3) was fit to the initial velocity data to yield KiO2, KmO2, and KmChol.
Secondary plots of Vmapp and (Vm/KmO2)app versus [cholesterol] were fit to equations (5) and (6), respectively to yield Vm, KiO2, KmO2, and KmChol. Vmapp and (Vm/KmO2)app were obtained by fitting initial velocity data for varied oxygen concentrations at fixed cholesterol concentrations to equation (4).
hmax was determined by plotting h versus [cholesterol]. Values reported are the mean of two independent measurements, and errors are the standard deviation of the mean.
The kinetic cooperativity of oxygen reaction was investigated with a poor substrate, dehydroepiandrosterone. In the wild-type catalyzed reaction, kcatapp is 20 times slower with dehydroepiandrosterone as substrate than with cholesterol at an ambient oxygen concentration of 256 μM. The wild-type catalyzed reaction has a hyperbolic dependence on oxygen concentration with a KmO2app = 342 ± 64 μM and kcatapp = 1.34 ± 0.09 s-1 (Figure 4). Importantly, the F359W catalyzed reaction is not cooperative with respect to oxygen when dehydroepiandrosterone is utilized as the substrate. The F359W catalyzed reaction also shows a hyperbolic dependence on oxygen concentration with a KmO2app = 544 ± 73 μM and kcatapp = 1.35 ± 0.08 s-1 (Figure 4). This result indicates that the kinetic cooperativity observed with cholesterol is lost when the rate(s) of kinetic step(s) subsequent to oxygen binding are reduced.
Figure 4.

Initial velocities for wild-type and F359W cholesterol oxidases were measured over a range of O2 concentrations with a fixed dehydroepiandrosterone concentration of 140 μM and fit to equation (1). ○, WT; ●, F359W. The data shown are the average of two independent experiments, and the errors are the standard deviation of measurement.
Atomic resolution structure of the F359W mutant
The crystal structure of the F359W mutant was refined to 0.95 Å resolution (Table 4). The mutant crystallizes in the same space group as the wild-type enzyme and the overall structure of the mutant is the same. At this resolution in the wild-type structure, multiple conformations of amino acid residues were resolved into two distinct populations. In population A, the tunnel between solvent and active site is closed (Figure 1A). In population B, the tunnel residues are rotated out of the tunnel and an open passage is formed (28) (Figure 1B).
In population A of the wild-type structure, the phenylalanine side-chain fills the tunnel, and in population B, it undergoes an 18° rotation about χ1 and the tunnel becomes solvent accessible. In the mutant structure, the indole ring of Trp359 is flipped into the tunnel in a position analogous to to the conformation of Phe359 in population A of the wild-type structure (Figure 1C, 1D and 5). A second conformation of Trp359 is not detected in the electron density maps (Figure 5) and as a result, no tunnel opening can be seen (Figure 1C, 1D). The conformations of the other tunnel residues as well as the active site residues differ little from those of the wild-type enzyme, with an RMSD = 0.144 Å between the two structures. The remaining residues that occupy two conformations in the wild-type structure also occupy two conformations in the F359W structure.
Figure 5.
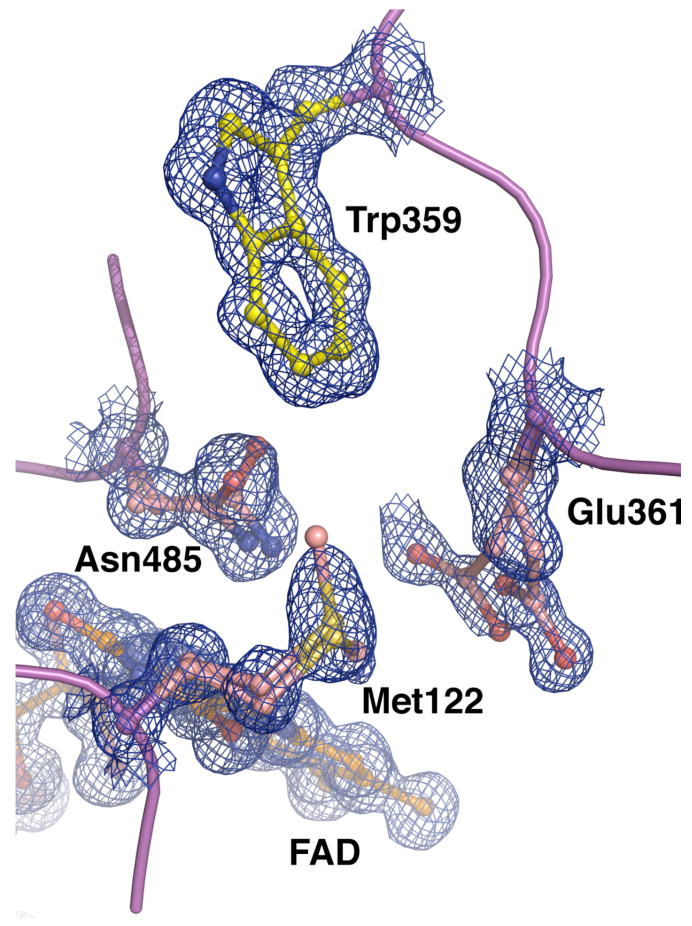
Electron density for a portion of the F359W active site, including Trp359. The density (blue mesh) was calculated using the coefficients 2Fo-Fc and contoured at 1.0 σ. Alternate conformations for Glu361, Met122 and Asn485 could clearly be resolved, while no alternate conformations for Trp359 were observed.
Discussion
The mutation of phenylalanine 359 to tryptophan results in a 13-fold decrease in catalytic turnover. Importantly, the reduced mutant enzyme is still oxidized by oxygen with multiple catalytic cycles. The atomic resolution structure of the mutant with a tryptophan at position 359 reveals the putative oxygen tunnel obstructed by the indole moiety. If the mutation blocked the tunnel to such an extent that oxygen can no longer traverse the tunnel, and oxygen is forced to access the active site via the steroid-binding site, a ternary complex of oxygen and steroid would no longer be formed. The steroid-binding site is not sufficiently large to accommodate oxygen in addition to steroid. Kinetically, a ping-pong mechanism of substrate binding would be observed. That is, the steroid binding, conversion, and release would have to occur before oxygen binding and turnover. However, the kinetic profile of the F359W mutant, as well as those of the other two mutants (Figure 3, Table 3), are inconsistent with this mechanism and the model was not considered further.
The reactions catalyzed by the F359W and G347N mutants do not fit a classical ternary complex mechanism either. Instead, the mutations introduce positive cooperativity with respect to oxygen concentration. Cooperativity is generally associated with multi-subunit proteins that relay binding occupancy and structural conformation information between subunits, for example, as occurs upon binding of oxygen to hemoglobin. Cholesterol oxidase is a monomeric enzyme, and thus, it is distinctly improbable that the cooperativity results from multi-site binding of oxygen with successive changes in association constants and quaternary structure. The proposed wild-type gas-binding tunnel is large enough to accommodate two oxygen molecules. One could be bound as substrate and the second as an allosteric effector. However, what remains of the tunnel in the F359W mutant is only large enough to accommodate half of one oxygen molecule (one water is observed in the electron density maps), and it is the mutant that shows positive cooperative behavior, not the wild-type enzyme. Furthermore, additional oxygen binding cavities were not observed in the mutant structure. Thus, a multi-site binding model was not consistent with either the structural data or the wild type kinetics.
Next, a kinetic cooperativity model was considered (44, 45). Monomeric enzymes display kinetic cooperativity under non-equilibrium conditions that are often due to a slow protein conformational change. In this model, two different protein conformations, E and E′, can bind O2 followed by cholesterol to form a ternary complex, E′•O2•cholesterol and the ternary complex reacts to form products (Scheme 1). The reaction of E′ is faster than the reaction of E. The protein conformation E′ persists after product release and at high oxygen concentrations, oxygen can bind to E′ before it has time to equilibrate to the more stable enzyme form, E. This kinetic trapping of E′ by O2 occurs because the two conformations, E and E′, interconvert slowly and the rate of binding of O2 is first-order in oxygen concentration. In this mechanism, the more active enzyme conformation E′ appears to be remembered because it relaxes back to the E form so slowly and this type of kinetic cooperativity is known as mnemonic (44, 45). There are two consequences of this mechanism. The first consequence is that the reaction kinetics only display cooperativity for the first or preferred substrate, and no cooperativity for the second substrate. In the case presented here, there is no cooperativity with respect to cholesterol. The second consequence is that the positive cooperativity will increase with increasing concentrations of the second substrate. Again, this is the case for the F359W cholesterol oxidase.
Scheme 1.

Mnemonic model for kinetically cooperative oxygen reaction in the mutant-catalyzed reactions. Adapted from the work of Cornish-Bowden and Ricard (44, 45).
The mnemonic model is consistent with both the F359W structure and kinetics. The tunnel is closed regardless of the conformation of the other tunnel residues (Figure 1C and 1D). As oxygen passage through the tunnel is sterically blocked by the mutation, it is assumed that oxygen must bind by another means and this route is slower than the native binding mechanism through the tunnel. One possibility is diffusion through the protein by many different paths. After chemical conversion of cholesterol and O2 to cholest-4-en-3-one and H2O2, respectively, the products are released. If the kinetic barrier to H2O2 diffusion through the protein is much higher than the barrier to H2O2 diffusion through the tunnel, the latter pathway will predominate. Alternatively, a second transient, and as yet undetected, pathway may open to release H2O2 as is postulated in the case of rabbit lipoxygenase (15). Upon H2O2 release, the exit path will be open. Because the open and closed tunnel conformations interconvert slowly and are not in equilibrium in the presence of high oxygen concentrations, oxygen can bind via the tunnel. This binding pathway is faster than diffusion through the protein and cooperativity in time is observed. This mnemonic mechanism suggests that the F359W tunnel or an alternate pathway can be in an open conformation, but this conformation is much higher in energy than the closed form, and the open-closed interconversion is kinetically slow.
The observation of a single tryptophan 359 conformation in the mutant structure suggests that the tryptophan conformation analogous to conformation B of phenylalanine 359 is not populated in the absence of substrate. In addition, rotation about χ1 of tryptophan 359 cannot occur without simultaneous movement in the surrounding protein structure because unfavorable van der Waals contacts would occur during rotation. The increased steric bulk of the indole ring over that of a phenyl ring must destabilize conformation B and raise the barrier to its formation. The precise structure of the F359W tunnel open conformation is not known.
An alternative model is that oxygen only binds by diffusion through the protein and that hydrogen peroxide is only released by diffusion. In this case, there is not a defined pathway of binding, but many paths would contribute to oxygen binding. If there were multiple accessible pathways, it would be very unlikely that the conformation of the enzyme (E′ in Scheme 1) released upon hydrogen peroxide exit would be in the optimal conformation for oxygen uptake, and kinetic cooperativity would not be observed upon mutation of Phe359 or Gly347.
We cannot completely exclude an alternative mechanism in which the substrates bind in random order, and the flux through the oxygen binding first pathway versus the cholesterol binding first pathway shifts upon changing oxygen concentration (46, 47). This mechanism requires that substrate binding not be at equilibrium, that is, steps subsequent to ternary complex formation are fast relative to substrate binding. The implication that results is that mutation of tunnel residues slows down oxygen binding and our conclusions about the importance of a protein tunnel through which oxygen binds are unchanged.
The hydrophobic tunnel in cholesterol oxidase, as well as other oxidases, might be functionally most important when used as an exit for hydrogen peroxide. Upon hydrogen peroxide reaction with amino acid side chains, the enzyme structures could be altered in ways deleterious to catalytic activity. It is possible that hydrophobic tunnels evolved to prevent the reactivity of the product from damaging the enzyme. Despite the relative hydrophilicity of hydrogen peroxide, a hydrophobic tunnel that lacked easily oxidized heteroatoms would be less susceptible to oxidative damage than a polar tunnel. In addition, channeling of oxygen to the reduced flavin via a tunnel would limit the peroxidation of the susceptible cholest-5-en-3-one intermediate.
The rest of the glucose-methanol-choline (GMC) oxidoreductase superfamily was surveyed for the presence of tunnel features in each of the structures. Few tunnel residues are conserved in the structure-based alignments except Phe 359 from cholesterol oxidase. Other non-GMC oxidoreductases have tunnels that connect the protein surface to the active site, e.g., D-amino acid oxidase (48), H. polymorpha copper-containing amine oxidase (18), and type II cholesterol oxidase (26). The conserved features are conformational flexibility, which is a feature not readily identified through sequence or structure alignments, and hydrophobicity, which can be satisfied by many constellations of hydrophobic amino acids. The multiplicity of solutions amongst oxidoreductases is similar to the structural diversity seen for ammonia transfer tunnels in different carbamoylphosphate synthetases (49) and oxygen tunnels in lipoxygenases (15, 16).
In addition, kinetic cooperativity has been observed in the steady-state reaction catalyzed by another glucose-methanol-choline oxidoreductase family member. Mutation of valine 464 in choline oxidase results in two conformers of the enzyme that slowly interconvert (S. Finnegan and G. Gadda, personal communication). The carbonyl of valine 464 forms a hydrogen bond to N5 of the flavin in the native enzyme structure (50) and thus it could act as a sensor of flavin oxidation state to gate oxygen access. Although a tunnel is not observed in the native structure, this may be due to the presence of a flavin adduct in the active site that would preclude a tunnel opening conformational change by asparagine 510, the ortholog of asparagine 485 in cholesterol oxidase that appears to the gate the conformation of other tunnel residues.
In conclusion, hydrophobic tunnels have been observed in many other protein structures, and their function as gas-binding tunnels hypothesized. The mutagenesis of three tunnel residues, and their kinetic and structural characterization presented here provide functional evidence for the utility of the hydrophobic tunnel that spans ∼18 Å between the solvent accessible surface and the active site of cholesterol oxidase. This tunnel provides oxygen access to the enzyme active site, and, in the reverse direction, hydrogen peroxide access to solvent. That is, there is a specific binding pathway for oxygen to reach the reduced flavin cofactor and for hydrogen peroxide release. Our observations are in accordance with the observations of functional oxygen binding tunnels in the lipoxygenases (15). Similar tunnels observed in other protein structures are likely to function in a similar fashion. Their evolutionary selection is presumably derived from the need for temporal control as well as regiospecific control over oxygen delivery to reactive intermediates in enzyme-catalyzed reactions and protection of the enzyme from reactive products like hydrogen peroxide.
Abbreviations
- chox
cholesterol oxidase
- choA
Streptomyces cholesterol oxidase
- LB
Luria broth
- IPTG
isopropyl α-D-thiogalactoside
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- HRP
horseradish peroxidase
- 2×YT
2× yeast-tryptone broth
- BSA
bovine serum albumin
- GMC
glucose-methanol-choline
- FAD
flavin adenine dinucleotide
- K.I.E.
kinetic isotope effects
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
Footnotes
This work was supported by the National Institutes of Health Grants HL53306 (N.S.S) and GM63262 (A.V.).
References
- 1.Tilton RF, Jr, Kuntz ID, Jr, Petsko GA. Cavities in proteins: structure of a metmyoglobin-xenon complex solved to 1.9 A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 2.Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- 3.Duff AP, Trambaiolo DM, Cohen AE, Ellis PJ, Juda GA, Shepard EM, Langley DB, Dooley DM, Freeman HC, Guss JM. Using xenon as a probe for dioxygen-binding sites in copper amine oxidases. J Mol Biol. 2004;344:599–607. doi: 10.1016/j.jmb.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 4.de Sanctis D, Dewilde S, Pesce A, Moens L, Ascenzi P, Hankeln T, Burmester T, Bolognesi M. Mapping protein matrix cavities in human cytoglobin through Xe atom binding. Biochem Biophys Res Commun. 2004;316:1217–1221. doi: 10.1016/j.bbrc.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Raushel FM, Thoden JB, Holden HM. Enzymes with molecular tunnels. Acc Chem Res. 2003;36:539–548. doi: 10.1021/ar020047k. [DOI] [PubMed] [Google Scholar]
- 6.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 7.Huang X, Raushel FM. Restricted passage of reaction intermediates through the ammonia tunnel of carbamoyl phosphate synthetase. J Biol Chem. 2000;275:26233–26240. doi: 10.1074/jbc.275.34.26233. [DOI] [PubMed] [Google Scholar]
- 8.Raushel FM, Thoden JB, Holden HM. The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry. 1999;38:7891–7899. doi: 10.1021/bi990871p. [DOI] [PubMed] [Google Scholar]
- 9.Fan Y, Lund L, Yang L, Raushel FM, Gao YQ. Mechanism for the transport of ammonia within carbamoyl phosphate synthetase determined by molecular dynamics simulations. Biochemistry. 2008;47:2935–2944. doi: 10.1021/bi701572h. [DOI] [PubMed] [Google Scholar]
- 10.Manjasetty BA, Powlowski J, Vrielink A. Crystal structure of a bifunctional aldolase-dehydrogenase: sequestering a reactive and volatile intermediate. Proc Natl Acad Sci U S A. 2003;100:6992–6997. doi: 10.1073/pnas.1236794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volbeda A, Fontecilla-Camps JC. Crystallographic evidence for a CO/CO(2) tunnel gating mechanism in the bifunctional carbon monoxide dehydrogenase/acetyl coenzyme A synthase from Moorella thermoacetica. J Biol Inorg Chem. 2004;9:525–532. doi: 10.1007/s00775-004-0565-9. [DOI] [PubMed] [Google Scholar]
- 12.Tan X, Volbeda A, Fontecilla-Camps JC, Lindahl PA. Function of the tunnel in acetylcoenzyme A synthase/carbon monoxide dehydrogenase. J Biol Inorg Chem. 2006;11:371–378. doi: 10.1007/s00775-006-0086-9. [DOI] [PubMed] [Google Scholar]
- 13.Knapp MJ, Klinman JP. Kinetic studies of oxygen reactivity in soybean lipoxygenase-1. Biochemistry. 2003;42:11466–11475. doi: 10.1021/bi0300884. [DOI] [PubMed] [Google Scholar]
- 14.Knapp MJ, Seebeck FP, Klinman JP. Steric control of oxygenation regiochemistry in soybean lipoxygenase-1. J Am Chem Soc. 2001;123:2931–2932. doi: 10.1021/ja003855k. [DOI] [PubMed] [Google Scholar]
- 15.Saam J, Ivanov I, Walther M, Holzhutter HG, Kuhn H. Molecular dioxygen enters the active site of 12/15-lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci U S A. 2007;104:13319–13324. doi: 10.1073/pnas.0702401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furse KE, Pratt DA, Schneider C, Brash AR, Porter NA, Lybrand TP. Molecular dynamics simulations of arachidonic acid-derived pentadienyl radical intermediate complexes with COX-1 and COX-2: insights into oxygenation regio- and stereoselectivity. Biochemistry. 2006;45:3206–3218. doi: 10.1021/bi052338h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee A, Brinkley DW, Chang KM, Roth JP. Molecular oxygen dependent steps in fatty acid oxidation by cyclooxygenase-1. Biochemistry. 2007;46:3975–3989. doi: 10.1021/bi602502j. [DOI] [PubMed] [Google Scholar]
- 18.Johnson BJ, Cohen J, Welford RW, Pearson AR, Schulten K, Klinman JP, Wilmot CM. Exploring molecular oxygen pathways in Hansenula polymorpha copper-containing amine oxidase. J Biol Chem. 2007;282:17767–17776. doi: 10.1074/jbc.M701308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilce MCJ, Dooley DM, Freeman HC, Guss JM, Matsunami H, McIntire WS, Ruggiero CE, Tanizawa K, Yamaguchi H. Crystal structures of the copper-containing amine oxidase from Arthrobacter globiformis in the holo and apo forms: implications for the biogenesis of topaquinone. Biochemistry. 1997;36:16116–16133. doi: 10.1021/bi971797i. [DOI] [PubMed] [Google Scholar]
- 20.Mims MP, Porras AG, Olson JS, Noble RW, Peterson JA. Ligand binding to heme proteins. An evaluation of distal effects. J Biol Chem. 1983;258:14219–14232. [PubMed] [Google Scholar]
- 21.Ostermann A, Waschipky R, Parak FG, Nienhaus GU. Ligand binding and conformational motions in myoglobin. Nature. 2000;404:205–208. doi: 10.1038/35004622. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt M, Nienhaus K, Pahl R, Krasselt A, Anderson S, Parak F, Nienhaus GU, Srajer V. Ligand migration pathway and protein dynamics in myoglobin: a time-resolved crystallographic study on L29W MbCO. Proc Natl Acad Sci U S A. 2005;102:11704–11709. doi: 10.1073/pnas.0504932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott EE, Gibson QH. Ligand migration in sperm whale myoglobin. Biochemistry. 1997;36:11909–11917. doi: 10.1021/bi970719s. [DOI] [PubMed] [Google Scholar]
- 24.Hiromoto T, Fujiwara S, Hosokawa K, Yamaguchi H. Crystal structure of 3-hydroxybenzoate hydroxylase from Comamonas testosteroni has a large tunnel for substrate and oxygen access to the active site. J Mol Biol. 2006;364:878–896. doi: 10.1016/j.jmb.2006.09.031. [DOI] [PubMed] [Google Scholar]
- 25.Moustafa IM, Foster S, Lyubimov AY, Vrielink A. Crystal structure of LAAO from Calloselasma rhodostoma with an L-phenylalanine substrate: insights into structure and mechanism. J Mol Biol. 2006;364:991–1002. doi: 10.1016/j.jmb.2006.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coulombe R, Yue KQ, Ghisla S, Vrielink A. Oxygen access to the active site of cholesterol oxidase through a narrow channel is gated by an Arg-Glu pair. J Biol Chem. 2001;276:30435–30441. doi: 10.1074/jbc.M104103200. [DOI] [PubMed] [Google Scholar]
- 27.Montet Y, Amara P, Volbeda A, Vernede X, Hatchikian EC, Field MJ, Frey M, Fontecilla-Camps JC. Gas access to the active site of Ni-Fe hydrogenases probed by X-ray crystallography and molecular dynamics. Nat Struct Biol. 1997;4:523–526. doi: 10.1038/nsb0797-523. [DOI] [PubMed] [Google Scholar]
- 28.Lario P, Sampson NS, Vrielink A. Sub-atomic resolution crystal structure of cholesterol oxidase: What atomic resolution crystallography reveals about enzyme mechanism and the role of the FAD cofactor in redox activity. J Mol Biol. 2003;326:1635–1650. doi: 10.1016/s0022-2836(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 29.Sampson NS, Kass IJ. Isomerization, but not oxidation, is suppressed by a single point mutation, E361Q, in the reaction catalyzed by cholesterol oxidase. J Am Chem Soc. 1997;119:855–862. [Google Scholar]
- 30.Ye Y, Lario P, Vrielink A, Sampson NS. The presence of a hydrogen bond betwen asparagine 485 and the π system of FAD modulates the redox potential in the reaction catalyzed by cholesterol oxidase. Biochemistry. 2001;40:13779–13787. doi: 10.1021/bi010843i. [DOI] [PubMed] [Google Scholar]
- 31.Kass IJ, Sampson NS. The importance of Glu361 position in the reaction catalyzed by cholesterol oxidase. Bioorg Med Chem Lett. 1998;8:2663–2668. doi: 10.1016/s0960-894x(98)00478-8. [DOI] [PubMed] [Google Scholar]
- 32.Stankovich MT. An anaerobic spectroelectrochemical cell for studying the spectral and redox properties of flavoproteins. Anal Biochem. 1980;109:295–308. doi: 10.1016/0003-2697(80)90652-1. [DOI] [PubMed] [Google Scholar]
- 33.Stankovich M. Potentiometric Measurements of Proteins. In: Wilson GS, editor. Bioelectrochemistry. Wiley-VCH; Weinheim: 2002. pp. 487–509. [Google Scholar]
- 34.Lyubimov AY, Lario PI, Moustafa I, Vrielink A. Atomic resolution crystallography reveals how changes in pH shape the protein microenvironment. Nat Chem Biol. 2006;2:259–264. doi: 10.1038/nchembio784. [DOI] [PubMed] [Google Scholar]
- 35.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Sheldrick GM, Schneider TR. SHELXL: High-resolution refinement. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology. Academic Press; Boston: 1997. pp. 319–343. [PubMed] [Google Scholar]
- 39.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 40.Project CC. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. The PyMOL Molecular Graphics System 2006 [Google Scholar]
- 42.Chen L. PhD Thesis. Department of Chemistry, Stony Brook University; Stony Brook, NY: 2007. The binding and release of oxygen and hydrogen peroxide are directed through a hydrophobic tunnel in cholesterol oxidase; p. 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyubimov A. PhD Thesis. Department of Chemistry, University of California; Santa Cruz, CA: 2006. A structural study of cholesterol oxidase at atomic and subatomic resoultion; p. 125. [Google Scholar]
- 44.Cornish-Bowden A, Cardenas ML. Co-operativity in monomeric enzymes. J Theor Biol. 1987;124:1–23. doi: 10.1016/s0022-5193(87)80248-5. [DOI] [PubMed] [Google Scholar]
- 45.Ricard J, Cornish-Bowden A. Co-operative and allosteric enzymes: 20 years on. Eur J Biochem. 1987;166:255–272. doi: 10.1111/j.1432-1033.1987.tb13510.x. [DOI] [PubMed] [Google Scholar]
- 46.Pettersson G. Mechanistic origin of the sigmoidal rate behaviour of glucokinase. Biochem J. 1986;233:347–350. doi: 10.1042/bj2330347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pettersson G. Mechanistic origin of the kinetic cooperativity of hexokinase type L1 from wheat germ. Eur J Biochem. 1986;154:167–170. doi: 10.1111/j.1432-1033.1986.tb09374.x. [DOI] [PubMed] [Google Scholar]
- 48.Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone MS, Ghisla S. The x-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc Natl Acad Sci U S A. 2000;97:12463–12468. doi: 10.1073/pnas.97.23.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weeks A, Lund L, Raushel FM. Tunneling of intermediates in enzyme-catalyzed reactions. Curr Opin Chem Biol. 2006;10:465–472. doi: 10.1016/j.cbpa.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Quaye O, Lountos GT, Fan F, Orville AM, Gadda G. Role of Glu312 in binding and positioning of the substrate for the hydride transfer reaction in choline oxidase. Biochemistry. 2008;47:243–256. doi: 10.1021/bi7017943. [DOI] [PubMed] [Google Scholar]