

Abstract
In this manuscript we report that oxidative cleavage reactions can be used to form oxocarbenium ions that react with pendent epoxides to form bicyclic epoxonium ions as an entry to the formation of cyclic oligoether compounds. Bicyclic epoxonium ion structure was shown to have a dramatic impact on the ratio of exo- to endo-cyclization reactions, with bicyclo[4.1.0] intermediates showing a strong preference for endo-closures and bicyclo[3.1.0] intermediates showing a preference for exo-closures. Computational studies on the structures and energetics of the transition states using the B3LYP/6-31G(d) method provide substantial insight into the origins of this selectivity.
Introduction
Cascade reactions, in which multiple bond forming processes occur in a single operation,1 have been shown to be effective for executing rapid increases in molecular complexity, as demonstrated by the biosyntheses of terpenes from polyolefins2 and cyclic ethers from polyepoxides.3 In these pathways strict regiocontrol must be achieved in each cyclization step to form products with high efficiency. Effective mimicry of these processes in the absence of enzymatic mediation requires an understanding of the inherent reactivity patterns of the relevant intermediates in these transformations and of the manner in which they can be manipulated. Of particular importance is determining the structural features that promote exo- and endo-cyclizations (Scheme 1).
Scheme 1.

Exo- and endo-cyclizations using alkene and epoxide groups.
Cyclic ether synthesis from polyepoxides has been demonstrated under acidic,4 basic,5 and oxidative6 conditions. While exo-cyclizations are most commonly observed, recently several reports have appeared in the literature that demonstrate favorable endo-cyclizations leading to fused polyethers. In a series of papers, McDonald and coworkers have convincingly proposed7 that bicyclic epoxonium ions, formed from nucleophilic attacks of epoxides on Lewis acid-activated epoxides, are key intermediates in the formation of fused polycyclic ethers. In these reactions endo-cyclizations were postulated to be favored due to the ring strain accrued during bicyclic epoxonium formation through exo-pathways. Holton proposed6b that endo-cyclizations can be promoted through the use of a polar solvent to promote the SN1-type transition state, in accord with computational studies.8 We have undertaken a program in which cascade cyclizations are initiated through intramolecular additions of epoxides to oxidatively generated oxocarbenium ions9 to determine whether other factors such as epoxonium ion structure and Lewis acid selection could also impact the outcomes of these processes. This activation protocol diminishes the potential for product isomerizations that can complicate mechanistic analyses of acid-mediated reactions,10 making it ideal for studying the inherent reactivity patterns of epoxonium ions with various structures.
To elucidate the epoxonium ion structural features that impact cyclization regiochemistry, we studied the effects of changing the degree of substitution in the precursor epoxide (disubstituted vs. trisubstituted), the ring size of the epoxonium ion (bicyclo[4.1.0] ions vs. bicyclo[3.1.0] ions), and the electrophilic alkyl group that reacts with the epoxide to form the epoxonium ion (oxocarbenium ion vs. carbenium ion). In this manuscript we show that each of these features can impact the partitioning of the reaction through exo- or endo-pathways. Transition structure energies of the exo- and endo-cyclizations were studied computationally at the B3LYP/6-31G(d) level, providing insight into the origins of the selectivity and a predictive model for the design of future cascade reactions.
Results and Discussion
Substrate Design and Synthesis
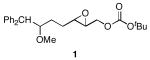
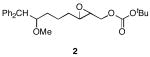
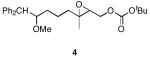
We designed a set of mono- and diepoxide cyclization substrates to determine the impact of changing several epoxonium ion structural features on cyclization regioselectivity (Figure 1). Monoepoxide substrates 1-4 were designed to probe the importance of bicyclic epoxonium ion ring size ([3.1.0] vs. [4.1.0]) and substitution pattern (disubstituted vs. trisubstituted) on cyclization efficiency and product distribution when oxocarbenium ions are used as activating groups (tethered Lewis acids). Diepoxide substrates 5-9 were designed to determine whether regioselectivity in the terminal cyclization event is sensitive to the identity of the cationic activating group since the relevant epoxonium ions are activated by unstabilized carbenium ions rather than oxocarbenium ions. This study was also designed to determine whether cyclization efficiency is affected by the relative stereochemical orientations of the epoxide groups. Figure 2 illustrates the structural features of the intermediate epoxonium ions that will be compared in this manuscript. The diphenylmethyl group was selected as the electroauxiliary11 because it is more reactive than an unsubstituted benzyl group,12 as required for cyclizations of weakly nucleophilic epoxycarbonates, and because it is easily introduced through the addition of Ph2CHLi (prepared from inexpensive diphenylmethane and nBuLi) to aldehydes.
Figure 1.

Cyclization substrates.
Figure 2.

Epoxonium ion substitution patterns to be compared in this study.
The monoepoxide substrates were synthesized according to the straightforward sequences shown in Scheme 2. These compounds were prepared as racemic mixtures since our sole objective was to determine the cyclization regiochemistry and relative stereochemical orientations in the reaction products. Disubstituted epoxides 1 and 2 were synthesized from unsaturated aldehydes 10 and 11 by identical pathways that proceeded through aldehydes 12 and 13. Alkenylation, reduction, epoxidation, and carbonate formation13 provided the desired substrates. The syntheses of trisubstituted epoxides 3 and 4 commenced by opening epoxide 1414 under Yamamoto's conditions15 and forming aldehyde 15 through a Claisen rearrangement/reduction sequence. This aldehyde was either taken directly to 3 or was homologated through methoxymethylenation and mercury-mediated enol ether hydrolysis.16 The resulting homologated aldehyde was converted to 4 in an identical manner to the conversion of 15 to 3.
Scheme 2.

Synthesis of monoepoxide substrates.
Reagents and conditions a) Ph2CH2, nBuLi, THF, 0 °C, 83% (n = 1), 70% (n = 2). b) NaH, DMF, 0 °C, then MeI, 97% (n = 1), 95% (n = 2). c) O3, CH2Cl2, −78 °C, then Ph3P, rt, 96% (n = 1 or 2). d) (EtO)2P(O)CH2CO2Et, NaH, THF, 0 °C, 94% (n = 1), 93% (n = 2). e) DIBAL-H, THF, −78 °C. f) m-CPBA, NaHCO3, CH2Cl2, 0 °C 70% (n = 1), 91% (n = 2), two steps. g) Boc2O, N-methylimidazole, PhMe, 0 °C, 90% (n = 1), 84% (n = 2). h) 2,2,6,6-Tetramethylpiperidine, nBuLi, Et2AlCl, benzene, 0 °C, 90%. i) (EtO)3CCH3, propionic acid, 145 °C, 96%. j) DIBAL-H, CH2Cl2, −78°C. k) Ph2CH2, nBuLi, THF, 0 °C, 70% (n = 1), 79% (n = 2). l) NaH, DMF, 0 °C, then MeI. m) Bu4NF, THF, 100% (n = 1), 97% (n = 2), two steps. n) m-CPBA, NaHCO3, CH2Cl2, 0 °C, 95% (n = 1), 99% (n = 2). o) Boc2O, N-methylimidazole, PhMe, 0 °C, 86% (n = 1), 89% (n = 2). p) Ph3P+CH2OMe Cl-, NaHMDS, THF, −78 °C, then Hg(OAc)2, THF, H2O, KI, 82%.
The synthesis of diepoxide substrates 5-8 (Scheme 3) parallels the synthesis of 3 and 4, with the exception that asymmetric epoxidation reactions were employed to assure high diastereocontrol. Known epoxide 1617 was converted to aldehyde 17, which could either be transformed to 5 and 6 or homologated and converted to 7 and 8. Substrates 5 and 7, with (2R, 3R, 6R, 7R) stereochemical orientations, were formed from a double Shi epoxidation of dienes 18 and 19,18 while substrates 6 and 8, with (2S, 3S, 6R, 7R) stereochemical orientations were prepared through sequential Sharpless19 and Shi epoxidations.
Scheme 3.

Synthesis of diepoxide cyclization substrates 5-8.
Reagents and conditions a) 2,2,6,6-Tetramethylpiperidine, nBuLi, Et2AlCl, benzene, 0 °C, 92%. b) (EtO)3CCH3, propionic acid, 145 °C, 94%. c) DIBAL-H, CH2Cl2, −78°C, 92%. d) Ph2CH2, nBuLi, THF, 0 °C, 80% (n = 1), 80% (n = 2). e) NaH, DMF, 0 °C, then MeI. f) Bu4NF, THF, 97% (n = 1), 98% (n = 2), two steps. g) Ph3P+CH2OMe Cl-, NaHMDS, THF, −78 °C, then Hg(OAc)2, THF, H2O, KI, 91%. h) Shi catalyst, KHSO5, (MeO)2CH2, CH3CN, H2O, 0 °C, 88% (5), 91% (6), 64% (7). i) Boc2O, N-methylimidazole, PhMe, 0 °C, 93% (5), 86% (6), 78% (7), 82% (8, two steps). j) (+)–Diisopropyl tartrate, tBuOOH, Ti(OiPr)4, CH2Cl2, −25 °C, 96% (n = 1), 97% (n = 2).
Substrate 9 was prepared (Scheme 4) through a convergent sequence that employed a Julia-Kocienski olefination20 between sulfone 20 (5 steps from valerolactone) and aldehyde 15. The resulting alkene was converted to the desired product through the sequence that was used for the preparation of 5 and 7.
Scheme 4.

Convergent synthesis of epoxide 9.
Reagents and conditions a) DIBAL-H, CH2Cl2, −78 °C. b) Ph2CH2, nBuLi, THF, 0 °C, 82% (2 steps). c) 1-Phenyl-1H-tetrazole-5-thiol, Ph3P, DIAD, THF, 97%. d) NaH, DMF, 0 °C, then MeI, 85%. e) m-CPBA, NaHCO3, CH2Cl2, 0 °C, 95%. f) KHMDS, DME, −78 °C, then 15, 63%. g) Bu4NF, THF, 95%. h) Shi catalyst, KHSO5, (MeO)2CH2, CH3CN, H2O, −5 °C, 94%. i) Boc2O, N-methylimidazole, PhMe, 83%.
Cyclization reactions
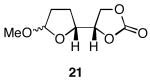
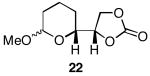
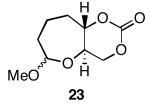
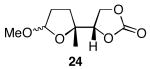
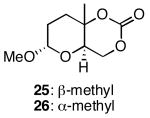
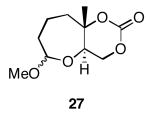
The results of the oxidative cyclization reactions of monoepoxides 1-4 are shown in Table 1. Essential components in these photochemical reactions (medium pressure mercury lamp, Pyrex filtration) were N-methylquinolinium hexafluorophosphate (NMQPF6) as a catalytic oxidant,21 O2 (from air) as the terminal oxidant,22 and toluene as a cosensitizer. Disubstituted epoxide 1 provided exo-product 21 as a mixture of anomers, as expected based on analogy to previous studies.9 Cyclization of the homologated substrate 2 yielded in a mixture of exo-product 22 and endo-product 23, with the exo-pathway being slightly favored. The formation of 23 was quite interesting since the difference between the reactions of 1 and 2 is simply that the initial cyclization of 1 forms a bicyclo[3.1.0] epoxonium ion and the cyclization of 2 proceeds through a bicyclo[4.1.0] epoxonium ion. Endo-cyclizations into bicyclo[4.1.0] epoxonium ions have also been observed by McDonald and coworkers23 from Lewis acid mediated processes in which epoxonium ions are formed by adding to non-stabilized carbenium ions and are opened by epoxide nucleophiles. Trisubstituted epoxide 3 also produced a complex product mixture upon oxidation, with both exo- and endo-cyclization products being formed. The stabilizing effect of the methyl group on the cationic intermediate modestly impacted the regioselectivity of the cyclization reaction but was insufficient to override the preference for the exo-pathway. Also of note, the endo-cyclizations proceeded from both inversion and retention of the epoxonium ion to form a mixture of trans- and cis-fused products. Homologous epoxide 4 proved to be the best substrate for this study, yielding endo-product 27 in 73% yield. Thus the combined impacts of the methyl substitution and the bicyclo[4.1.0] intermediate lead to a complete reversal of the exo-preference that was observed in the cyclization of 1.
Table 1.
Cyclization reactions of monoepoxide substrates.
Yields are reported for isolated materials unless otherwise stated. See the Supporting Information for procedures.
Yields are based on NMR analyses of product mixtures.
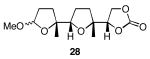
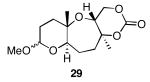
The results of the cyclization reactions of the diepoxide substrates generally followed those of the monoepoxide substrates, though some subtle but noteworthy differences were observed. Diepoxides 5 and 6, in which the initial cyclization reaction produces a bicyclo[3.1.0] epoxonium ion, yield only the products of consecutive exo- (28 and 30) or consecutive endo-cyclizations (29 and 31), with the all exo-pathway being preferred. No products from an exo-cyclization followed by an endo-cyclization or vice versa were isolated, indicating that bicyclo[3.1.0] epoxonium ions that form from the addition of epoxides to carbenium ions show a higher fidelity toward opening through the exo-pathway than those that form from the addition of epoxides to oxocarbenium ions. Syn-products from the endo-cyclizations were not observed, indicating either that epoxides are superior nucleophiles to carbonates or that carbonates react with epoxonium ions reversibly prior to tert-butyl cation loss. Reactions of substrates 7 and 8, in which the initial cyclizations produce bicyclo[4.1.0] epoxonium ions, proceed exclusively through consecutive endo-pathways to provide fused tricycles 32 and 33. The formation of 33 was remarkably efficient in consideration of the complexity of the product. The higher yield for the cyclization of 8 relative to 7 presumably reflects diminished steric interactions in the cyclization reactions. As expected, when disubstituted epoxide 9 was used in the initial cyclization the level of regioselectivity dropped, though all endo-product 34 was formed in a slight excess over all exo-product 35. Product regiochemical and stereochemical outcomes for the cyclizations of 7 and 9 were confirmed by treating the acetals with BF3•OEt2, and m-CPBA, then adding Et3N (Scheme 5).24 The structures of the resulting lactones 36 and 37 were unambiguously confirmed through crystallographic analysis.25
Scheme 5.

Lactone formation from acetals.
Computational studies
The results of the cyclization reactions demonstrate that the regiochemical outcomes of intramolecular nucleophilic additions into bicyclic epoxonium ions are highly structurally dependent. Activating epoxides through coordinating to oxocarbenium ions rather than unstabilized carbenium ions erodes the strong preference for the exo-pathway in bicyclo[3.1.0] epoxonium ions. Trisubstituted epoxide substrates show a higher preference for endo-cyclizations relative to diepoxide substrates, as predicted by their enhanced ability to stabilize positive charge, though the magnitude of effect is small. Most notable, however, is the dramatic impact of ring size on cyclization regioselectivity. Bicyclo[3.1.0] epoxonium ions profoundly favor reaction through the exo-pathway while bicyclo[4.1.0] epoxonium ions favor the endo-pathway. While the exo-cyclizations can be understood based on analogies to Baldwin's rules,26 the endo-cyclizations are difficult to rationalize. This led us to initiate computational studies using the B3LYP/6-31G(d) method27 to explore the origin of the structural basis for our experimental observations. The method applied here is known to overestimate the exothermicity of the ring openings of the epoxonium ions by 2 to 3 kcal/mol when compared to more accurate CCSD(T) or CBS-Q calculations, respectively.28 The calculations were carried out by using Gaussian0329 program. All of the geometries were fully optimized and ground state structures were verified to have no imaginary frequencies. The transition state geometries with one imaginary frequency were verified to connect the desired reactants to the desired products via IRC (imaginary rection coordinates) calculations.
Transition structures for the nucleophilic addition of dimethyl carbonate to the 1,2,2,3-tetramethyloxiranium ion were computed to reveal the geometry of the transition state in the absence of geometrical constraints. The nucleophile, dimethyl carbonate, can add either to the secondary or tertiary carbon of the epoxonium ion in this model reaction. These two competing pathways for the nucleophilic addition of the dimethyl carbonate to the 1,2,2,3-tetramethyloxiranium ion are shown in Scheme 6.
Scheme 6.

Bimolecular epoxonium ion opening.
The transition structures (TS) for the addition of dimethyl carbonate to the 1,2,2,3-tetramethyloxiranium ion are shown in Figure 3. TS1 in Figure 3 corresponds to nucleophilic addition to the secondary carbon and TS2 corresponds to the addition to the tertiary carbon. The forming and breaking C-O distances at the transition states for TS1 and TS2 vary between 2.0 and 2.2 Å, indicating that both reactions proceed through SN1-like transition states. TS2 has longer C-O bond distances, with greater SN1 character resulting from the formation of a partial tertiary carbocation. The greater stabilization that arises from the partial tertiary carbocation results in TS2 being lower in energy than TS1 by 4.3 kcal/mol. The bond lengths in Figure 3 are taken as reference points and the transition state structures that are shown in subsequent studies with shorter bond lengths than the ones given in Figure 3 can be considered to be indicative of SN2-like transition structures.
Figure 3.

TS1 and TS2 for the addition of the dimethyl carbonate to the 1,2,2,3-tetramethyloxiranium ion. Relative energies are given in kcal/mol and the distances are given in Å.
Bicyclo[4.1.0] epoxonium ions preferentially undergo intramolecular cyclization through the 6-endo cyclization pathway. The intramolecular cyclization/epoxide opening of the bicyclo[4.1.0] epoxonium ion was mimicked with a model reaction shown in Scheme 7. The difference between the model reaction and the actual reaction (4 → 27, Table 1) is the replacement of the anomeric methoxy group with a hydrogen atom and the tert-butyl carbonate with a methyl carbonate. This model is also relevant to the terminal ring forming events in the cyclizations of 7 and 8 (Table 2).
Scheme 7.

Model reactions for bicyclo[4.1.0] epoxonium ion opening.
Table 2.
Cyclization reactions of diepoxide substrates.
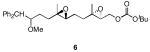
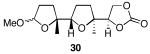
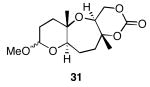
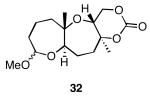
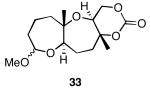
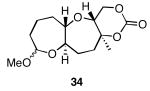
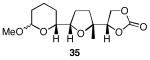
Yields are reported for isolated materials unless otherwise stated. See the Supporting Information for procedures.
See the Supporting Information for isolation protocols.
The nucleophilic attack by the oxygen of the carbonyl group to the epoxide carbon generates either 5- or 6-membered cyclic carbonates. The transition structures for the 5-exo (TS3_exo) and 6-endo (TS3_endo) modes of the attack are shown in Figure 4 for the reaction shown in Scheme 7. TS3_endo is lower in energy than TS3_exo by 4.6 kcal/mol. TS3_endo is an SN1-like TS with relatively longer forming carbonyl oxygen–epoxide carbon, and breaking epoxide oxygen–epoxide carbon distances due to the formation of the partial tertiary carbocation at the transition state. TS3_exo, however, adapts a more SN2-like TS with shorter forming and breaking bonds. The oxygen–carbon–oxygen bond angle in TS3_exo is perturbed to a greater extent from the unconstrained system (161.1° vs. 147.4°) than TS3_endo (135.5° vs. 143.3°). The energy difference between TS3_exo and TS3_endo is nearly identical to the energy difference in the unconstrained model.
Figure 4.

5-exo and 6-endo transition structures for the intramolecular cyclization of the bicyclo[4.1.0] epoxonium ion. The energies are given in kcal/mol and the distances are given in Å.
The effect of the anomeric alkoxy group on the reaction energetics was explored with a second model reaction, as shown in Scheme 8. In this model reaction, the methoxy group can be either cis or trans to the epoxonium ring. The relative energies of the both cis- and trans-methoxy isomers and the corresponding TSs for the 5-exo and 6-endo modes of cyclization for both isomers have been modeled.
Scheme 8.

Model reaction with methoxy group incorporation.
The gas phase transition structures for the 5-exo and 6-endo modes are shown in Figure 5. The cis-methoxy epoxonium ion is 0.3 kcal/mol more stable than the trans-methoxy epoxonium ion for the bicyclo[4.1.0] epoxonium ion. The activation enthalpy for the 6-endo cyclization mode for the cis-methoxy isomer is 0.2 kcal/mol whereas the activation enthalpy for the trans-methoxy isomer for the same mode is 1.4 kcal/mol in the gas phase. In general, methoxy group incorporation results in negligible geometrical perturbations on the transition structures, though TS4_trans_endo shows slightly enhanced leaving group departure and bond formation relative to TS3_endo. The trans-methoxy isomer favors the 6-endo cyclization mode by 4.6 kcal/mol and the cis-methoxy isomer favors the same mode by 4.8 kcal/mol, also indicating that the impact of methoxy group incorporation is negligible for bicyclo[4.1.0] epoxonium ions.
Figure 5.

Transition structures for the 5-exo and 6-endo cyclization modes of bicyclo[4.1.0] epoxonium ion for the cis- and trans-methoxy isomers. The energies are given in kcal/mol and the distances are given in Å.
To determine the extent to which the methyl group on the epoxide promotes the 6-endo cyclization mode, as studied in the conversion of 2 to 22 and 23 (Table 1) a third model reaction was studied (Scheme 9). In this model reaction, the methyl group on the epoxonium ion was replaced by a hydrogen atom to equalize the capacity for carbocation stabilization for both 5-exo and 6-endo cyclization transition structures.
Scheme 9.

Model reaction for the study of a methoxy-substituted bicyclo[4.1.0] epoxonium ion derived from a disubstituted epoxide.
The absence of the methyl group leads to significant perturbations of the 6-endo-transition structures (Figure 6). The oxygen–carbon–oxygen bond angle widens significantly. Greater bond formation and less leaving group departure are observed, indicating that the transition structures have increased SN2 character. The absence of the methyl group has a negligible impact on the transition structures of the 5-exo-transition structures. Calculations show that the 6-endo cyclization mode is favored over 5-exo cyclization mode by 1.4 kcal/mol for both cis- and trans-methoxy isomers, presumably because strain is released in the opening of the bicyclo[4.1.0] system while the 5-exo TS retains the strain generated by the forming 1,3-dioxolane ring. Although the experimental results show that the reactions proceed through the exo- and endo-pathways to a nearly identical extent, these results support the observation that bicyclo[4.1.0] epoxonium ions are more highly disposed toward the endo-pathway than bicyclo[3.1.0] epoxonium ions.
Figure 6.

Transition structures for the 5-exo and 6-endo cyclization modes of bicyclo[4.1.0] epoxonium ion for the cis- and trans-methoxy isomers in the absence of the methyl group. The energies are given in kcal/mol and the distances are given in Å.
Consistent with Baldwin's rules, and as observerved in the terminal ring forming steps in the cyclizations of 7 and 8 (Table 2), intramolecular cyclization/epoxide ring openings of bicyclo[3.1.0] epoxonium ions preferentially produce 5-exo cyclization products. This reaction was mimicked with the model reaction shown in Scheme 10.
Scheme 10.

Model reaction for the study of bicyclo[3.1.0] epoxonium ion ring opening.
As shown in Figure 7, TS6_exo is nearly identical to the corresponding transition structure for the bicyclo[4.1.0] epoxonium ion with respect to bond formation, leaving group departure, and the oxygen-carbon-oxygen bond angle. TS6_endo, while showing a similar degree of bond formation and leaving group departure relative to the transition structure for the bicyclo[4.1.0] epoxonium ion, has a significantly diminished oxygen-carbon-oxygen bond angle (123.6° vs. 135.5°). This energetically costly perturbation results in the 5-exo cyclization mode being preferred over 6-endo cyclization mode (TS6_endo) by 2.7 kcal/mol in the gas phase.
Figure 7.

5-exo and 6-endo transition structures for cyclizations of the bicyclo[3.1.0] epoxonium ion.
To elucidate the role of the methoxy group on the regioselectivity, relevant to the cyclization of 3 (Table 1) an additional model reaction (Scheme 11) was studied in the presence of cis- and trans-methoxy group. For these epoxonium ion structures, the trans-methoxy isomer is 1.0 kcal/mol lower in energy than the substrate for the cis-methoxy isomer because the lone pair of its epoxide oxygen is antiperiplanar to the σ* orbital of the methoxy group.
Scheme 11.

Model reaction for the bicyclo[3.1.0] epoxonium ion ring opening in the presence of an anomeric methoxy group.
For the cis-isomer, methoxy group incorporation led to enhanced leaving group departure for the exo-transition structure (TS7_cis_exo), but bond formation and the oxygen-carbon-oxygen bond angle were essentially unchanged (Figure 8). TS7_cis_endo, however, has enhanced bond formation and an oxygen-carbon-oxygen bond angle that is close to those that were observed for endo-cyclizations in reactions of bicyclo[4.1.0] epoxonium ions. For the trans-isomer, methoxy group incorporation had no impact on transition structure TS7_trans_exo. The 6-endo transition structure (TS7_trans_endo), while geometrically similar with respect to bond formation and leaving group departure, has the expanded oxygen-carbon-oxygen bond angle that was seen in TS7_cis_endo. Energetically TS7_trans_endo is the lowest energy transition structure, presumably because this transition structure allows the electron withdrawing methoxy group to adopt an axial orientation with respect to the forming pyran ring, thereby benefiting from a developing anomeric effect. Transition structures for exo-cyclizations were found to be slightly higher in energy, and TS7_cis_endo, which does not benefit from developing anomeric stabilization, was the highest energy transition structure. The fascinating role of the methoxy group in these reactions explains the diminished selectivity for exo-cyclizations when bicyclo[3.1.0] epoxonium ions form from the addition of epoxides into oxocarbenium ions rather than non-stabilized carbenium ions. This is also interesting in consideration of the isolation of endo-product 25 as a single anomer in which the methoxy group has an axial orientation. Of note, while anomeric stabilization in the transition state accounts for the formation of the endo-product for trisubstituted epoxide substrates, the effect is not strong enough to promote endo-cyclization for disubstituted epoxides such as 1.
Figure 8.

Transition structures for intramolecular cyclization/epoxide ring opening of bicyclo[3.1.0] epoxonium ion with methoxy group incorporation. The energies are given in kcal/mol and the distances are given in Å.
Conclusions
We have conducted an extensive study of intramolecular nucleophilic additions into bicyclic epoxonium ions to determine the structural features that promote exo- and endo-selective cyclization reactions. These results will serve as the basis for designing cascade processes that lead to complex polycyclic ethers. In particular the effects of epoxide substitution pattern, the structure of the carbocation that activates the epoxide, and the ring size of the bicyclic epoxonium ion were examined. We observed that ring size has a significant impact on these processes, with endo-cyclizations being preferred for bicyclo[4.1.0] epoxonium ions and exo-cyclizations being preferred for bicyclo[3.1.0] epoxonium ions. This effect can be attributed to the larger ring size accommodating a looser transition state that has significant SN1 character, thereby promoting the endo-process, regardless of solvent polarity. The smaller ring size, however, distorts endo-transition structure, thereby increasing its energy to an extent that allows the exo-process to become favorable. Thus the superior efficiency for reactions that proceed through bicyclo[4.1.0] epoxonium ions arises from the accessibility of the low energy endo-pathway and not from the difficulty in forming bicyclo[3.1.0] epoxonium ions.30 The substitution pattern of the epoxide also plays a substantial role in determining the regiochemical outcome of the reactions, with the endo-pathway being promoted for cyclizations that result from tertiary leaving group displacement due to the greater ability to stabilize the SN1-like transition state. While the structure of the cationic activating group appears to have little impact on the regioselectivity of reactions that proceed through bicyclo[4.1.0] epoxonium ions, epoxide activation through addition into unstabilized carbenium ions to form bicyclo[3.1.0] epoxonium ions leads to a strong preference for exo-cyclizations while activation by addition into oxocarbenium ions leads to a diminished preference for the exo-pathway. This was shown to result from an intriguing anomeric stabilization that can develop in endo-cyclization transition states when the alkoxy group has a pseudo axial orientation. While the effect is modest, this result provides evidence that bicyclic epoxonium ion structure can potentially be modified to promote tetrahydropyran formation through endo-cyclizations, as will be required to design cascade processes that form ladder polyether structures. The results from these studies clearly demonstrate that epoxonium ion structure is a significant determinant of kinetic cyclization regioselectivity, and efforts to alter the outcomes of these reactions must exploit thermodynamic differences in product energy or employ reactions that proceed through different mechanisms.
Supplementary Material
Experimental procedures for all cyclization reactions, spectral characterization of all new products, crystallographic data for compounds 38 and 39, absolute energies (in Hartree) and optimized geometries (as Cartesian coordinates) for all calculated transition structures, and the full reference for Gaussian 03. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health (GM36700 to K. N. H. and GM62924 to P. E. F.) and the National Science Foundation (CHE-0139851 to P. E. F.) for generous support of this work. We thank Drs. Lijun Wang and Christopher Lee for preliminary studies on this project and Dr. Steven Geib (University of Pittsburgh) for crystal structure analysis.
References
- 1.(a) Nicolaou KC, Montagnon T, Snyder SA. Chem Commun. 2003:551. doi: 10.1039/b209440c. [DOI] [PubMed] [Google Scholar]; (b) Tietze LF. Chem Rev. 1996;96:115. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stork G, Burgstahler AW. J Am Chem Soc. 1955;77:5068. [Google Scholar]; (b) Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv Chim Acta. 1955;38:1890. [Google Scholar]
- 3.(a) Cane DE, Celmer WD, Westley JW. J Am Chem Soc. 1983;105:3594. [Google Scholar]; (b) Lee MS, Qin G, Nakanishi K, Zagorski MG. J Am Chem Soc. 1989;111:6234. [Google Scholar]; (c) Krishna Prasad AV, Shimizu Y. J Am Chem Soc. 1989;111:6476. [Google Scholar]
- 4.(a) Still WC, Romero AG. J Am Chem Soc. 1986;108:2105. [Google Scholar]; (b) Schreiber SL, Sammekia T, Hulin B, Schulte G. J Am Chem Soc. 1986;108:2106. [Google Scholar]; (c) Xiong Z, Corey EJ. J Am Chem Soc. 2001;122:9328. [Google Scholar]
- 5.(a) Nicolaou KC, Claremon DA, Barnette WE. J Am Chem Soc. 1980;108:6611. [Google Scholar]; (b) Dolle RE, Nicolaou KC. J Am Chem Soc. 1985;107:1691. [Google Scholar]; (c) Hoye TR, Suhadolnik JC. J Am Chem Soc. 1985;107:5312. [Google Scholar]; (d) Simpson GL, Heffron TP, Merino E, Jamison TF. J Am Chem Soc. 2006;128:1056. doi: 10.1021/ja057973p. [DOI] [PubMed] [Google Scholar]
- 6.(a) Alvarez E, Díaz MT, Rodríguez ML, Martín JD. Tetrahedron Lett. 1990;31:1629. [Google Scholar]; (b) Zakarian A, Batch A, Holton RA. J Am Chem Soc. 2003;125:7822. doi: 10.1021/ja029225v. [DOI] [PubMed] [Google Scholar]; (c) Bravo F, McDonald FE, Neiwert WA, Hardcastle KI. Org Lett. 2004;6:4487. doi: 10.1021/ol048212e. [DOI] [PubMed] [Google Scholar]
- 7.(a) Valentine JC, McDonald FE. Synlett. 2006:1816. [Google Scholar]; (b) McDonald FE, Bravo F, Wang X, Wei X, Toganoh M, Rodríguez JR, Do B, Neiwert WA, Hardcastle KI. J Org Chem. 2002;67:2515. doi: 10.1021/jo0110092. [DOI] [PubMed] [Google Scholar]
- 8.(a) Na J, Houk KN, Shevlin CG, Janda KD, Lerner RA. J Am Chem Soc. 1993;115:8453. [Google Scholar]; (b) Na J, Houk KN. J Am Chem Soc. 1996;118:9204. [Google Scholar]
- 9.(a) Kumar VS, Aubele DL, Floreancig PE. Org Lett. 2002;4:2489. doi: 10.1021/ol0261074. [DOI] [PubMed] [Google Scholar]; (b) Kumar VS, Wan S, Aubele DL, Floreancig PE. Tetrahedron: Asymmetry. 2005;16:3570. [Google Scholar]
- 10.(a) Nakata T, Schmid G, Vranesic B, Okigawa M, Smith-Palmer T, Kishi Y. J Am Chem Soc. 1978;100:2933. [Google Scholar]; (b) Hayashi N, Fujiwara K, Murai A. Tetrahedron. 1997;53:12425. [Google Scholar]
- 11.Yoshida Ji, Nishikawa KJ. Chem Soc, Dalton Trans. 1998:2589. [Google Scholar]
- 12.(a) Seiders JR, II, Wang L, Floreancig PE. J Am Chem Soc. 2003;125:2406. doi: 10.1021/ja029139v. [DOI] [PubMed] [Google Scholar]; (b) Wang L, Seiders JR, II, Floreancig PE. J Am Chem Soc. 2004;126:12596. doi: 10.1021/ja046125b. [DOI] [PubMed] [Google Scholar]
- 13.Basel Y, Hassner A. J Org Chem. 2000;65:6368. doi: 10.1021/jo000257f. [DOI] [PubMed] [Google Scholar]
- 14.Maruoka K, Murase N, Bureau R, Ooi T, Yamamoto H. Tetrahedron. 1994;50:3663. [Google Scholar]
- 15.Yasuda A, Tanaka S, Oshima K, Yamamoto H, Nozaki H. J Am Chem Soc. 1974;96:6513. doi: 10.1021/ja00823a042. [DOI] [PubMed] [Google Scholar]
- 16.Ansell MF, Caton MPL, Stuttle KAJJ. Chem Soc, Perkin Trans 1. 1984:1069. [Google Scholar]
- 17.Hamilton JGC, Hooper AM, Ibbotson HC, Kurosawa S, Mori K, Muto S, Pickett JA. Chem Commun. 1999:2335. [Google Scholar]
- 18.Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J Am Chem Soc. 1997;119:11224. [Google Scholar]
- 19.Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J Am Chem Soc. 1987;109:5765. [Google Scholar]
- 20.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26. [Google Scholar]
- 21.Kumar VS, Floreancig PE. J Am Chem Soc. 2001;123:2842. doi: 10.1021/ja015526d. [DOI] [PubMed] [Google Scholar]
- 22.Kumar VS, Aubele DL, Floreancig PE. Org Lett. 2001;3:4123. doi: 10.1021/ol016996f. [DOI] [PubMed] [Google Scholar]
- 23.Valentine JC, McDonald FE, Neiwert WA, Hardcastle KI. J Am Chem Soc. 2005;127:4586. doi: 10.1021/ja050013i. [DOI] [PubMed] [Google Scholar]
- 24.(a) Grieco PA, Oguri T, Yokoyama Y. Tetrahedron Lett. 1978;19:419. [Google Scholar]; (b) Masaki Y, Nagata K, Kaji K. Chem Lett. 1983:1835. [Google Scholar]
- 25.CCDC620345 and CCDC620346 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Structures are included in the Supporting Information.
- 26.Baldwin JE. J Chem Soc, Chem Commun. 1976:738. [Google Scholar]
- 27.(a) Becke A. Phys Rev A. 1988;38:3089. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (b) Lee C, Yang W, Parr R. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 28.(a) Carlier PR, Deora N, Crawfold TD. J Org Chem. 2006;71:1592. doi: 10.1021/jo052303n. [DOI] [PubMed] [Google Scholar]; (b) Zhao Y, Truhlar DG. J Org Chem. 2007;72:295. doi: 10.1021/jo061863z. [DOI] [PubMed] [Google Scholar]
- 29.Gaussian 03, Revision C.02, Frisch, M. J.; et al Gaussian, Inc., Wallingford CT, 2004.
- 30.For several reactions that proceed efficiently through bicyclo[3.1.0] epoxonium ions, see reference 9.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures for all cyclization reactions, spectral characterization of all new products, crystallographic data for compounds 38 and 39, absolute energies (in Hartree) and optimized geometries (as Cartesian coordinates) for all calculated transition structures, and the full reference for Gaussian 03. This material is available free of charge via the Internet at http://pubs.acs.org.