

Abstract
Methylation of histone 3 lysine 4 (H3K4) by yeast Set1-COMPASS requires prior monoubiquitination of histone H2B. To define whether other residues within the histones are also required for H3K4 methylation, we systematically generated a complete library of the alanine substitutions of all of the residues of the four core histones in Saccharomyces cerevisiae. From this study we discovered that 18 residues within the four histones are essential for viability on complete growth media. We also identified several cis-regulatory residues on the histone H3 N-terminal tail, including histone H3 lysine 14 (H3K14), which are required for normal levels of H3K4 trimethylation. Several previously uncharacterized trans-regulatory residues on histones H2A and H2B form a patch on nucleosomes and are required for methylation mediated by COMPASS. This library will be a valuable tool for defining the role of histone residues in processes requiring chromatin.
Chromatin is an essential platform for almost all DNA-templated processes, including replication, repair, recombination and transcription1-3. Nucleosomes, the fundamental unit of chromatin, consist of 147 base pairs (bp) of DNA wrapped around an octamer of histones consisting of two H2A-H2B dimers and a single H3-H4 tetramer, forming two nearly symmetrical halves in their tertiary structure4. The core histones are highly conserved proteins from yeast to humans, indicating the importance of each amino acid residue within the histones. Nucleosomes present a degree of structural constraint, and to circumvent these restrictions chromatin structure may be locally or globally altered either through the post-translational modification of histones1-3, interactions with other proteins such as ATP-dependent chromatin-remodeling complexes, or by replacement of core histones with histone variants5-7. Covalent modifications of histones include acetylation, methylation, ubiquitination, sumoylation and phosphorylation8,9. Most of the identified histone modifications are located in the histone tail, where modifying enzymes or modification-targeting proteins can gain relatively easy access. However, post-translational modifications within the core histones have also been reported2,10-14.
A prevalent modification of histones that is associated with active transcription is H3K4 methylation15-24. More importantly, the lysine residues on histones can be mono-, di- or trimethylated by histone methyltransferases (HMTases), with each pattern of modification having a specific biological outcome2,17,23. Chromatin immunoprecipitation (ChIP)-based studies revealed that COMPASS and H3K4 trimethylation is found at the 5′ end of the coding region15,16. Several studies also demonstrated that the acetylation of H3K9 and H3K14 are also found at the 5′ ends of actively transcribed genes25,26. However, the relationship between H3 acetylation and H3 methylation is yet to be determined.
Methylation of H3K4 is catalyzed by the enzymatic activity of the macromolecular complex COMPASS, which contains the methyltransferase Set1 (ref. 2). Following the identification of Set1-COMPASS as the first H3K4 HMTase15,18,19,21, it was demonstrated that the human homologs of Set1, the mixed lineage leukemia (MLL) proteins, MLL2-4, and Set1A and Set1B, were also found in COMPASS-like complexes capable of methylating H3K4 (ref. 27). Previously, we, and others, demonstrated that monoubiquitination of histone H2B on lysine 123 (H2BK123) is required for the proper methylation of H3K4 by COMPASS28,29. We now know that H2B monoubiquitination regulates H3K4 methylation via regulation of COMPASS catalytic activity. The molecular mechanism identified in yeast that regulates the implementation and removal of H3K4 methylation is conserved in humans30-32. Therefore, the lessons learned from yeast chromatin are highly valuable in defining the chromatin and transcriptional machinery in humans.
Not only is H2B monoubiquitination required for H3K4 methylation by COMPASS, but methylation of histone H3 arginine 2 (H3R2) also has an important role in regulating COMPASS’s activity33,34. To determine how many other residues within the histones are required for proper H3 methylation by COMPASS, we systematically generated a library of alanine mutants of all residues of the four core histones in yeast S. cerevisiae. We call this library the scanning histone mutagenesis with alanine (SHIMA) library. This is an unbiased approach, which will facilitate determination of the importance and functional significance of all of the residues within histones. Given the conservation of residues between histones from yeast to humans, we initially predicted that many mutated residues within the histones would be required for viability, and therefore, be lethal. To our surprise, only 18 residues were found to be essential for viability on complete growth medium, three of which represented an extreme slow-growth phenotype. Recently, Matsubara and colleagues developed a strategy called global analysis of surfaces by point mutation (GLASP), where they mutated the surface residues of the four histones35. Their study resulted in the identification of eight essential residues within the four histones. The identification of additional essential residues here suggests the utility of SHIMA as a comprehensive and systematic mutant collection that will allow characterization of the functional importance of almost all residues within the core histones.
Using this entire comprehensive histone-mutant collection and our global proteomic screen (GPS) in S. cerevisiae, we then explored the network of histone cross-talk between histone H3K4 methylation and other residues within the histones36. With GPS, we have examined the extracts from the histone-mutant collection by western analysis using antibodies directed toward modified histones. We have identified several previously uncharacterized residues within histones acting either in cis or trans to regulate proper H3K4 methylation. Here we provide evidence for the existence of possible cis-cross-talk between histone H3K4 trimethylation and histone H3K14 acetylation. We have also identified residues that act in trans to regulate the pattern of histone H3K4 methylation, and they map to a patch on nucleosomes. This patch contains His112 and Arg119 of histone H2B, and Glu65, Leu66, Asn69 and Asp73 of histone H2A, all of which reside near H2BK123 when visualized on the three-dimensional structure of the nucleosome. Two of the residues are required for H2BK123 monoubiquitination, whereas the other residues may regulate COMPASS’s activity independently of H2B monoubiquitination. This comprehensive library of histone mutants has been instrumental in defining the global regulation of histone cross-talk for H3K4 methylation. This collection will be a useful resource to the chromatin community for defining the role of histone residues in numerous processes involving chromatin.
RESULTS
Generation of the histone-mutant library
We have generated a complete library of alanine mutants at all residues of the four core histones, except at the naturally occurring alanine residues in yeast S. cerevisiae (Fig. 1a). We have named this library SHIMA. Plasmids containing alanine point mutations within the histone genes were generated by site-directed mutagenesis (Methods). Each plasmid was then sequenced for confirmation. The entire collection of the histone alanine mutant library in S. cerevisiae was generated by transforming the plasmids into yeast histone shuffle strains, either YBL574 containing hht2/hhf2 encoding H3 and H4, or Y131 (Osley’s lab) containing hta1/htb1, encoding H2A and H2B37. We carried out a strain selection for transformants, followed by a second single-colony selection on 5-fluoroorotic acid (5-FOA; Fig. 1b) to remove wild-type histone plasmids containing the URA gene. At this point, we identified the residues that are essential for cell survival under normal growth conditions (Fig. 1b). We carried out three additional selections by YPD-5-FOA to ensure the complete removal of wild-type histone plasmids before making the final stock of the library. Individual strains in the final yeast histone mutation library were sequenced for confirmation of the mutation and to determine the absence of the corresponding wild-type histone copy. The key for each plate is shown in Figure 2. This complete yeast mutation collection is now available to our colleagues.
Figure 1.

Schematic representation of the experimental procedure. (a) Scanning histone mutagenesis with alanine (SHIMA). A library of alanine mutants at all residues of the four core histones, except the wild-type alanine residues in yeast S. cerevisiae, was systematically generated. (b) High-throughput yeast transformation and global proteomic screen (GPS) of S. cerevisiae histone mutants. The plasmids containing alanine point mutations within histone genes were generated by site-directed mutagenesis (Methods). The entire collection of histone alanine mutant libraries in S. cerevisiae was generated by transforming yeast shuffle strains (YBL 574 for H3 and H4, 4131 for H2A and H2B) and selection for the transformants, followed by a second selection on 0.1% (w/v) 5-FOA to remove wild-type histones. The collection of histone mutants was further analyzed by GPS to identify the amino acid residues required for proper H3 methylation.
Figure 2.

Key to the organization of the SHIMA library. Each mutant histone is stored in the indicated cell block.
Essential amino acid residues of yeast histones
Histone residues within nucleosomes are highly evolutionarily conserved. As a matter of fact, the amino acid residues in histones H2A and H2B are more than 70% conserved from yeast to humans, and in histones H3 and H4 more than 90% are conserved. When we decided to generate a complete alanine scanning collection of the histones, we considered its high evolutionary conservation and predicted that many of the mutants would be lethal. To our surprise, point mutations of less than 5% of the total residues of the four histones were lethal. Our comprehensive histone alanine scanning analysis identified only 18 residues within all of the core histones that were required for viability under normal growth conditions (Fig. 3a). All of the essential residues are found within the globular domains of histones (Fig. 3b). A few of these residues, marked by asterisk in Figure 3, are either slow growers or generate revertants as judged by growth after 5 d of slow or no growth. Although most of the post-translational modifications occur in histone tail domains, tail-less H3-H4, H2A and H2B histones are still capable of nucleosomal assembly in vitro. The histone H4 N-terminal tail is required for chromatin folding; however, it is dispensable for growth38. We have found that there are nine residues in histone H3 that are essential for viability: Tyr41, Leu48, Ile51, Gln55, Glu97, His113, Arg116, Thr118 and Asp123 (Fig. 3a). Five residues in histone H4 are also required for viability: Arg39, Arg40, Arg45, Tyr72 and Leu90. We have found that histone H2A and H2B together have only four residues required for yeast viability under normal growth conditions: three in H2A (Tyr58, Glu62 and Asp91) and one in H2B (Leu109) (Fig. 3a).
Figure 3.

Identification of amino acid mutations essential for viability under normal growth conditions. (a) Viability of histone alanine mutants. Strains expressing histones containing each single alanine mutation in the presence of wild-type histones (either HTA1 and HTB1, or HHT2 and HHF2) were plated with an initial OD600 of 0.5, followed by a four-fold serial dilution onto SC-Trp in the absence or presence of 5-FOA. * indicates that these mutants started to form revertants after 5 days of incubation at 30 °C; however, the appearance of the colonies of these mutants was repeatedly delayed and the number of colonies was extremely low. (b) Mapping of lethal residues on the nucleosome crystal structure as determined previously60. The locations of lethal residues are shown in red and numbers indicate the corresponding histone mutants shown in Figure 2a. Representation of nucleosome was generated using PyMOL. (c,d) Mapping of lethal residues in the surface of the yeast nucleosome on the crystal structure of the nucleosomes as determined previously (PDB 1ID3)60. H2A, H2B, H3 and H4 lethal residues are shown in orange, pink, blue and green, respectively. Red circles show H2A and H2B lethal residues. Representations of nucleosomes were generated using PyMOL (http://pymol.sourceforge.net/).
Most of the H3 and H4 essential residues are clustered on the lateral surface near the dyad axis or at the DNA entry-exit points (Fig. 3c,d), as mapped onto the crystal structure of the nucleosome determined previously60. These are the regions where histones make contact with DNA. Alanine substitutions at three of the five Sin (SWI/SNF-independent) mutation sites previously identified by genetic screens39, H3 R116A, H3 T118A and H4 R45A, are lethal. These residues are all located at the protein-DNA interaction interface formed by the L1L2 loops of the H3-H4 tetramer40. Additionally, H3 Arg116 makes a salt bridge with H3 Asp123 (ref. 41), which is also identified as an essential residue (Fig. 3a). Mutation of these residues caused a loss of the salt bridge and small changes in the conformation of the L2 loop. It has been demonstrated that H3 His113 also forms a hydrogen bond with H3 Asp123 (ref. 41). H3-H4 and H2A-H2B quasisymmetric heterodimers can be superimposed by rotating 180° around the axis (ref. 4). Indeed, lrs (loss of rDNA silencing) mutation sites are located at the other end of the H3-H4 heterodimer40,42. Even though these sites are structurally equivalent to the Sin mutation sites, none of these residues are required for viability (Fig. 3). H2A Arg42, H2B Thr85 and H2B Arg83 are the residues in the H2A-H2B heterodimer that are equivalent to H4 Arg45, H3 Thr118 and H3 Arg116, respectively40. However, none of these residues is required for viability (Fig. 3). This may be explained by the fact that the interactions between DNA and the histones are strongest at the nucleosome dyad4. Mutations in these regions may have more of an impact on histone-DNA interactions. The H2A and H2B essential residues are positioned on the surface of nucleosomes (Fig. 3c, red circle).
Cross-talk between histone residues
It has already been well established that the monoubiquitination of H2BK123 is required for proper H3K4 and H3K79 methylation by COMPASS and Dot1, respectively2. This mode of regulation is highly conserved from yeast to human. Yeast has had a fundamental role in paving the way to then defining the pathway of H3K4 methylation in mammalian cells. Almost all aspects of regulation of H3K4 methylation found in yeast2 were later shown to be similar in mammalian cells30-32,43. Rad6 is the E2 conjugating enzyme and was first reported in yeast to be required for proper H2BK123 monoubiquitination28,29,44,45. Bre1 is the E3 ligase for Rad6, physically associates with it and is required for its recruitment to chromatin45, and for cross-regulation from monoubiquitination to methylation45,46. In addition to Rad6 and Bre1, several other factors including the Paf1 complex, the Bur1-Bur2 kinase and the Ctk complex are required for proper H2BK123 monoubiquitination and H3K4 and H3K79 methylation16,17,47-52. Recently, it was demonstrated that the H2B monoubiquitination signal is translated via the Cps35 subunit of COMPASS53. Cps35 interacts with chromatin in an H2BK123 monoubiquitination-dependent manner, and its interaction with chromatin brings the COMPASS core and Cps35 together to activate the enzyme’s trimethylase activity on chromatin53. This model describes how the interaction of COMPASS with Cps35 on chromatin will allow the activation of the enzyme and the methylation of the histones on chromatin, but not on the soluble histones53.
So far, we know that two identified residues (H2BK123 and H3R2) have important roles in proper methylation by COMPASS. Therefore, we wanted to determine whether any other residues within histones are required for proper H3K4 methylation. We performed a GPS analysis of the extracts from the entire histone-mutant collection. Our analysis has resulted in the identification of several previously uncharacterized key regulatory residues within the histones required for normal levels of H3K4 methylation (Figs. 4-6). The key for the position of the mutants is shown in Figure 2. First and foremost, we have identified a few consistently reproducible cis-regulatory residues within histone H3 regulating H3K4 trimethylation (Fig. 4). These include Arg2, Lys4, Gln5, Thr3 and Lys14 of H3, indicated on Figure 4a,b as residues I, II, III, IV and V, respectively. Of these, Lys4 mutated to alanine can no longer be methylated. Arg2 has already been shown to be required for substrate recognition by COMPASS33,34. Thr3 could be phosphorylated, and its phosphorylation could regulate COMPASS’s activity; however, it is also possible that H3T3A mutation may result in epitope masking and therefore loss of immunoreactivity by the H3K4 polyclonal antibodies. Further studies should clarify these possibilities. Mutations in H3Q5 also result in the loss of the mono-, di- and trimethylation of H3K4. As many of these residues lay near the H3K4 site, their mutation could result in a defect in epitope recognition by the antibodies or defect in the substrate recognition by Set1-COMPASS.
Figure 4.
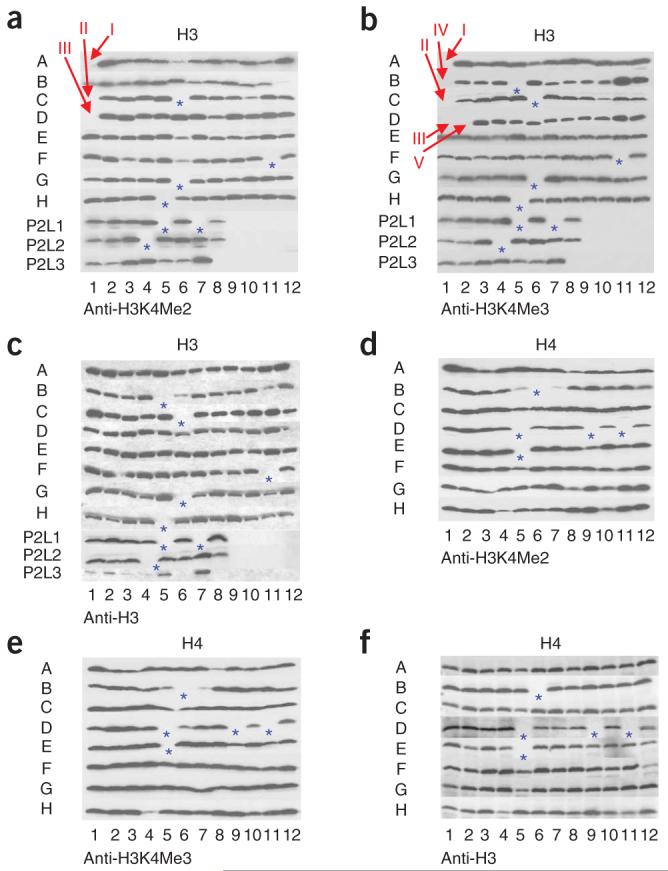
GPS analyses to defining amino acid residues of histone H3 and H4 required for proper H3K4 methylation. Cell extracts prepared from the entire histone-mutant collection (a-c from H3 mutants, d-f from H4 mutants) were subjected to SDS-PAGE and western blot analysis and tested for the presence of dimethylated lysine 4 of histone H3 (H3K4Me2) and trimethylated lysine 4 of histone H3 (H3K4Me3) as indicated below panel. As the loading control, an antibody to histone H3 was also used. Red arrows indicate the possible hits, and blue asterisks (*) indicate the positions of empty wells (lethal residues). For the key to the organization of the strains within each plate, see Figure 2. Positions I, II, III, IV and V indicate histone H3 Arg2, Lys4, Gln5, Thr3 and Lys14, respectively. P2 indicates plate 2.
Figure 6.

Identification of a nucleosomal patch regulating the H3K4 methylation pattern. (a,b) Titration analyses of histone mutants identified as defective in proper H3K4 methylation. Titration analysis was performed to confirm the positive hits obtained from GPS analysis. Extracts were analyzed by SDS-PAGE followed by western blot analysis, and testing for the presence of monomethylated lysine 4 of histone H3 (H3K4Me1), dimethylated lysine 4 of histone H3 (H3K4Me2) and trimethylated lysine 4 of histone H3 (H3K4Me3). As a positive control, extracts from the strain carrying a plasmid containing wild-type histone (either HTA1 and HTB1 or HHT2 and HHF2) were loaded onto each individual gel. (c) The locations of residues required for normal levels of H3K4 methylation were mapped onto nucleosomes of the crystal structure (PDB 1ID3)60. Ac, acetylation; Ub, monoubiquitination. (d) Identification of histone amino acid residues required for H2B monoubiquitination. Extracts from H2A and H2B mutants identified as defective in Lys4 methylation were tested for the presence of H2B monoubiquitination.
Another cis-regulatory residue identified in this screen is H3K14 (Figs. 4a,b and 6). The H3K14A strain showed a specific loss of H3K4 trimethylation with no effect on H3K4 mono- or dimethylation (Fig. 6b). This residue is far from H3K4, and therefore its effect on H3K4 trimethylation cannot result from a defect in epitope recognition by the H3K4 trimethylation-specific antibodies. As H3K14 is a known site of acetylation, we tested the effect of the H3K14Q mutation to mimic a neutral, acetylated lysine, and an H3K14R mutant to mimic the basic, nonacetylated lysine (Fig. 6b, lanes 19-27). Both mutant strains lacked specific H3K4 trimethylation (Fig. 6b, lanes 19-27), suggesting that the regulation of H3K4 methylation is not simply electrostatic in nature. Both the H3K14 acetylation and the H3K4 global methylation patterns show a similar localization to the 5′ ends of actively transcribed genes25,26, and the presence of H3K4 trimethylation can be correlated with the hyperacetylation of histone H3 (ref. 54). Two enzyme complexes, GCN5-SAGA and Sas3-NuA3, are known to acetylate H3K14 (ref. 55). Interestingly, the deletion of Gcn5 results in decreased H3K4 methylation in the coding region of the ARG1 ORF56, suggesting H3K14 acetylation levels could affect H3K4 methylation. Our finding that the H3K14A mutation abrogates H3K4 trimethylation indicates that H3K14 acetylation might regulate H3K4 trimethylation globally. Notably, the NuA3 subunit, Yng1, binds trimethylated H3K4 and facilitates H3K14 acetylation57,58, suggesting that these two modifications stabilize each other. Similarly, an acetyllysine binding protein such as the bromodomain-containing protein Rsc4 (ref. 59), could bind and stabilize acetylated H3K14, whereas Yng1 stabilizes trimethylated H3K4 by COMPASS. Indeed, we have identified COMPASS in a complex containing the RSC complex (data not shown), suggesting a possible physical link between these modifications. Our unbiased approach to understanding H3K4 methylation has thus generated hypotheses for future investigations into the mechanistic link between two highly studied histone modifications.
In addition to cis-regulatory residues identified within histone H3, we have also identified several trans-regulatory residues that specifically regulate H3K4 di- and trimethylation (Figs. 5 and 6). These include four residues within histone H2A—Glu65, Leu66, Asn69 and Asp73 (indicated on Fig. 5a,b as I, II, III and IV, respectively)—and three residues within histone H2B—Lys123, Arg119 and His112 (indicated on Fig. 5e,f as V, VI and VII, respectively). When we localized these residues on the crystal structure of the nucleosomes, they formed a patch near Lys123 of H2B, the site of monoubiquitination required for H3K4 methylation by COMPASS (Fig. 6c). We tested whether the mutation of residues within this patch could alter H2B monoubiquitination levels and found that only mutations in H2BH112 and H2AL66 result in a defect in H2B monoubiquitination (Fig. 6d). The other residues within the patch, H2AN69, H2AD73 and H2AE65, regulate COMPASS’s activity independently of H2B monoubiquitination. We recently found that the Cps35 subunit of COMPASS interacts with chromatin in a monoubiquitination-dependent manner53 and that Cps35 is required for H3K4 methylation by COMPASS. Indeed, Cps35 loss phenocopies either Rad6 and/or H2BK123R mutations in regard to H3K4 di- and trimethylation. Conceivably, the residues within this patch that still have high levels of H2B monoubiquitination could be a part of a binding surface for proteins that recognize monoubiquitinated H2B in its nucleosomal context, or other factors, such as the Paf1 complex, that are also required for the association of COMPASS with transcribing polymerase.
Figure 5.
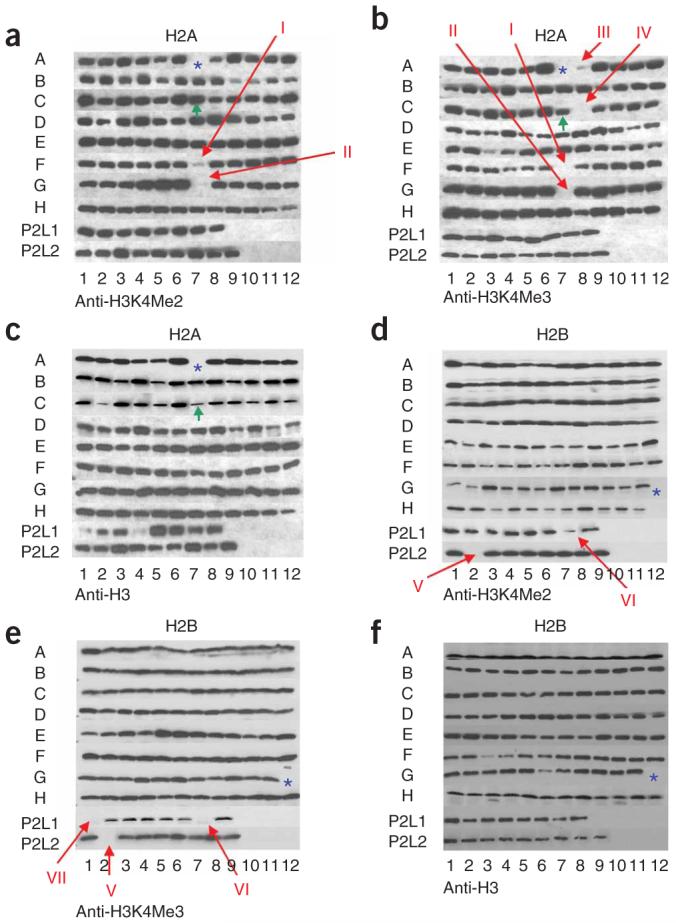
GPS analyses in defining amino acid residues of histones H2A and H2B required for proper H3K4 methylation. Cell extracts prepared from the entire histone-mutant collection (a-c from H2A mutants, d-f from H2B mutants) were subjected to SDS-PAGE and western blot analysis, and tested for the presence of dimethylated lysine 4 of histone H3 (H3K4Me2) and trimethylated lysine 4 of histone H3 (H3K4Me3) as indicated under panel. As the loading control, an antibody to histone H3 was also used. Red arrows indicate the possible hits, and blue asterisks (*) indicate the positions of empty wells (lethal residues), green arrows (C8) indicate the positions where the wild-type was used in place of a lethal residue-bearing strain as a positive control for growth in these regions. For the key to the organization of the strains within each plate, please see Figure 2. Positions I, II, III and IV indicate histone H2A Glu65, Leu66, Asn69 and Asp73, respectively. Positions V, VI and VII indicate histone H2B Lys123, Arg119 and His112, respectively. P2 indicates plate 2.
DISCUSSION
In this study, we have generated a comprehensive library of all of the four core histones mutated at every single residue with an alanine. This library is now available to our colleagues who are interested in defining the functional and physiological relevance of histone residues in many processes requiring chromatin. As a proof of principle, using this library we have identified 18 residues within all four of the core histones that are essential for viability and proper growth under normal growth conditions. We have also identified several cis-regulatory residues on histone H3, including H3K14, which are required for the implementation of normal levels of H3K4 trimethylation. H3K14 is acetylated by both Gcn5 and Sas3, and this modification is associated within the early transcribed regions of active genes, similarly to the pattern of H3K4 trimethylation. Our study raises the possibility of a communication between H3K14 acetylation and H3K4 trimethylation.
Using this comprehensive library of histone mutants, we have also identified a trans-regulatory patch on chromatin containing several residues within histones H2A and H2B regulating H3K4 methylation. Given the high conservation of core histones from yeast to human, we predict that many of the corresponding residues in the human core histones will also be required for proper H3K4 methylation by the MLL complexes and the Set1A/B-containing complexes. Considering that translocations of MLL are associated with the pathogenesis of hematological malignancies, the identification of the role of these residues in the regulation of the enzymatic activity of the MLL complex will be of great interest.
We envision a plethora of studies that can take advantage of the availability of this comprehensive point-mutated histone collection. This collection can be used to define the role of different histone residues under many growth conditions, including sporulation. It is of great interest to define how signaling through chromatin allows chromatin compaction and spore formation. Analysis of epigenetic memory may also be facilitated by using this mutant collection, especially given that there are currently no identified, direct links between the known modified histone residues and this process. An unbiased approach using the entire histone-mutant collection will allow the identification of such possible residues. This collection can also be used for defining the role of individual histone residues and telomere-associated gene silencing. Application of this collection to methods such as the synthetic genome array (SGA) or just a simple synthetic analysis would allow one to determine the role of each histone residue with the deletion in a specific gene or the entire yeast deletion collection. In conclusion, the availability of this comprehensive histone-mutant collection is likely to facilitate numerous new insights into chromatin.
METHODS
Yeast strains and plasmids
We used plasmids pWZ414-F12, carrying HHT2 and HHF2, and pZS145 (ref. 29), carrying HTA1-Flag-HTB1 CEN HIS3 for this study. Previously described yeast shuffle strains, YBL 574 (ref. 37) and YBL 397 (Y131)29, were a gift from J. Workman.
Generation of histone-mutant library
Plasmids bearing alanine mutations in the HTA1, HTB1, HHT2 and HHF2 genes were systematically generated by site-directed mutagenesis. We performed site-directed mutagenesis in 96-well plates using the QuickChange II Site-Directed Mutagenesis Kit (Stratagene). Products were transformed into E.cloni 10G ELITE electrocompetent cells (Lucigen), using a 96-well electroporator, BTX model ECM 630 electro cell manipulator (Harvard Apparatus). Plasmids were prepared with a BioMekFX (Beckman-Coulter) using the CosMCPrep Kit (Agencourt). Mutated targets were confirmed by sequencing, using the following primers: HTA1seqF: 5′-cgaagcc agccagtggatcg-3′; HTA1seqR: 5′-gaagcagtttagttccttccgcct-3′; HTBseqF: 5′-ggca aatactaccttggttgg-3′; HTBseqR: 5′-tttcgagaacacaattttacaaccga-3′; HHT2seqF: 5′-gcgtgataacagcgtgttgtgc-3′; HHT2seqR: 5′-catgtcgttaaaagcattgcgaatag-3′; HHF2 seqF: 5′-gttgttcactcgcgcctggg-3′; and HHF2seqR: 3′-atatcgaattctggaggagca-5′.
Each plasmid was then transformed manually into yeast shuffle strains, Y131 for HTA1 and HTB1, and YBL 574 for HHT2 and HHF2, using a standard yeast-transformation protocol with minor modifications, and strains grown on a synthetic dropout medium lacking histidine, SC-His (for hta1 and htb1 mutants), or a medium lacking tryptophan, SC-Trp (for hht2 and hhf2 mutants). After 2 days of incubation, each transformant (20 μL) was plated onto 48-segment bioassay trays (Genetix) containing: either SC-HIS plus 5-FOA or SC-Trp plus 5-FOA to single-colony select cells that had lost the plasmid containing the wild-type histones. Each colony was inoculated into YPD medium plus 5-FOA in 96-well plates. All histone-mutant strains were again confirmed by sequencing, and the glycerol stocks of the histone-mutant library were generated.
Viability assays for the identification of essential residues within histones
Mutant and wild-type strains were grown to an optical density at 600 nm (OD600) of 0.5. Next, four-fold serial dilutions of these strains were spotted on SC-Trp plates in the presence or absence of 5-FOA. The dilutions of the hta1 and htb1 mutant strains were spotted on SC-His plates in the presence or absence of 5-FOA. The plates were incubated at 30 °C for 72 h before being photographed.
Global proteomic screening for the identification of the residues required for proper methylation of H3K4
We carried out GPS as described previously36 using antibodies specific for H3K4 di- and trimethylation (Millipore).
Detection of H2B monoubiquitination
We prepared extracts from the histone H2A and H2B mutants that resulted in defects in H3K4 methylation, and subjected the extracts to western blot analysis. The plasmid used for the mutagenesis of HTA1 and HTB1 contains a Flag-tagged HTB1 gene. Therefore, an antibody against the Flag epitope was used to detect H2B and its monoubiquitination form.
ACKNOWLEDGMENTS
We thank L. Shilatifard for editorial assistance and E. Smith for critical reading of the manuscript. We are grateful for M. Ruhlman and N. Dillon for large-scale preparation of polyacrylamide gels, and C. Wimberly and H. Strobietto for large-scale plasmid preparation and DNA sequencing. We are also grateful to B. Li from the Workman laboratory at the Stowers Institute for providing YBL574 and Y131 strains and shuffling plasmids. This work was supported by the US National Institute of Health grants 2R01CA89455 and 2R01GM069905 to A.S.
References
- 1.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 2.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 5.Mito Y, Henikoff JG, Henikoff S. Histone replacement marks the boundaries of cis-regulatory domains. Science. 2007;315:1408–1411. doi: 10.1126/science.1134004. [DOI] [PubMed] [Google Scholar]
- 6.Polo SE, Roche D, Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127:481–493. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 7.Lacoste N, Almouzni G. Epigenetic memory: H3.3 steps in the groove. Nat. Cell Biol. 2008;10:7–9. doi: 10.1038/ncb0108-7. [DOI] [PubMed] [Google Scholar]
- 8.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 9.Berger SL. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 10.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 11.Ng HH, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider J, Bajwa P, Johnson FC, Bhaumik SR, Shilatifard A. Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem. 2006;281:37270–37274. doi: 10.1074/jbc.C600265200. [DOI] [PubMed] [Google Scholar]
- 13.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 14.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc. Natl. Acad. Sci. USA. 2002;99:90–94. doi: 10.1073/pnas.221596698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krogan NJ, et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol. Cell. 2003;11:721–729. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 17.Schneider J, et al. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol. Cell. 2005;19:849–856. doi: 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 18.Miller T, et al. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA. 2001;98:12902–12907. doi: 10.1073/pnas.231473398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roguev A, et al. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001;20:7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krogan NJ, et al. COMPASS, a histone H3 (lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 2002;277:10753–10755. doi: 10.1074/jbc.C200023200. [DOI] [PubMed] [Google Scholar]
- 22.Krogan NJ, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2003;23:4207–4218. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 24.Xiao T, et al. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein BE, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Liu CL, et al. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 28.Dover J, et al. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J. Biol. Chem. 2002;277:28368–28371. doi: 10.1074/jbc.C200348200. [DOI] [PubMed] [Google Scholar]
- 29.Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol. Cell. 2005;20:759–770. doi: 10.1016/j.molcel.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Zhu B, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol. Cell. 2005;20:601–611. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 32.Pavri R, et al. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125:703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 33.Kirmizis A, et al. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007;449:928–932. doi: 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guccione E, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–937. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- 35.Matsubara K, Sano N, Umehara T, Horikoshi M. Global analysis of functional surfaces of core histones with comprehensive point mutants. Genes Cells. 2007;12:13–33. doi: 10.1111/j.1365-2443.2007.01031.x. [DOI] [PubMed] [Google Scholar]
- 36.Schneider J, Dover J, Johnston M, Shilatifard A. Global proteomic analysis of S. cerevisiae (GPS) to identify proteins required for histone modifications. Methods Enzymol. 2004;377:227–234. doi: 10.1016/S0076-6879(03)77013-X. [DOI] [PubMed] [Google Scholar]
- 37.Carrozza MJ, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 38.Kayne PS, et al. Extremely conserved histone H4 N terminus is dispensable for growth but essential for repressing the silent mating loci in yeast. Cell. 1988;55:27–39. doi: 10.1016/0092-8674(88)90006-2. [DOI] [PubMed] [Google Scholar]
- 39.Kruger W, et al. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995;9:2770–2779. doi: 10.1101/gad.9.22.2770. [DOI] [PubMed] [Google Scholar]
- 40.Luger K. Structure and dynamic behavior of nucleosomes. Curr. Opin. Genet. Dev. 2003;13:127–135. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 41.Muthurajan UM, et al. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004;23:260–271. doi: 10.1038/sj.emboj.7600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park JH, Cosgrove MS, Youngman E, Wolberger C, Boeke JD. A core nucleosome surface crucial for transcriptional silencing. Nat. Genet. 2002;32:273–279. doi: 10.1038/ng982. [DOI] [PubMed] [Google Scholar]
- 43.Zhu B, et al. The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev. 2005;19:1668–1673. doi: 10.1101/gad.1292105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robzyk K, Recht J, Osley MA. Rad6-dependent ubiquitination of histone H2B in yeast. Science. 2000;287:501–504. doi: 10.1126/science.287.5452.501. [DOI] [PubMed] [Google Scholar]
- 45.Wood A, et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell. 2003;11:267–274. doi: 10.1016/s1097-2765(02)00802-x. [DOI] [PubMed] [Google Scholar]
- 46.Hwang WW, et al. A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol. Cell. 2003;11:261–266. doi: 10.1016/s1097-2765(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 47.Wood A, Schneider J, Dover J, Johnston M, Shilatifard A. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J. Biol. Chem. 2003;278:34739–34742. doi: 10.1074/jbc.C300269200. [DOI] [PubMed] [Google Scholar]
- 48.Wood A, Schneider J, Dover J, Johnston M, Shilatifard A. The Bur1/Bur2 complex is required for histone H2B monoubiquitination by Rad6/Bre1 and histone methylation by COMPASS. Mol. Cell. 2005;20:589–599. doi: 10.1016/j.molcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 49.Wood A, Schneider J, Shilatifard A. Cross-talking histones: implications for the regulation of gene expression and DNA repair. Biochem. Cell Biol. 2005;83:460–467. doi: 10.1139/o05-116. [DOI] [PubMed] [Google Scholar]
- 50.Wood A, et al. Ctk complex-mediated regulation of histone methylation by COMPASS. Mol. Cell. Biol. 2007;27:709–720. doi: 10.1128/MCB.01627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laribee RN, et al. BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr. Biol. 2005;15:1487–1493. doi: 10.1016/j.cub.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 52.Xiao T, et al. The RNA polymerase II kinase Ctk1 regulates positioning of a 5′ histone methylation boundary along genes. Mol. Cell. Biol. 2007;27:721–731. doi: 10.1128/MCB.01628-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee JS, et al. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 54.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howe L, et al. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 2001;15:3144–3154. doi: 10.1101/gad.931401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Govind CK, Zhang F, Qiu H, Hofmeyer K, Hinnebusch AG. Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol. Cell. 2007;25:31–42. doi: 10.1016/j.molcel.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 57.Martin DG, et al. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell. Biol. 2006;26:7871–7879. doi: 10.1128/MCB.00573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taverna SD, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kasten M, et al. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 2004;23:1348–1359. doi: 10.1038/sj.emboj.7600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White CL, Suto RK, Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]