

Abstract
The Runx genes play paradoxical roles in cancer where they can function either as dominant oncogenes or tumor suppressors according to context. We now show that the ability to induce premature senescence in primary murine embryonic fibroblasts (MEFs) is a common feature of all 3 Runx genes. However, ectopic Runx-induced senescence contrasts with Ras oncogene-induced senescence, as it occurs directly and lacks the hallmarks of proliferative stress. Moreover, a fundamental role for Runx function in the senescence program is indicated by the effects of Runx2 disruption, which renders MEFs prone to spontaneous immortalization and confers an early growth advantage that is resistant to stress-induced growth arrest. Runx2−/− cells are refractory to H-RasV12 induced premature senescence, despite the activation of a cascade of growth inhibitors and senescence markers, and are permissive for oncogenic transformation. The aberrant behaviour of Runx2−/− cells is associated with signaling defects and elevated expression of S/G2/M cyclins and their associated cyclin dependent kinase (CDK) activities that may override the effects of growth inhibitory signals. Coupling of stress responses to the cell cycle represents a novel facet of Runx tumor suppressor function and provides a rationale for the lineage-specific effects of loss of Runx function in cancer.
INTRODUCTION
The 3 members of the Runx family of mammalian transcription factors are related to Runt, the Drosophila pair rule gene (1), sharing a highly conserved DNA binding domain and a common DNA-binding cofactor (Cbfβ). Their products regulate multiple cell fate decisions and have been implicated in a wide range of cancers where there is unequivocal evidence that members of this family can act as oncogenes or as tumor suppressors according to context (2).
The identification of somatic and germ-line loss-of-function mutations associated with acute myeloid leukaemia (AML) strongly supports the view that RUNX1 operates as a tumor suppressor gene in this lineage. Further evidence that the RUNX genes can inhibit tumor development comes from a number of reports suggesting that methylation and down-regulation of RUNX3 is etiologically associated with the development and progression of a spectrum of epithelial cancers ((2) and refs therein). However, it is clear that the tumor suppressor role of the RUNX gene family is expressed in a lineage restricted manner. Functional redundancy and partially overlapping expression patterns of the three family members have been proposed to account for these observations. Functional similarity between the Runx genes is also suggested by the finding that all three genes act as a single complementation group by retroviral activation in experimental models of hematological malignancy (3-5).
The mechanisms underlying these contrasting manifestations in cancer are not fully understood, although it appears that ectopic Runx expression can inhibit apoptosis, particularly in transgenic mice over-expressing the Myc gene. Moreover, the growth suppressive features of Runx2 are evident in this T-cell transgenic model, as pre-lymphomatous mice display a proliferative defect in immature thymocytes which is overcome by ectopic Myc expression (6). Loss of Runx1 appears to result in increased proliferation and failure of hematopoietic precursor cells to differentiate (7), suggesting that homeostatic control of hematopoiesis is a central function of this gene, while RUNX3 down-regulation and hypermethylation in epithelial cells has been linked to loss of growth inhibitory responses to TGF-β (reviewed in (8,9)). Together, these observations are consistent with growth inhibitory Runx functions that are selectively lost in cancers, or overcome by complementing oncogenic mutations affecting collaborating genes such as Myc or p53 which serve to reveal the oncogenic side of their dual nature (10,11). However, it is not known whether these various inhibitory phenomena represent shared functions across family members and in different lineages, or whether they can fully account for the tumor suppressor activities of this gene family.
A striking demonstration of RUNX induced negative growth regulation is the senescence-like phenotype observed following ectopic expression of RUNX1 in primary MEFs (11,12). Premature senescence or growth stasis shares many of the characteristics of replicative senescence but is independent of telomere shortening and occurs in response to activation of stress pathways that detect or heighten cellular responses to DNA damage (13). Thus the aberrant expression of oncogenes such as activated Ras can result in a transient round of cell proliferation followed by permanent withdrawal from the cell cycle and other phenotypic features indicative of cellular senescence. Moreover, it is now clear that this is not merely a feature of in vitro conditions and that this phenomenon limits tumor development in vivo (14,15). While species and cell type differences exist, many of the essential components of the Ras-induced senescence pathway have been identified and there is considerable overlap between these components and genes with established tumor suppressor function. In accord with this hypothesis activation of the major tumor suppressor pathways mediated by p53 and/or pRb family members is a hallmark of premature senescence (16).
We now report that induction of premature senescence in MEFs is a common feature of all three Runx genes. However, this process differs markedly from Ras-induced senescence as it occurs without an initial aberrant proliferative phase. Moreover, MEFs lacking Runx2 display an early growth advantage that is resistant to in vitro culture stress, and are susceptible to H-RasV12 induced cellular transformation. Our results suggest that Runx genes play a central role in regulating the cell cycle in response to stress signals, including ectopic oncogene expression, offering new insights into the mechanisms of Runx tumor suppression.
MATERIALS and METHODS
Cells, constructs and retroviral transductions
Littermate wild type (WT) and Runx2−/− MEFs were prepared from E14.5 embryos derived from mating Runx2+/− mice (17). The virus packaging line Phoenix (ecotropic) was obtained from G Nolan, Stanford University, Palo Alto, CA, USA. All cultures were maintained in DMEM (Invitrogen) with 10% heat inactivated fetal calf serum (FCS), glutamine (2mM) and penicillin/streptomycin (200U/ml/100μg/ml). The retroviral vectors were all based on the pBabe plasmid (18), carrying the puromycin selectable marker. pBabe-PURO Runx2, kindly provided by M. Stewart, comprises a 2.3kb cDNA fragment encoding the Cbfa1-G1 isoform (3) subcloned into the polylinker region of pBabe-PURO. pBabe-PURO Ras was kindly provided by S. Lowe (16). Retroviral transductions were performed as previously described (11).
Growth curves, senescence staining and 3T3 passage culture
Early passage (p3-p4) MEFs were plated at 2.5 × 104/well in 12 well plates in growth medium or in selection medium containing 2μg/ml puromycin. Live cell counts were carried out in triplicate using trypan blue as a vital stain. Triplicate wells were pooled from each time point for analysis of protein expression. Media changes were carried out every 3-4 days. For UVC irradiation early passage MEFs were plated overnight at 5.0 × 105/60mm dish. The media was then removed and the cells irradiated using an XL-1500 UV crosslinker (Spectrolinker). The cultures were incubated in fresh media and live/dead cell counts performed after 24h by trypan blue exclusion. Graphs were plotted using Sigma plot and significance values determined by Student's t-test. Error bars relate to standard deviations. Senescence staining was assayed on a parallel plate after 7-10 days using a solution of X-gal (Invitrogen) at pH6.0 to detect SA-β-gal activity as described previously (16). 3T3 passage culture was essentially performed according to the protocol of Todaro and Green (19).
Cell cycle analysis
MEFs plated at 3 × 105 cells/10cm dish were sampled for flow cytometry in parallel with growth curves. The protocol for cell cycle analysis has been described previously (20). For simultaneous labelling with bromodeoxyuridine (BrdUrd), cells were incubated at 37°C with 10μM BrdUrd (Sigma) and then rinsed three times in warmed PBS. After harvesting, cells were washed in 2ml cold PBS, resuspended in 0.2ml cold PBS and fixed for at least 30 minutes in 2mls 70% ethanol at 4°C. Cells were denatured in 4M HCl for 20 min, rinsed twice in cold PBS, incubated for 10 mins in PBS/0.5% BSA/0.1% Tween 20 and labeled with FITC-conjugated anti-BrdUrd antibody (Roche). Samples were washed twice in PBS and resuspended in PBS containing 10μg/ml propidium iodide (PI). Analysis was carried out on a Beckman Coulter Epics XL using EXPO32 software.
Western blotting and antibodies
Preparation of whole cell protein extracts was performed as described previously (11). Samples equivalent to 30μg total protein (Biorad protein assay) were resolved on 8%, 10% or 17% SDS-polyacrylamide gels and transferred to ECL nitrocellulose membranes (Amersham). The antibodies used were α p16INK4a , αp21WAF1, α actin (Santa Cruz sc-1207, sc-471, sc 1616), α p19ARF (Abcam ab80), α p53, α phospho p38MAP kinase, α p38MAP kinase (Cell Signalling Tech. IC12, 9211, 9212), α c-H-ras (Oncogene Ab-1) and α RUNX2 (Stratech D130-3). Western blots were developed using ECL (Amersham) according to the manufacturer's protocol. Positive controls: p16INK4a and p19ARF, SV3T3 cell extract; p21WAF1, p53, UVC treated WT MEF extract and p38MAP kinase, Jurkat cell extract. Negative controls: p16INK4a and p19ARF, NIH3T3 cell extract; p21WAF1, RAT 1 fibroblast extract and p53, p53 null MEF extract.
In vivo transplantation studies
Runx2-null and WT MEFs were transduced with either the constitutive H-RasV12 or PURO-vector control and expanded in culture for 7 days. 106 cells in 0.2ml PBS were injected subcutaneously into the left flank of 5-6 MFI nu/nu mice (Harlan, UK) for each cell group. Animals were housed in sterile filter-top cages, monitored thrice weekly and humanely sacrificed when tumor masses reached a predetermined size or became ulcerated. All animal work was undertaken in line with the UK Animals (Scientific Procedures) Act of 1986.
Colony assays
For soft agar colony assays cells were diluted into 0.3% agar (Noble Difco) in DMEM containing 20% FCS and seeded in triplicate onto solidified 0.6% agar containing culture medium at 103, 104 or 105 cells/6cm plate (Bibby Sterilin). The colonies were fed every 3-4 days and evaluated after 5 weeks.
RNA extraction and cDNA preparation
Runx2−/− and WT MEFs were transduced with either constitutive H-RasV12 or the PURO-vector control and plated on 6 well plates at 7.5×104 cells/well. The cells were harvested from triplicate wells at day 3, 7, 10 and 13 into 1ml RNA-Bee (ams biotechnology) and RNA prepared according to the manufacturers protocol. RNA pellets were dissolved in DEPC-treated water (Ambion) and the concentration determined using an Agilent Bioanalyzer. cDNA was prepared from 1μg aliquots of RNA using a Quantitect Reverse Transcription Kit (Qiagen) and diluted 1 in 20 in DEPC-treated water to give a working stock.
Quantitative real time PCR
5μl aliquots of cDNA were amplified in triplicate on an ABI 7500 Real Time PCR System using Power SYBR Green PCR master Mix (Applera UK) and primers for murine cyclin A2, cyclin B1, cyclin E1, (Qiagen Quantitect Primer Assays QT00102151, QT00152040, QT00103495) or endogenous control hprt (106F 5′agcgtcgtgattagcgatgat 3′ and 253R 5′ccttcatgacatctcgagcaag 3′). Relative quantification was carried out and calibrated to day 0 WT sample.
In vitro kinase assays
Immunoprecipitation-kinase assays for cyclin A, cyclin B1 and cyclin E associated kinase activities with Histone H1 as a substrate were performed as described previously (21). Essentially Runx2−/− and WT MEFs were plated at 1.5×106 cells / 150mm dish and and grown to 80-90% confluence for 4-5 days. The contents of one dish were extracted into 0.5ml lysis buffer and the protein concentrations determined as for western extraction. 500μg of total cellular extract was analysed per reaction. Monoclonal antibodies against cyclin A (Chemicon, MAB3682) and cyclin B1 (Santa Cruz sc-245) were used to immunoprecipitate cyclin A and cyclin B1 associated kinase complexes directly. For cyclin E associated kinase activity cell lysates were sequentially immunoprecipitated with α cyclin A and α CDK2 (Santa Cruz sc-163). Kinase reaction were transferred to nitrocellulose membranes for autoradiography.
RESULTS
Induction of premature senescence in primary mouse fibroblasts is a common feature of the Runx family, but is distinct from H-RasV12 induced senescence
We previously demonstrated that RUNX1 mediates senescence-like growth arrest in primary MEFs (11). To determine whether this response is a general property of the Runx family we tested the effect of introducing ectopic Runx2 into WT cells by retroviral transduction. Western blot analysis revealed high intracellular levels of ectopic Runx2 and a corresponding dramatic reduction in cell numbers over a 7-day culture period (Figure 1A). In contrast, cells carrying the control vector (PURO control) showed at least a 10 fold increase in cell number over the same period. Moreover cells expressing ectopic Runx2 displayed a senescent-like phenotype characterized by an enlarged flattened appearance and senescence associated β-galactosidase staining. These results clearly demonstrate that ectopic Runx2 can induce a profound growth arrest in cultured cells and implicate Runx2 as a direct mediator of this response. Parallel experiments with Runx3 gave essentially identical results (Figure 1B). However, this phenomenon differed from premature senescence induced by H-RasV12 in primary MEFS which produced an early proliferative response followed by a sustained growth arrest with retention of fibroblastic and spindle shapes in a proportion of the culture (Figure 1C). While H-RasV12 induced senescence appears to be activated in response to aberrant cellular proliferation ((22) and refs therein), Runx-induced senescence is a more direct process reflecting negative growth regulation.
Figure 1.

Runx2 and Runx3 induce a senescence-like growth arrest in WT MEFs that is distinct from H-RasV12. (A) Cells were transduced with a vector containing Runx2 or a control vector containing the PURO-selectable gene. Growth curves showing viable cell numbers of Runx2 (dashed line) and vector control (solid line) cells are shown. Similar results were obtained in 2 independent experiments. Senescence-associated (SA) β-galactosidase staining of matched day 8 cultures are displayed. Photographs are at the same magnification. (B) Parallel growth curves and senescence-associated (SA) β-galactosidase staining in WT MEFs transduced with a vector containing Runx3 (dashed line) and (C) mutant H-RasV12 (dashed line).
Runx2−/− fibroblasts are prone to spontaneous immortalization and display an early growth advantage in vitro.
Analysis of Runx gene family expression in primary MEFs by western blot and gene expression microarray revealed that Runx2 is the most abundantly expressed family member, and this observation led us to consider the possibility that this gene has a non-redundant role in their growth regulation. Under typical culture conditions, primary MEFs exit the cell cycle in increasing numbers during passage resulting in eventual growth stasis (19). The use of a 3T3 passage protocol can occasionally permit the accumulation of secondary events that serve to immortalize primary cells in vitro. Using this approach we observed cessation of growth in 6/6 cultures of WT MEFs (Figure 2A). In contrast, 5/6 lines derived from Runx2−/− embryos exhibited an enhanced growth rate around passage 8-10 indicating the emergence of clones with an increased proportion of proliferating cells (Figure 2A). Further study revealed that these cells were immortalized and unlike their WT counterparts could grow beyond passage 25 with little or no sign of reduced proliferation. However, these cells remained non-tumorigenic in immunocompromised mice.
Figure 2.
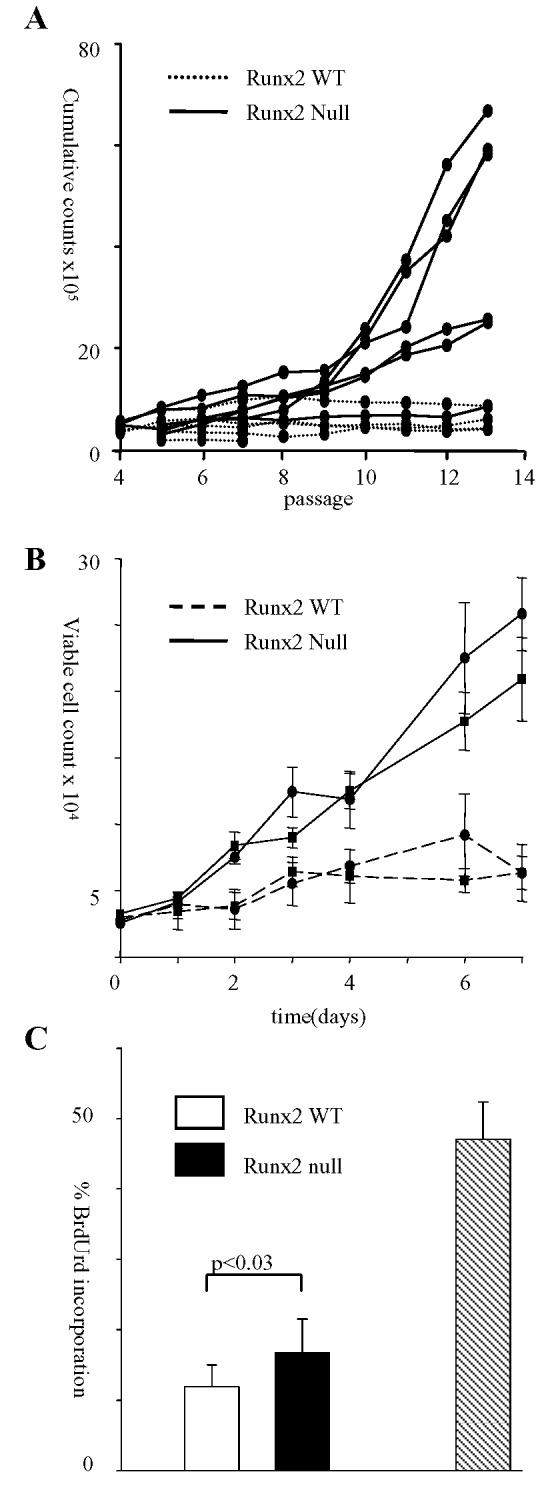
Runx2−/− fibroblasts are prone to secondary events that predispose to immortalisation. (A) 3T3 passage culture was performed on five WT and six Runx2−/− lines. Cells were plated at 3.0 × 105/T25 flask and passaged every third day reseeding 3.0 × 105 cells into a fresh flask. The increase in total viable cell numbers was calculated at every passage and added to a cumulative count over time. Secondary events that predispose to immortalisation occurred between passage 8 and 10. (B) Runx2−/− fibroblasts display an early growth advantage in vitro. Growth curve of two Runx2−/− lines compared to two WT lines seeded at passage 4. The experiment was repeated on six littermate matched Runx2−/− and WT lines with essentially identical results. (C) The percentage of BrdUrd incorporation in three Runx2−/− and three WT lines seeded at passage 5 and labelled for 3h. A comparison is shown for NIH3T3 cells labelled for 1h (hatched bar).
To further investigate the nature of their growth deregulation, we examined the properties of early passage Runx2−/− MEFs. As shown in Figure 2B, Runx2−/− cells expanded significantly faster than litter-matched controls over 7 days in culture. Flow cytometry analysis of BrdUrd accumulation indicated that the increase in total cell count was due, at least in part, to increased cell division in the Runx2−/− cultures (Figure 2C). However, we noted that the overall rates of proliferation of early MEF cultures were very low compared to established 3T3 fibroblasts (Figure 2C), with only 10% of control cells taking up BrdUrd over a 3 hr period.
Primary rodent fibroblasts display a progressive loss in proliferative capacity in culture that has been attributed to culture stress and may account for the low proliferative rates of primary MEFs observed in this study (23,24). To determine whether the growth advantage of Runx2−/− fibroblasts was a consequence of reduced sensitivity to “culture shock” cells of both genetic background were plated at passages 3, 5 & 7 and their growth compared over 7 days in culture. As shown in Figure 3A Runx2−/− fibroblasts retain a greater proliferative capacity than their WT counterparts with successive passages but have not lost their sensitivity to replication-induced growth inhibition. In contrast differential susceptibility was seen in the response to low dose UVC, where Runx2−/− cells showed increased viability relative to their wild-type counterparts (Figure 3B). In view of evidence that the Runx genes play important roles in negative growth regulation by TGF-β in a range of cell types (9,25,26) we also tested the effects of Runx2 disruption on this response. Surprisingly, we saw no impairment in TGF-β growth arrest over a range of concentrations (Figure 3C).
Figure 3.
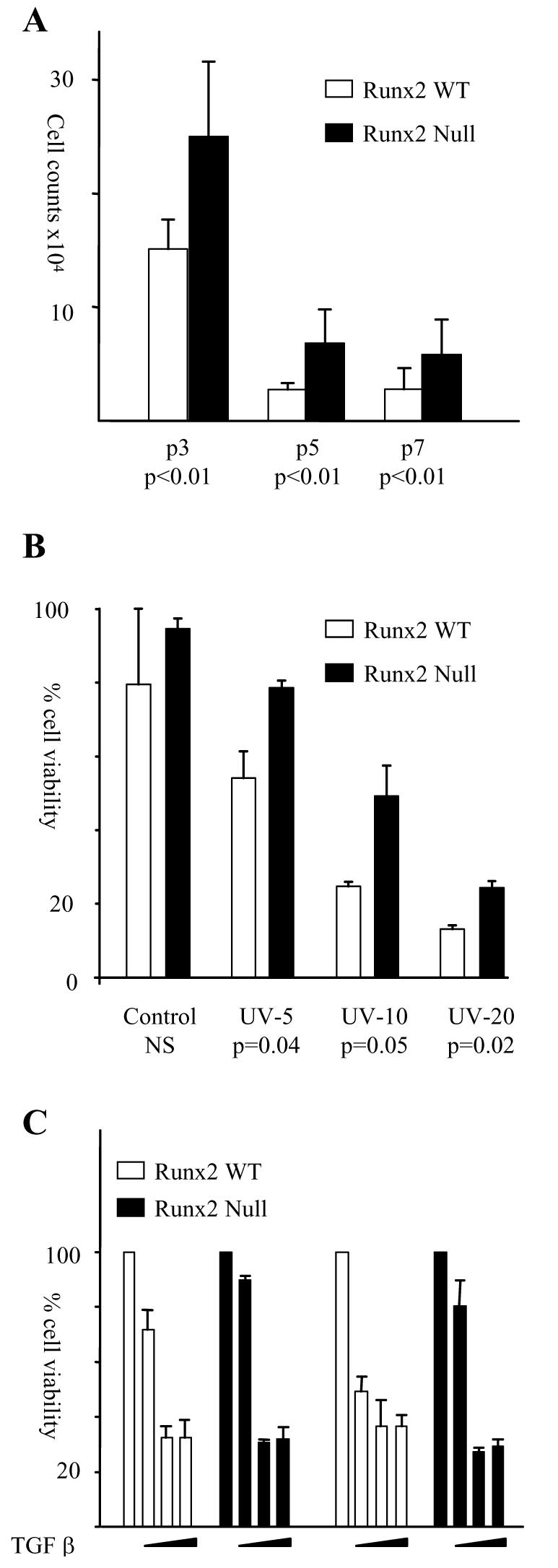
Loss of Runx2 conveys a growth advantage that is resistant to certain cellular stresses but not to TGFβ mediated growth inhibition. (A) Total viable cell counts at day seven from six WT and six Runx2−/− lines plated in triplicate at passages 3, 5 and 7. (B) Runx2−/− fibroblasts are more resistant to UVC induced cell death than WT controls. Two Runx2−/− and two WT cultures were plated in triplicate in 60mm dishes and irradiated with 5, 10 and 20 J/m2 UVC irradiation. Live / dead cell counts were performed by trypan blue exclusion after 24h in culture. Similar results were obtained in 2 independent experiments. (C) The growth of Runx2−/− and WT fibroblasts was inhibited by TGFβ. 5ng/ml, 0.5ng/ml or 0.05ng/ml TGFβ was added to triplicate wells on days 1, 3, 5 & 6 and total viable cell counts calculated on day 7 by trypan blue exclusion. Control cultures were fed fresh media in parallel. The histogram is a representative of two independent experiments. Replicate experiments on 3 independent Runx2−/− and WT lines performed with 5ng/ml TGFβ gave essentially identical results.
Loss of Runx2 permits escape from H-RasV12-induced senescence and oncogenic transformation
Since the Runx genes are potent agonists of premature senescence and Runx2 disruption confers differential sensitivity to certain stress signaling pathways in primary MEFs, we investigated the effect of Runx2 loss on the well-characterised phenomenon of Ras oncogene-induced senescence. As shown in Figure 4A expression of H-RasV12 in the WT cell background induced growth stasis after an initial proliferative burst that was comparable to the vector transduced controls. In contrast, Runx2−/− lines expressing H-RasV12 displayed a twenty-forty fold expansion in cell number over the same period and unlike WT cells were negative for senescence-associated β-galactosidase staining (Figure 4A). The contrasting effects were not due to differential levels of H-RasV12 expression as WT and Runx2−/− lines expressed comparable levels of H-RasV12 by western blotting. Also, Runx2−/− cells were not intrinsically resistant to growth stasis as reintroduction of Runx2 induced senescent-like growth arrest and β-galactosidase staining in a manner indistinguishable from wild-type controls (Figure 4B).
Figure 4.

Runx2−/− cells expressing H-RasV12 fail to undergo senescence. (A) Runx2−/− and WT cells were transduced with a vector encoding H-RasV12 or a control vector containing only the PURO selectable gene. Passage 4 growth curves over 14 days showing viable cell numbers. Similar results were obtained in 4 independent experiments using littermate matched Runx2−/− and WT lines. B) Senescence associated β-galactosidase activity of Runx2−/− cells transduced with a vector encoding murine Runx2. Transduction with the control vector containing only the PURO selectable gene failed to generate positively stained cultures. Photographs are at the same magnification. (C) Colony formation of Runx2−/− fibroblasts retrovirally transduced with H-RasV12. Cells were transduced with a retrovirus encoding H-RasV12 or a control vector containing only the PURO selectable gene and analysed for oncogenic transformation. Numbers were determined for agar colonies > 0.1mm derived from plating 103 cells per 60mm dish and fed every 3-4 days for 5 weeks. Error bars indicate the average number of colonies derived from 3 independent dishes.
To determine whether Runx2 loss and H-RasV12 can collaborate in the oncogenic transformation of MEFs, H-RasV12 expressing Runx2−/− and WT cells were seeded in soft agar. As shown in Figure 4C, anchorage independent growth was significantly enhanced in H-RasV12/Runx2−/− cells as compared to H-RasV12/WT cells. The enhanced growth potential of H-RasV12/Runx2−/− cells was also seen in colony formation assays where these cells showed a much higher frequency of colony growth following low density plating (data not shown). To determine whether loss of Runx2 renders primary MEFs susceptible to tumorigenesis in vivo H-RasV12/Runx2−/−, H-RasV12/WT and vector control cells were transplanted into athymic mice. Whilst no tumor growth occurred with cells carrying the empty vector (control cells = 0/6, Runx2−/− cells = 0/6) or WT cells expressing H-RasV12 (0/6), those cells null for Runx2 and expressing H-RasV12 invariably produced tumors (6/6). Although most tumors appeared within 2-4 weeks, it was notable that these generally remained small and dormant before a later phase of expansion and vigorous growth. Examination of the rapidly growing tumors revealed a heterogeneous pattern with loss of expression of p16, p21 and/or p53 (not shown). While Runx2 loss is clearly sufficient to allow Ras-induced tumors to arise, it appears that the transformed cells are not immortalized and that additional events are necessary for tumor progression.
Loss of Runx2 does not block induction of cell cycle inhibitors, but nevertheless disables cell cycle checkpoint controls in response to H-RasV12
H-RasV12 induced premature senescence is accompanied by activation of p38MAP kinase (27,28) and the induction of p53, p19ARF, p16INK4a and p21WAF1 (16). To determine whether these responses are intact in Runx2−/− MEFs we examined the levels of activated p38MAP kinase using an antibody that specifically recognizes phospho Thr180 and Tyr182 p38 MAP kinase, previously shown to represent the active kinase (29). As shown in Figure 5A phospho-p38MAP kinase was readily detectable in WT MEFs. The levels exceeded that of our positive control but could not be further induced by H-RasV12 indicating high endogenous activation of this stress signalling cascade. Activation of p38 MAP kinase was similarly high in Runx2−/− fibroblasts but in contrast to WT controls could be further activated by H-RasV12 (Figure 5A). In vitro kinase assays using ATF2 as a substrate confirmed it to be fully functional (data not shown) indicating that signalling between H-RasV12 and p38 MAP kinase remained intact in Runx2−/− MEFs. An examination of the expression levels of p53, p19ARF, p16INK4a and p21WAF1 revealed that despite lower basal levels in the Runx2−/− genetic background, H-RasV12 induced p53, p19ARF, p16INK4a and p21WAF1 to a comparable level in both Runx2−/− and WT control cells (Figure 5B). Therefore, although Runx2 is essential for H-RasV12 to exert its growth suppressing function it does not appear to be required for the induction of a panel of genes that have been shown to contribute to the growth arrest.
Figure 5.

Loss of Runx2 does not block the induction of growth arrest pathways by Ras but facilitates transit through S/G2/M. (A) Total protein was extracted from early passage (p4) littermate matched Runx2−/− and WT lines transduced with H-RasV12 (R) or the PURO (P) vector control and probed against antibodies to phospho-p38MAP kinase or p38MAP kinase as a loading control. The western blot is representative of two independent experiments. (B) Western blot analysis as in (A) probed against antibodies to p16INK4a, p19ARF, p21WAF1 and p53. Actin was used as a loading control. (C) Cell cycle analysis of H-RasV12 transduced WT and Runx2−/− MEFs is shown as the percentage of propidium iodide stained cells with 2N (top) and 4N (bottom) DNA content at day 13 following transduction. Two independent littermate matched WT and Runx2−/− cultures are grouped (1 & 2). Error bars represent triplicate samples. Similar results from two independent experiments. (D) Cytometry density plots of Runx2−/− (top) and WT (bottom) cells transduced with H-RasV12 which were labelled with BrdUrd for 15 hours. Percentage of BrdUrd incorporation is given based on triplicate samples. Identical results were seen with 2 independent WT and Runx2−/− samples.
In contrast, cell cycle analysis confirmed and elucidated a profound difference in the growth response of the Runx2−/− cells to H-RasV12. Propidium iodide staining revealed a higher proportion of cells with a 2N (G1) DNA content and a significantly lower proportion of cells with a 4N (G2/M) DNA content in Runx2−/− cells compared to controls (Figure 5C). Dual staining with PI and BrdUrd demonstrated higher rates of BrdUrd incorporation in Runx2−/− H-RasV12 cultures, consistent with their observed expansion in cell numbers during the culture period (Figure 5D). Of these cells a significant proportion displayed a 2N DNA content indicating successful transit through G2/M. A striking difference was seen in the WT cultures which displayed a much lower number of BrdUrd positive cells with a 2N DNA content. Together these results demonstrate that the H-RasV12 expressing Runx2−/− cells are actively cycling whilst a significant proportion of WT cells are delayed or arrested during transit through S/G2/M.
Loss of Runx2 confers increased expression of S/G2/M cyclins and associated cyclin-dependent kinase activities
The continued cycling of Runx2−/− cells in the presence of a cascade of growth inhibitors suggested a compensatory mechanism possibly involving constitutive expression of positive growth mediators. We therefore compared the levels of cyclin gene expression in the WT and Runx2−/− genetic backgrounds. Quantitative RT-PCR analysis showed that cyclin D1 expression was unaffected by Runx2 status although it was upregulated by H-RasV12 as previously reported (Figure 6A). However, the expression levels of cyclins A2 (Figure 6B), B1 (Figure 6C) and E1 (Figure 6D) mRNA were all markedly up-regulated in Runx2−/− fibroblasts as compared to WT controls (Figure 6A). Moreover, unlike WT cells where expression of these cyclins declined steadily throughout the culture period, high levels of cyclin E1, cyclin A2 and cyclin B1 mRNA were sustained in Runx2−/− MEFs and persisted even as the cells reached confluence. Strikingly, deregulation of cyclin levels was observed in Runx2−/− cells whether or not they expressed H-RasV12.
Figure 6.
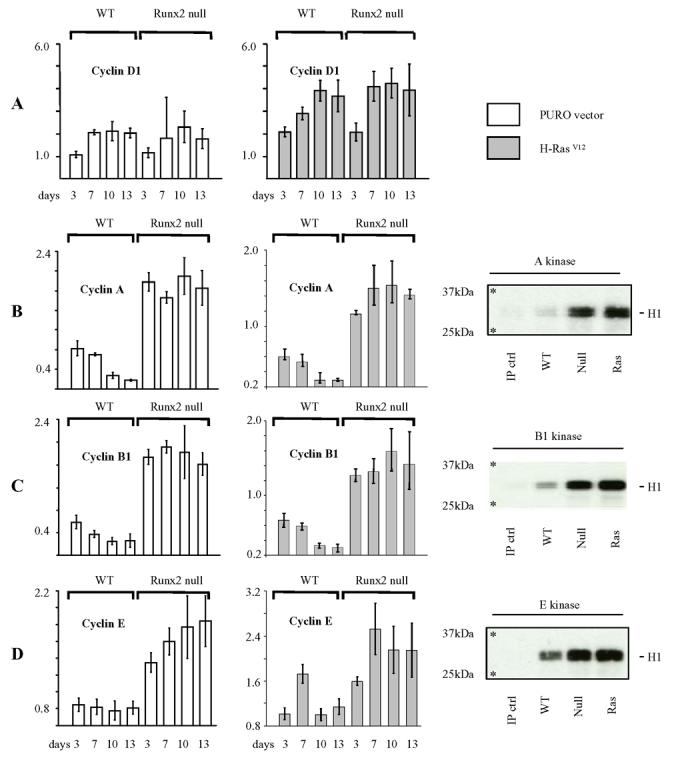
Loss of Runx2 confers increased expression of S/G2/M cyclins and associated cyclin-dependent kinase activities. (A) Cyclin D1 expression is refractory to loss of Runx2 but remains sensitive to induction by H-RasV12. cDNA was prepared from littermate matched Runx2−/− and WT cultures transduced with H-RasV12 or the PURO control vector and plated for 0 to 13 days in culture. Cyclin D1 expression was assayed at each timepoint by relative quantification to endogenous control hprt and calibrated to the day 0 WT sample. (B) – (D) Loss of Runx2 confers increased expression of cyclin A2 (B), cyclin B1 (C) and cyclin E1 (D) and sustains downstream cyclin dependent kinase activity. cDNA was analysed from the same data set as described in (A). The data is displayed as raw RQ values and is representative of two independent experiments on two littermate matched Runx2−/− and WT lines. Parallel cultures were harvested for immunoprecipitation (IP) kinase assays of total cyclin A, cyclin B1 and cyclin E associated kinase activities. Cell extracts were immunoprecipitated with antibodies specific for the relevant cyclin and the precipitates analysed for kinase activity using Histone H1(32kDa) as a substrate. The 25kDa and 37kDa molecular weight markers are marked with an asterisk. The data is representative of 2 independent experiments.
To determine whether cyclin dependent kinase activity was also deregulated in the absence of Runx2, in vitro kinase assays were performed using Histone H1 as a substrate. As shown in the parallel panels elevated levels of A, B1 and E dependent kinase activities were observed in Runx2−/− cells relative to WT controls after co-immunoprecipitation with the corresponding antibody. Comparable results were also obtained upon retroviral transduction with H-RasV12. A direct analysis of the kinase complex composition was problematic due to co-migration of cyclin proteins with the IgG heavy chain moiety but examination of cyclin protein expression by western blotting confirmed the qRT-PCR data, thereby supporting a direct relationship between cyclin gene expression levels and S/G2/M cyclin dependent kinase activities in Runx2−/− cells.
DISCUSSION
In this study we have demonstrated that Runx2 can induce premature senescence in primary MEFs, revealing a further functional attribute shared by the Runx gene family. This process is markedly different from Ras-induced senescence that occurs in response to aberrant proliferation and DNA damage signaling ((22) and refs therein) and it appears that Runx signaling induces a senescence cascade downstream of these responses. However, the process remains dependent on p53, and our finding that p53 is required for Runx2-induced senescence recapitulates previous results for RUNX1 (11). More surprising is our finding that Runx2 has a non-redundant role in MEFs where gene disruption promotes immortalization, disables Ras-induced senescence and facilitates oncogenic transformation. These findings offer a new perspective on the lineage-specific tumor suppressor activities of the Runx gene family.
Runx2−/− MEFs display enhanced proliferation, but it is important to note that this phenomenon was observed on the background of primary cultures which display low proliferative rates compared to established cell lines. The early growth advantage was refractory to the non-physiological conditions of in vitro culture (23,24), but was amplified by low-dose UVC irradiation suggesting that loss of Runx2 alters sensitivity to a subset of stress signaling pathways. The early growth advantage of Runx2−/− cells may be sufficient to account for their increased susceptibility to become immortalized at much higher rates than WT cultures, although a more direct role in regulating cellular lifespan or replicative senescence in response to telomere shortening cannot be formally excluded. However, it is clear that immortalized Runx2−/− cells remain non-tumorigenic, unlike their p53−/− counterparts, suggesting that this tumor suppressor-like property of Runx2 is likely to be less potently manifested in vivo. Another interesting contrast is that Runx2−/− cells retain full sensitivity to the growth inhibitory effects of TGF-β while p53−/− cells are refractory (30). Whether Runx function is dispensable for TGF-β inhibition in primary MEFs or this process is mediated by low level expression of another family member remains to be determined.
Escape from senescence in response to exogenous H-RasV12 is the most striking attribute of Runx2−/− cells. We have established that this phenotype is not due to an intrinsic loss of capacity to undergo senescence as Runx2−/− lines remain susceptible to senescence induced by ectopic expression of Runx2. However, attempts to identify the basis of their resistance to H-RasV12 revealed some unanticipated results. In wild-type MEFs, H-RasV12 induced senescence is accompanied by activation of the p38 MAP kinase signaling cascade (28) and the induction of p16INK4a, p19ARF, p21WAF1 and p53 (16). We found no obvious defect in these responses in Runx2−/− MEFs which express high levels of the active p38 MAP kinase and accumulate all four proteins to similar levels in response to H-RasV12. In fact, there was evidence of hyper-activation of p38 MAP kinase in Runx2−/− cells, suggestive of the loss of a feedback control mechanism. These findings were somewhat surprising as at least two of the genes involved have been implicated as direct targets for Runx regulation (12,31,32), and Runx2−/− MEFs expressed slightly lower basal levels of all of these markers. It appears that Runx2 is dispensable for the induction of this cascade of inhibitory signals by H-RasV12 and that the explanation for the continued proliferation of Runx2−/− cells must lie elsewhere downstream.
Analysis of other components of the cell cycle machinery provided us with a clear rationale for the aberrant growth properties of Runx2−/− cultures, as these cells were found to express high levels of cyclin A2, B1 and E1 that were refractory to down-regulation by Ras. Moreover, the corresponding cyclin-associated CDK activities were markedly elevated on the Runx2−/− background. While earlier studies of H-RasV12-induced senescence have described a G1 arrest as the predominant feature (16,33) cell cycle analysis of our MEF cultures revealed stronger evidence of a block at G2/M in wild-type cells, with accumulation of cells with a 4N DNA content, while Runx2−/− cells continued to cycle. Our observations do not exclude the existence of multiple blocks to cell cycle progression in H-RasV12 transduced cells, but are in accord with recent studies showing that oncogene-induced senescence and associated DNA damage can result in activation of S/G2/M checkpoints (34,35).
A precedent for the ability of deregulated cyclin expression to overcome the Ras-induced inhibitory cascade has been provided by two genes discovered in screens for mediators of escape from Ras-induced senescence. Thus, hDRIL binds E2F1 and activates the cyclin E1 promoter (36) while repression of BTG2 leads to upregulation of cyclins D1 and E1 (37). Similarly, it now appears that the constitutive expression and/or lack of silencing of several cyclin genes underlies the resistance of Runx2−/− cells to culture stress and Ras-induced growth arrest. It is interesting to note that cyclin A2 is among the E2F-regulated genes that are sequestered in senescence-associated heterochromatin foci (38,39), which appear in the course of a multi-step process leading to irreversible loss of replicative capacity in human fibroblasts. The possibility that Runx functions are required in this critical program merits further investigation, particularly in light of evidence implicating Runx2 in SWI/SNF chromatin remodelling in osteoblast differentiation (40). It is conceivable that other defects in Runx2 null cells also contribute to their resistance to Ras-induced senescence. A recent study in human fibroblasts has shown that Ras-induced senescence requires attenuation of MAPK and PI3K signalling in addition to induction of cell cycle inhibitors (41) and it is intriguing to note that we observed elevated p38 MAPK signalling in Runx2 null cells in the presence of Ras.
This study also has wider implications for the role of the Runx genes in cancer. Since all three genes induce premature senescence when expressed ectopically, the prospect of a similar redundant role for the endogenous Runx genes in the induction of the senescence program in response to oncogene-induced growth arrest merits further investigation. It may be expected that manifestations of loss of Runx expression or function will be limited to those tissues where a single family member plays a decisive role, as we have found for Runx2 in MEFs. Whether Runx2 loss or silencing is associated with specific cancers in vivo is an interesting question. As this gene plays a critical role in vivo in the development of chondrogenic and osteogenic tissues, its tumor protective action might be manifested in mesenchymal tissues. With regard to other family members, it is notable that RUNX1 loss of function mutations have been associated with activated Ras signaling in AML and MDS/AML (42,43). In addition epithelial cancers that have been associated with RUNX3 down-regulation ((2), and refs therein) display Ras pathway mutations at significant frequencies suggesting the possibility of parallel roles for the other Runx family members. It will also be interesting to explore the Runx2-dependent growth arrest mechanism uncovered here for its relevance to the normal physiological roles of the Runx genes in cell fate determination, particularly where these entail checks to the proliferation of stem cells or early progenitors to permit lineage-specific differentiation (44).
References
- 1.Gergen JP, Butler BA. Isolation of the Drosophila Segmentation Gene Runt and Analysis of Its Expression During Embryogenesis. Genes Dev. 1988;2:1179–93. doi: 10.1101/gad.2.9.1179. [DOI] [PubMed] [Google Scholar]
- 2.Blyth K, Cameron ER, Neil JC. The Runx gene family : gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–87. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- 3.Stewart M, Terry A, Hu M, et al. Proviral insertions induce the expression of bone-specific isoforms of PEBP2alphaA (CBFA1): evidence for a new myc collaborating oncogene. Proc Natl Acad Sci USA. 1997;94:8646–51. doi: 10.1073/pnas.94.16.8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart M, Mackay N, Cameron ER, Neil JC. The common retroviral insertion locus Dsi1 maps 30kb upstream of the P1 promoter of the murine Runx3/Cbfa3/Aml2 gene. J Virol. 2002;76:4364–9. doi: 10.1128/JVI.76.9.4364-4369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wotton S, Stewart M, Blyth K, et al. Proviral insertion indicates a dominant oncogenic role for Runx1/AML1 in T-cell lymphoma. Cancer Res. 2002;62:7181–5. [PubMed] [Google Scholar]
- 6.Blyth K, Vaillant F, Mackay N, et al. Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res. 2006;66:2195–201. doi: 10.1158/0008-5472.CAN-05-3558. [DOI] [PubMed] [Google Scholar]
- 7.Growney JD, Shigematsu H, Li Z, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106:494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyazono K, Maeda S, Imamura T. Coordinate regulation of cell growth and differentiation by TGF-beta superfamily and Runx proteins. Oncogene. 2004;23:4232–7. doi: 10.1038/sj.onc.1207131. [DOI] [PubMed] [Google Scholar]
- 9.Ito Y, Miyazono K. RUNX transcription factors as key targets of TGF-beta superfamily signaling. Curr Opin Genet Dev. 2003;13:43–7. doi: 10.1016/s0959-437x(03)00007-8. [DOI] [PubMed] [Google Scholar]
- 10.Blyth K, Terry A, Mackay N, et al. Runx2: a novel oncogenic effector revealed by in vivo complementation and retroviral tagging. Oncogene. 2001;20:295–302. doi: 10.1038/sj.onc.1204090. [DOI] [PubMed] [Google Scholar]
- 11.Wotton S, Blyth K, Kilbey A, et al. RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene. 2004;23:5476–86. doi: 10.1038/sj.onc.1207729. [DOI] [PubMed] [Google Scholar]
- 12.Linggi B, Muller-Tidow C, van de LL, et al. The t(8;21) fusion protein, AML1 ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nat Med. 2002;8:743–50. doi: 10.1038/nm726. [DOI] [PubMed] [Google Scholar]
- 13.Hemann MT, Narita M. Oncogenes and senescence: breaking down in the fast lane. Genes Dev. 2007;21:1–5. doi: 10.1101/gad.1514207. [DOI] [PubMed] [Google Scholar]
- 14.Michaloglou C, Vredeveld LCW, Soengas MS, et al. BRAF(E600)-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 15.Chen ZB, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a) Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 17.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 18.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–96. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todaro GJ, Green H. Quantitative Studies of Growth of Mouse Embryo Cells in Culture and Their Development Into Established Lines. J Cell Biol. 1963;17:299. doi: 10.1083/jcb.17.2.299. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blyth K, Stewart M, Bell M, et al. Sensitivity to Myc-induced apoptosis is retained in spontaneous and transplanted lymphomas of CD2-mycER mice. Oncogene. 2000;19:773–82. doi: 10.1038/sj.onc.1203321. [DOI] [PubMed] [Google Scholar]
- 21.Clark W, Black EJ, MacLaren A, et al. v-Jun overrides the mitogen dependence of S-phase entry by deregulating retinoblastoma protein phosphorylation and E2F-pocket protein interactions as a consequence of enhanced cyclin E-cdk2 catalytic activity. Mol Cell Biol. 2000;20:2529–42. doi: 10.1128/mcb.20.7.2529-2542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell. 2007;128:233–4. doi: 10.1016/j.cell.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Sherr CJ, DePinho RA. Cellular senescence: Mitotic clock or culture shock? Cell. 2000;102:407–10. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 24.Ramirez RD, Morales CP, Herbert BS, et al. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001;15:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torquati A, O'Rear L, Longobardi L, Spagnoli A, Richards WO, Beauchamp RD. RUNX3 inhibits cell proliferation and induces apoptosis by reinstating transforming growth factor beta responsiveness in esophageal adenocarcinoma cells. Surgery. 2004;136:310–6. doi: 10.1016/j.surg.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Chi XZ, Yang JO, Lee KY, et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor beta-activated SMAD. Mol Cell Biol. 2005;25:8097–107. doi: 10.1128/MCB.25.18.8097-8107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang WP, Chen JX, Liao R, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22:3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulavin DV, Kovalsky O, Hollander MC, Fornace AJ. Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of gadd45a. Mol Cell Biol. 2003;23:3859–71. doi: 10.1128/MCB.23.11.3859-3871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raingeaud J, Gupta S, Rogers JS, et al. Pro-Inflammatory Cytokines and Environmental-Stress Cause P38 Mitogen-Activated Protein-Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. J Biol Chem. 1995;270:7420–6. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 30.Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–14. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 31.Lutterbach B, Westendorf JJ, Linggi B, Isaac S, Seto E, Hiebert SW. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J Biol Chem. 2000;275:651–6. doi: 10.1074/jbc.275.1.651. [DOI] [PubMed] [Google Scholar]
- 32.Westendorf JJ, Zaidi SK, Cascino JE, et al. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol Cell Biol. 2002;22:7982–92. doi: 10.1128/MCB.22.22.7982-7992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mason DX, Jackson TJ, Lin AW. Molecular signature of oncogenic ras-induced senescence. Oncogene. 2004;23:9238–46. doi: 10.1038/sj.onc.1208172. [DOI] [PubMed] [Google Scholar]
- 34.Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 35.Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 36.Peeper DS, Shvarts A, Brummelkamp T, et al. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002;4:148–53. doi: 10.1038/ncb742. [DOI] [PubMed] [Google Scholar]
- 37.Boiko AD, Porteous S, Razorenova OV, Krivokrysenko VI, Williams BR, Gudkov AV. A systematic search for downstream mediators of tumor suppressor function of p53 reveals a major role of BTG2 in suppression of Ras-induced transformation. Genes Dev. 2006;20:236–52. doi: 10.1101/gad.1372606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narita M, Nunez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang RG, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. 2007;27:2343–58. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young DW, Pratap J, Javed A, et al. SWI/SNF chromatin remodeling complex is obligatory for BMP2-induced, Runx2-dependent skeletal gene expression that controls osteoblast differentiation. J Cell Biochem. 2005;94:720–30. doi: 10.1002/jcb.20332. [DOI] [PubMed] [Google Scholar]
- 41.Courtois-Cox S, Williams SMG, Reczek EE, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roumier C, Lejeune-Dumoulin S, Renneville A, et al. Cooperation of activating RAS/rtk signal transduction pathway mutations and inactivating myeloid differentiation gene mutations in M0 AML: a study of 45 patients. Leukemia. 2006;20:433–6. doi: 10.1038/sj.leu.2404097. [DOI] [PubMed] [Google Scholar]
- 43.Niimi H, Harada H, Harada Y, et al. Hyperactivation of the RAS signaling pathway in myelodysplastic syndrome with AML1/RUNX1 point mutations. Leukemia. 2006;20:635–44. doi: 10.1038/sj.leu.2404136. [DOI] [PubMed] [Google Scholar]
- 44.Pratap J, Galindo M, Zaidi SK, et al. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 2003;63:5357–62. [PubMed] [Google Scholar]