

Abstract
In vivo incorporation of isotopically labeled unnatural amino acids into large proteins drastically reduces the complexity of nuclear magnetic resonance (NMR) spectra. Incorporation is accomplished by co-expressing an orthogonal tRNA/aminoacyl-tRNA synthetase pair specific for the unnatural amino acid added to the media and the protein of interest with a TAG amber codon at the desired incorporation site. To demonstrate the utility of this approach for NMR studies, 2-amino-3-(4-(trifluoromethoxy) phenyl) propanoic acid (OCF3Phe), 13C/15N-labeled p-methoxyphenylalanine (OMePhe), and 15N-labeled o-nitrobenzyl-tyrosine (oNBTyr) were incorporated individually into 11 positions around the active site of the 33 kDa thioesterase domain of human fatty acid synthase (FAS-TE). In the process, a novel tRNA synthetase was evolved for OCF3Phe. Incorporation efficiencies and FAS-TE yields were improved by including an inducible copy of the respective aminoacyl-tRNA synthetase gene on each incorporation plasmid. Using only between 8 and 25 mg of unnatural amino acid, typically 2 mg of FAS-TE, sufficient for one 0.1 mM NMR sample, were produced from 50 mL of E. coli culture grown in rich media. Singly labeled protein samples were then used to study the binding of a tool compound. Chemical shift changes in 1H-15N, 1H-13C HSQC and 19F NMR spectra of the different single site mutants consistently identified the binding site and the effect of ligand binding on conformational exchange of some of the residues. OMePhe or OCF3Phe mutants of an active site tyrosine inhibited binding; incorporating 15N-Tyr at this site through UV-cleavage of the nitrobenzyl-photocage from oNBTyr re-established binding. These data suggest not only robust methods for using unnatural amino acids to study large proteins by NMR but also establish a new avenue for the site-specific labeling of proteins at individual residues without altering the protein sequence, a feat that can currently not be accomplished with any other method.
Introduction
NMR spectroscopy is an established and very powerful biophysical method to study the structure, dynamics and function of proteins.1–3 In principle, molecular processes such as binding or conformational changes can be deciphered by NMR with atomic resolution. However, the complexity of NMR spectra of large proteins in practice often represents a formidable challenge. Simplification of spectra by amino acid-type specific isotope labeling4 including labeling with fluorinated analogs of natural amino acids,5–8 by incorporation of stereo-array isotope labeling (SAIL) amino acids9 or selective isotope labeling of methyl groups in perdeuterated proteins10,11 is very helpful for many applications and required for larger systems. Recently developed methods for the in vivo incorporation of unnatural amino acids into proteins12–18 provide an unique opportunity to introduce NMR-active labels at a single designated amino acid residue.19
Using an orthogonal amber tRNA/tRNA synthetase (tRNA/RS) pair that specifically aminoacylates an amber tRNA with the desired unnatural amino acid and incorporates it into a protein at the amber nonsense codon UAG, any desired protein residue can, at least in principle, be substituted in vivo by an unnatural amino acid. The incorporation of the unnatural amino acid at the desired location instantly provides an “assignment” for the NMR signal of the unnatural amino acid. Monitoring the chemical shift change of a single resonance opens an avenue for focused, site-directed screening for binders. This is particularly interesting for drug development since it can reduce the number of screening hits that bind to protein pockets of little interest. Furthermore unnatural amino acids incorporated at different sites in multiple samples can be used to establish binding within an active site or could be used to triangulate binding of a small molecule or a biomacromolecule via NOE measurements. Similarly, saturation transfer techniques that utilize selective labeling strategies to study the structures of protein-ligand complexes20 can utilize the specificity afforded by unnatural amino acid incorporation to determine both the protein residues and regions of the molecule that are in close proximity. Protein conformational changes and dynamics can also be monitored at specific sites of interest with minimal or no perturbation of sequence. Mixtures of proteins labeled with different unnatural amino acids can simplify the NMR characterization of symmetric oligomers. Selectively incorporating methyl labels can be very useful for examining the millisecond time scales associated with many biological processes, such as enzyme catalysis, product release, and protein folding.21 The unnatural amino acid labeling technique presented here has the potential to extend NMR studies to many proteins that are currently not amendable because of their complexity, size, or physical characteristics.
Previously, we have shown that protein amounts sufficient for NMR studies can be produced in E. coli using 15N-labeled O-methyl-phenylalanine (OMePhe) (Figure 1).19 Since the yields were modest one of the primary goals of the current study was to improve the incorporation efficiency and consequently increase protein yields. This was accomplished by constructing a plasmid that overexpresses the RS specific for the unnatural amino acid at the time of induction. The second goal was to expand the number of NMR-active unnatural amino acids. Producing 15N and 13C-labeled OMePhe was an obvious first step (Figure 1). Developing an orthogonal tRNA/RS pair for the incorporation of a tri-fluorinated analog was our second choice. Fluorine represents an attractive NMR label5–8 with high intrinsic sensitivity, 100% natural abundance, and the absence of any natural background. The chemical shift of 19F is extremely sensitive to the local environment and can detect changes in van der Waals contacts, electrostatic fields and hydrogen bonding. In contrast to Mehl and coworkers who recently selected a specific tRNA/RS pair for trifluoromethyl-phenylalanine and demonstrated its utility for 19F protein NMR studies,22 we opted to develop such a system for OCF3Phe (Figure 1) because of its structural similarity to OMePhe. As a third unnatural amino acid, we tested the incorporation of 15N-labeled o-nitrobenzyl-tyrosine (oNBTyr) (Figure 1). It has been demonstrated that UV-photo-cleavage of this unnatural amino acid after incorporation converts oNBTyr back to the natural amino acid tyrosine.23 In contrast to the other unnatural amino acids, photo-caged tyrosine facilitates the introduction of an NMR label at specific tyrosine residues without altering the protein sequence.
Figure 1.
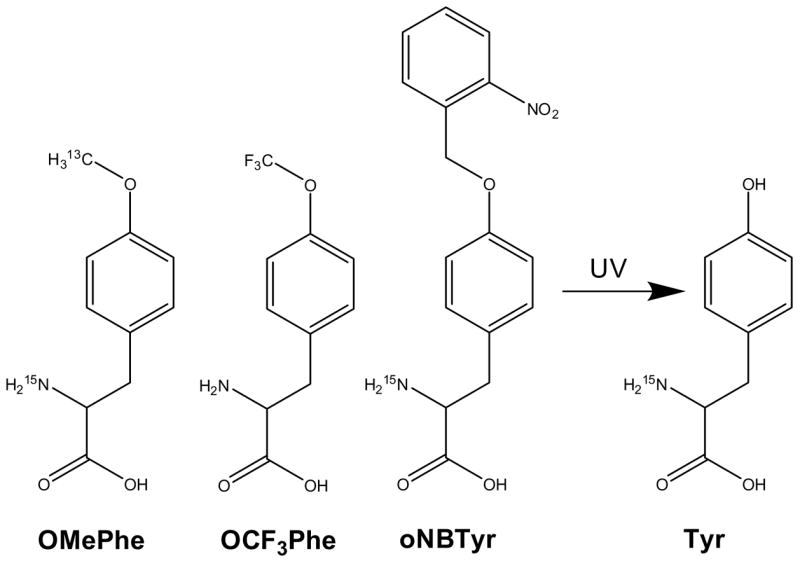
Isotopically labeled unnatural amino acids O-methyl-phenylalanine (OMePhe), its trifluorinated analog OCF3Phe and o-nitrobenzyl-tyrosine (oNBTyr) that is readily converted into tyrosine upon UV illumination.23
The three different NMR-active unnatural amino acids were used to study the binding of a small molecule ligand to the thioesterase domain of fatty acid synthase (FAS-TE), a 33 kDa protein of pharmaceutical interest. Fatty acid synthase (FAS) is a large, multi-domain enzyme essential for the synthesis of long-chain fatty acids.24 Because the sequence and architecture of FAS differ significantly between bacteria and mammals, bacterial FAS has long been considered a valuable target for the development of novel antibiotics (see ref. 25 and references herein). In humans, FAS is overexpressed in many cancers26 and is a drug target for obesity and related diseases27. Orlistat, an approved obesity drug, exhibits antitumor activity by inhibiting FAS-TE.28,29 The molecular details of orlistat’s interactions with FAS-TE have only been revealed in a recent X-ray crystallographic study.30 Here we use a more soluble tool compound to evaluate the utility of unnatural amino acids for the characterization of protein-ligand interactions. Successful incorporation of three different NMR-active unnatural amino acids at 11 different positions around the proposed binding site in FAS-TE demonstrates the general utility of the approach to studies of protein structure, dynamics, function and binding of small molecules in particular.
Materials and Methods
Unnatural amino acids, tool compound and other chemicals
OCF3-DL-Phe was purchased from JRD Fluorochemicals (Leatherhead, Surrey, U.K.) and used without further purification. OMe-L-Phe was purchased from Sigma-Aldrich and used without further purification. 13C/15N-labeled OMePhe and 15N-labeled oNBTyr were prepared from 15N-labeled L-tyrosine (Cambridge Isotopes Laboratory) according to Scheme I and II, respectively. Tool compound 17 was synthesized according to Scheme III (vide infra). Deuterated imidazole and D2O were obtained from Cambridge Isotopes Laboratory and TCEP was obtained from Pierce. All other chemicals were obtained from Sigma.
Plasmids for the expression of FAS-TE and generation of mutants
The coding sequence of our model protein, FAS-TE, was inserted into pMH4 (JCSG) using the PIPE cloning method.31 TAG codons were mutated in at 11 specific sites using the same cloning technique. The sites of incorporation were chosen based on the solvent accessibility of the side chain in the FAS-TE apo-structure (1XKT.pdb).32 To lower termination efficiency and subsequently reduce truncation at the TAG stop codon, the codon of the residue C-terminal of the incorporation site was optimized33 for Tyr-2351 (Arg-2352: CGG to AGG, silent mutation) and for Leu-2222 (Leu-2223: CTG to ATT, resulting in a Leu-2223-Ile mutation). Cys-2359 was mutated to Ser to prevent disulphide mediated aggregation.
Plasmids for the incorporation of OMePhe and oNBTyr and improving the expression plasmid
Initial incorporation tests were performed with the previously described pSUP-OMePhe-3TRN plasmid.13 This plasmid contains three copies of an amber tRNA and the mutant M. jannaschii tyrosyl-tRNA synthetase selected for OMePhe under the control of a glnS promotor. To co-express the tRNA synthetase and therefore enhance amino acylation activity during protein production, we modified the plasmid as follows: For each synthetase of interest, the coding sequence was inserted into pMH4 by PIPE cloning.31 The resulting plasmid was then used as template for a standard PCR amplification using the oligos KpnRS_F (5′-GGAGGTACCCAATTATGACAACTTGACGGCTACATC-35′) and SacRS_R (55′-GGAACGAGCTCACAAACAACAGATAAAACGAAAGGCCC-35′). The PCR product, containing the entire arabinose operon, was then prepared by restriction digest and inserted by ligation between the KpnI and SacI sites in the pSUP-RS-3TRN vector13 with the same synthetase gene. The result is pSUPAR3-RS containing all of the elements of the pSUP-RS-3TRN vector plus an additional arabinose-inducible copy of the synthetase (Figure 2).
Figure 2.

pSUPAR3 plasmid. Plasmid contains two copies of the amino-acyl tRNA synthetase specific for the unnatural amino acid (Uaa-RS), one under the control of a constitutively-on glnS promoter and the second under the control of an arabinose-inducible ara promoter. The plasmid also encodes three copies of the unnatural amino acid specific amber tRNA with a proK promoter, a p15A origin and a chloramphenicol CmR resistance marker.
Plasmids and Libraries for the selection of a OCF3Phe-specific tRNA synthetase
All plasmids used in the selection of a OCF3Phe-specific tRNA synthetase were described previously16,34. pREP2-YC-JYCUA encodes the mutated tRNATyrCUA from M. jannaschii, a chloramphenicol resistance gene containing a TAG stop codon, and a tetracycline resistance gene. pNEG encodes the mutated tRNATyrCUA, barnase with three TAG stop codons in the coding sequence, and β-lactamase. pBK-MJYRS-L1 (positions Tyr32, Leu65, His70, Gln155, Asp158, and Leu162 randomized), pBK-MJYRS-L234 (positions Tyr32, Leu65, Phe108, Gln109, Asp158, and Leu162 randomized), and pBK-MJYRS-L3D35 (positions Tyr32, Leu65, His70, Phe108, Gln109, Gln155, Asp158, Ile159, and Leu162 randomized) encode the tyrosyl tRNA synthetase (aa RS) from M. jannaschii with different positions of the active site randomized to all 20 amino acids, as previously described34,35. pLeiZ encodes the Z-domain protein with a TAG codon at position 7 and C-terminal His6-tag, the MjtRNATyrCUA, and β-lactamase36.
Selection of a novel OCF3Phe-specific tRNA synthetase
In the first round of selection, frozen aliquots, equivalent to ~1010 cfu, of DH10B cells transformed with pREP2-YC-JYCUA and each of the pBK library plasmids were plated on GMML-Agar supplemented with 0.04% glucose; 50 μg/ml kanamycin; 0.25 mM OCF3Phe; and 50μg/ml chloramphenicol or on LB-Agar with 50 μg/ml kanamycin; 0.5 mM OCF3Phe; and 50μg/ml chloramphenicol. After 3 days, cells were scraped from the plates and the plasmids purified by Spin Miniprep Kit (Qiagen). The pREP2 and pBK plasmids were separated by gel electrophoresis on a 1% agarose gel, and the pBK plasmid pools were repurified by Minelute Gel Extraction Kit (Qiagen). For negative selections, the pBK pools were transformed by electroporation into HK100 cells (JCSG; derived from Genehogs by Invitrogen) harboring the pNEG plasmid and plated on LB-Agar supplemented with 50 μg/ml kanamycin, 100 μg/ml ampicillin, and 0.2% arabinose. After 12–14 hours of growth, the pBK plasmid pools were isolated as before and transformed into HK100 cells with the pREP2 plasmid. After a total of four positive rounds (with 50, 50, 75, and 100 μg/ml chloramphenicol, with 0.25, 0.25, 0.5, and 0.5 mM OCF3Phe, and with all defined media plates after the first round) and three negative rounds, colonies were picked from the final positive round for further study. Following the positive selection recipe, a total of 296 colonies were replica plated onto agar plates either without OCF3Phe but with 20, 35, or 50 μg/ml chloramphenicol; with 0.5 mM OCF3Phe and 100, 125, or 150 μg/ml chloramphenicol; or with 1 mM OCF3Phe and 100, 125, or 150 μg/ml chloramphenicol. The plasmids from 37 of these colonies that showed high growth with unnatural amino acid and poor growth without it were isolated and sequenced, revealing 14 novel sequences (Table S1 and S2). Five clones (A6, B7, B10, F6 and H4) were chosen for further evaluation based on the results of pooled replica plates and the number of occurrences of the clone.
Test incorporation of OCF3Phe into Z-domain protein and evaluation of misincorporation
HK100 cells co-transformed with pLeiZ and each of the five pBK-OCF3Phe-RS were grown in 50 ml cultures of TB supplemented with 50 μg/ml kanamycin, 100 μg/ml ampicillin, and with or without 1 mM OCF3Phe. Cells were harvested after 5 hours induction with 1 mM IPTG at 30°C. Lysates were prepared in 6 M guanidine by sonication and clarified by centrifugation at 20,000 g for 20 minutes. The His-tagged proteins from each culture were purified by Ni-NTA (Qiagen) columns according to the manufacturer’s protocol. Denaturant was removed by PD-10 columns (GE Healthcare). The protein samples were next evaluated by SDS-PAGE, Bradford assay (Pierce), and LC-ESI-MS (Supplemental Figure S2 and S3). To test for miscorporation of natural amino acids at the TAG incorporation site, protein samples of Z-domain expressed in the absence or presence of OCF3Phe with the five evolved OCF3Phe-RS were digested with trypsin and subjected to LC-ESI-MS to evaluate the presence of the N-terminal peptide TSVDNXINK, where X represents the mutated position. The peptide masses for incorporation of OCF3Phe, Tyr, Phe, and Trp were all monitored in MS experiments to evaluate misincorporation levels of the natural aromatic amino acids. The Z-domain protein produced with OCF3Phe-RS clone A6 in the presence of 1 mM OCF3Phe was used to verify the sequence of the peptide by MS-MS. In a separate experiment, the incorporation fidelity of the OCF3Phe RS (clone F6) was further investigated at the Thr-26 site of FKBP1237 by multiple-reaction monitoring as described previously.38 The homogeneity of the protein was found to be better than 99.5%.
Expression of FAS-TE mutants containing OMePhe, OCF3Phe and oNBTyr
OCF3Phe-RS clone F6 was inserted into the pSUPAR3 plasmid. For each mutant, the expression plasmid and pSUPAR3-OMePhe, pSUPAR3-OCF3Phe or pSUPAR3-oNBTyr plasmid were co-transformed into HK100 cells. Multiple single colonies were picked for growth in 50 ml TB cultures (in 250 mL shaker flasks) with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol. At OD595 = 1.0, cultures were moved to 30 °C and supplemented with 2.5 mM 13C/15N-labeled OMePhe, 1 mM OCF3Phe (both from 0.5 M stock solutions in 1 N HCl) or 0.5 mM 15N-labeled oNBTyr (from a fresh 0.5 M stock in DMSO). Thirty minutes later, expression was induced by addition of 0.2% arabinose. After 20 hours, cells were harvested. Lysates were prepared by sonication, clarified by centrifugation, and purified by Ni-NTA under native conditions according to the manufacturer’s protocol. Protein samples were evaluated by SDS-PAGE, Bradford assay, and ESI-MS before being exchanged into NMR buffer (20 mM deuterated imidazole, 100 mM NaCl, 0.5 mM TCEP, pH 6.5) via PD-10 column and concentrated using Amicon-ultra units (Millipore). Expression yields after purification are given in Table 1. Photocleaveage of oNBTyr was accomplished by exposing the NMR samples for 10 mins to UV light generated by an Oriel 500 W Hg light source (Newport Corporation; Stratford, CT).
Table 1.
Incorporation of three unnatural amino acids into FAS-TE at 11 positions using pSUPAR3 and 2.5 mM 13C/15N -labeled OMePhe, 1 mM Yields after OCF3Phe and 0.5 mM 15N-labeled oNBTyr. purification are given in milligram per 50 mL E.coli culture. The concentrations for the NMR samples shown in Figure 4, 5 and 6 respectively are also listed.
| FAS-TE | Yield per 50 mL culture[mg/50 mL] | NMR sample concentration[mM] | ||||
|---|---|---|---|---|---|---|
| Mutant | OMePhe | OCF3Phe | oNBTyr | OMePhe | OCF3Phe | oNBTyr |
| Leu-2222 | 2.7 | 2.2 | 1.2 | 0.15 | 0.12 | 0.11 |
| Thr-2255 | 2.2 | 3.1 | 2.1 | 0.12 | 0.17 | 0.080 |
| Tyr-2307 | 1.6 | 2.0 | 0.94 | 0.088 | 0.11 | 0.10 |
| Tyr-2343 | 0.47 | 2.2 | 1.9 | 0.051 | 0.12 | 0.13 |
| Tyr-2347 | 1.3 | 2.3 | 2.5 | 0.072 | 0.13 | 0.27 |
| Tyr-2351 | 1.7 | 1.4 | 1.9 | 0.095 | 0.077 | 0.16 |
| Gln-2373 | 0.67 | 0.42 | 0.25 | 0.055 | 0.034 | 0.16 |
| Phe-2375 | 2.4 | 1.8 | 4.5 | 0.096 | 0.096 | 0.11 |
| His-2408 | 2.0 | 2.7 | 5.0 | 0.11 | 0.15 | 0.14 |
| Thr-2450 | 0.84 | 2.5 | 1.9 | 0.099 | 0.14 | 0.052 |
| Tyr-2454 | 2.1 | 1.2 | 7.2 | 0.11 | 0.066 | 0.12 |
Expression of FAS-TE mutants selectively labeled with 15N-Tyrosine
The tyrosine residues of wild-type and mutant FAS-TE were selectively labeled using the following protocol: 20 mL of an overnight TB culture was added per 1 L M9 media (Sigma) supplemented with 0.5% glycerol and 0.1 mM FeCl3. Thirty minutes prior to induction a sterile 100 mL solution containing 100 mg 15N-labeled tyrosine; 500 mg each of glutamic acid, aspartic acid, glutamine and asparagine; 300 mg each of alanine, valine, isoleucine, leucine, tryptophan, phenylalanine, serine and glycine; and 200 mg of each of the remaining amino acids was added. The culture was induced as described above and allowed to proceed for approximately one fifth of the normal expression time to minimize scrambling.
NMR sample preparation and data collection
As a lock solvent, 50 μL of D2O were added to 500 μL of protein solution resulting in a final buffer composition of 18 mM d-imidazole, 91 mM NaCl, 0.45 mM TCEP, pH 6.5. The final protein concentrations of all NMR samples are given in Table 1. Aliquots of the tool compound were added from a 50 mM stock in deuterated DMSO to a final concentration of 1.4 molar equivalents unless mentioned otherwise. The final DMSO concentration was between 0.10% and 0.76% v.v. for all samples but typically less than 0.5% v.v. resulting in negligible chemical shift changes. All spectra were recorded at 300 K. 19F spectra were recorded on an Avance 400 MHz instrument (Bruker Biospin, Billerica, MA) equipped with a 1H/13C/19F/31P-QNP-cryoprobe. Spectra were typically recorded with 1024 scans, a recycle delay of 2 s, 8192 complex data points with a sweep width of 20 ppm. Proton decoupling was accomplished using Waltz-16. All other spectra were recorded on an Avance 600 MHz instrument equipped with a 1H/13C/15N-TXI-cryoprobe. 1H-13C HSQC39 were typically recorded with 32 scans, 128 t1 experiments at 300 K using a spectral width of 50 ppm in the carbon dimension and 13.97 ppm in the proton dimension. Similarly, 1H-15N HSQC spectra40 were recorded using 48 scans, 256 t1 experiments using a spectral width of 32 ppm and 13.97 ppm in the nitrogen and proton dimension respectively.
Results
Improving incorporation efficiency
A first incorporation test of OMePhe into the thioesterase domain of fatty acid synthase (FAS-TE) was performed in E. coli with the previously described mutant M. jannaschii tyrosyl tRNA/tRNA synthetase (tRNA/RS) pair encoded by the pSUP-OMePhe-6TRN plasmid.13 50 mL bacterial cultures were grown in rich media at OMePhe concentrations between 2.5 and 30 mM (between 25 mg and 300 mg per 50 mL) (Figure S1A and S1B). Only between ~0.4 and 1.2 mg full-length protein was produced respectively compared to ~3.8 mg for wild-type protein produced under the same conditions. The lower yield is caused by a high-rate of translation termination at the UAG amber codon as large amounts of truncated FAS-TE were detected in the insoluble fraction by SDS-gel and MS analysis. To increase the concentration and activity of the mutant tRNA synthetase, we constructed a plasmid pSUPAR3 that features an additional RS gene that is under the control of an arabinose inducible promoter (Figure 2). Simultaneous overexpression of RS and of the target protein dramatically reduced truncation and enhanced production of full-length FAS-TE (Figure S1C and S1D). Optimization of the expression conditions showed that growth in TB media and induction at 30°C for 20 hours further increased yields of both wild-type and mutant proteins. Incorporation tests with the pSUPAR3 plasmid at different concentrations of OMePhe indicated saturation of protein production above 5 mM suggesting that unnatural amino acid and synthetase are not limiting under these conditions. Comparisons of different versions of pSUPAR3 revealed that expression of full-length FASTE Tyr-2454-OMePhe was maximized by having 3 copies of the tRNA (vs. 1 or 6) and both the inducible and the non-inducible copies of the synthetase together (vs. either copy alone). Changing the number of tRNA copies did not enhance yields suggesting that additional factors, that will be optimized in the future, are limiting to achieve wild-type yield. Using the pSUPAR3-OMePhe-3TRN plasmid and only 2.5 mM OMePhe in 50 mL TB media, mutant FAS-TE with a single OMePhe incorporated can currently be produced with yields sufficient for a single 0.1 mM NMR sample. Subsequently, existing orthogonal tRNA synthetase for oNBTyr and a newly evolved tRNA synthetase for OCF3Phe described below were also transferred to the pSUPAR3 plasmid and successfully used for protein expression.
Expanding the number of NMR-active unnatural amino acids
Gram quantities of isotope labeled analogs of unnatural amino acids, namely a 13C/15N-labeled analog of OMePhe and a 15N-labeled analog of oNBTyr (Figure 1) were synthesized. 13C/15N-labeled OMePhe 4 was prepared according to Scheme I while 15N-labeled oNBTyr 8 was prepared according to Scheme II following procedures for the synthesis of o-benzyl-L-tyrosine ether.41
In addition, an incorporation system for a fluorinated unnatural amino acid was developed. Fluorinated analogs of natural amino acids, especially of Phe, Tyr and Trp, can be incorporated into proteins at all respective aromatic residues and used for protein NMR studies.5–8 A tRNA/RS pair for the site-specific in vivo incorporation of a fluorinated unnatural amino has so far been developed for only trifluoromethyl-phenylalanine.22 Independently, we chose to develop such a system for OCF3Phe (Figure 1) because a) its structure is a conservative modification of the unnatural amino acid OMePhe that can be introduced without adverse effects on the structure of our test protein (see below), b) three fluorine atoms virtually decoupled from a ring (5JHF < 1 Hz, data not shown) should experience minimal fluorine-proton relaxation resulting in high intrinsic sensitivity for applications in large proteins, and c) the tRNA/RS pair specific for OMePhe should be a good starting point for the evolution of a OCF3Phe-specific RS. Initial tests to incorporate OCF3Phe into proteins using the OMePhe-specific tRNA/RS pair failed and demonstrates the high selectivity of tRNA synthetases for their substrates which is consistent with previous suggestions that a CF3 group should be bulkier and more hydrophobic than a CH3 group.42
Randomizing amino acid residues in M. Jannaschii TyrRS that are in contact with the substrate tyrosine side chain as illustrated in the x-ray structure43 enabled the selection of a RS specific for OMePhe.34 We successfully used similar libraries to select a mutant TyrRS specific for OCF3Phe and illustrate that this RS selectively and specifically incorporates OCF3Phe into Z-domain protein and into FAS-TE. Three libraries of mutants of the M. Jannaschii tyrosyl-tRNA synthetase were subjected to alternating rounds of positive and negative selection in order to isolate clones that specifically recognized OCF3Phe and not any natural amino acids. Five promising RS clones, A6 (Val32, Ala65, Gln108, Trp109, Ala158, Lys162), B7 (Val32, Ala108, Trp109, Gly158, Gln162), B10 (Ala32, Ala65, Trp108, Met109, Gly158, Asn159), F6 (Ala32, Ser65, Gln108, Ala109, Ala158, Tyr162) and H4 (Ile26, Val32, Gly65, His108, Tyr109, Ala158A, His162) (Table S1 and S2), were used to express Z-domain protein. In all five cases, the Z-domain was expressed only in the presence of OCF3Phe, as measured by SDS-PAGE and LC-MS (Supplemental Figure S2). The expressed protein was confirmed to contain OCF3Phe at the expected position by peptide sequencing. Based on LC-ESI-MS, the ratio of expressed protein containing a correct OCF3Phe amino acid compared to a misincorporated Tyr, Phe, or Trp was greater than 300:1 for two of the amino acyl-RS clones, A6 and F6 (Supplemental Figure S3). Clone F6 was subsequently chosen for future use because it consistently produced higher expression yields.
FAS-TE binding tool compound
Since orlistat is very insoluble in aqueous solutions, our binding experiments were performed with a FAS-TE binder 17 that was discovered in a high throughput screen (Caldwell et al., unpublished results). Compound 17 was synthesized according to Scheme III. Briefly, Garner’s aldehyde 9 was treated with a Grignard reagent, affording benzyl alcohol 10 quantitatively, followed by the global deprotecion by aqueous HCl. The amino group of 11 was masked as azide by trifluoromethanesulfonyl azide; the configuration of the stereo center of the amino group was retained under the condition.44 The primary hydroxyl group of 12 was selectively converted to the nosylate, and then to the cyclohexylamino group by nucleophilic displacement. The resulting intermediate, 14, was elaborated to the final compound, 17, by following the sequence of benzoylation, azide reduction, and ureation. Although we were unable to determine the diastereo selectivity of the first reaction (9 to 10), the H-NMR and LC-MS analysis of 13 indicated that 13 comprizes mainly one diastereomer. Since the synthesis started with homochiral aldehyde 9 and no steps were considered to involve racemization, we assume the final compound, 17, also comprises mainly one species regarding the benzilic and homobenzylic positions.
Incorporation of OMePhe, OCF3Phe and oNBTyr into 11 FAS-TE mutant proteins
Using pSUPAR3 plasmids, 11 FAS-TE mutants were expressed with three unnatural amino acids substituted at single sites. Positions Leu-2222, Thr-2255, Tyr-2307, Tyr-2343, Gln-2373, Phe-2375, His-2408, and Thr-2450 were selected in order to place the unnatural amino acid in solvent exposed positions surrounding the active site. Tyr-2347, Tyr-2351 and Tyr-2454 were selected as additional incorporation sites to probe adjacent protein regions that are disordered in the crystal structures of apo-protein32 and the orlistat complex.30 Cells transformed with an expression plasmid for each mutant and pSUPAR3-OMePhe, pSUPAR3-OCF3Phe or pSUPAR3-oNBTyr were fed the respective unnatural amino acid and induced to express the model proteins as described in Materials and Methods. Each mutant was purified using Ni-affinity and size-exclusion chromatography. The average yield of purified protein was ~2 mg per 50 ml culture (Table 1). With the exception of Gln-2373-OMePhe, Gln-2373-OCF3Phe, Tyr-2307-oNBTyr, Tyr-2347-oNBTyr, Tyr-2351-oNBTyr, and Gln-2373-oNBTyr, purified protein from only one 50 ml culture was transferred to NMR buffer resulting in NMR samples of typically 0.1 mM in concentration (total volume 0.55 ml). The predicted molecular weight of each mutant protein was confirmed by ESI-MS. Generally, there is little difference in the incorporation yield for each of the three unnatural amino acids at the same incorporation site. However, protein yields of the different mutants varied significantly possibly because of differences in incorporation efficiencies and effects on protein stability during expression and purification.
Effects of unnatural amino acid incorporation on protein structure
NMR spectra of FAS-TE mutants individually labeled at 11 positions with three different unnatural amino acids were recorded in the absence and presence of a FAS-TE binding tool compound as described below. Individual incorporation of an unnatural amino acid does not grossly perturb the structure of any of these FAS-TE mutants as indicated by only small changes in the 1H NMR spectra (Supplemental Figure S4, S5 and S6). All mutants retain well dispersed 1D 1H NMR spectra indicative of well folded proteins. For some but not all proteins, structural integrity was further confirmed by 1H-15N HSQC spectra of uniformly 15N-labeled mutant protein expressed in the presence of the unlabeled unnatural amino acid (Figure 3 and Supplemental Figure S7). For example, the “reverse-labeled” (15N-labeled at all nitrogens except for oNBTyr) Tyr-2454-oNBTyr mutant differences in the amide cross peaks are restricted to a few resonances (Figure 3A) as would be expected for any well tolerated single site mutation. Upon UV cleavage, oNBTyr is converted to Tyr and as expected the 1H-15N HSQC spectrum becomes identical to that of wild-type FAS-TE (Figure 3B). The resonance of Tyr-2454 is, as discussed below, exchange broadened and consequently not visible as an additional peak in the wild-type spectrum. Both wild-type and reverse labeled Tyr-2454-oNBTyr show identical chemical shift changes upon tool compound binding. Furthermore, a Tyr-2454-oNBTyr (unlabeled) mutant sample selectively labeled with 15N-tyrosine gives rise to a 1H-15N spectrum identical to a 15N-tyrosine wild-type protein sample (Supplemental Figure S8). Additional data for reverse labeled samples are shown in Supplemental Materials (Figure S7) and all support the conclusion that incorporation of unnatural amino acids at the 11 sites chosen appears not to negatively affect the structural integrity of these FAS-TE mutants.
Figure 3.

Incorporation of unnatural amino acids preserves structure of FAS-TE. 1H-15N HSQC of A) “reverse-labeled” (unlabeled oNBTyr incorporated into uniformly 15N-labeled protein) FAS-TE Tyr-2454-oNBTyr before (blue) and after UV cleavage (red); B) the UV-cleaved sample (red) overlaid with uniformly 15N-labeled wild-type FAS-TE (black); C) Overlay of wild-type protein before (black) and after addition of tool compound (green) and D) overlay of reverse labeled and UV-cleaved Tyr-2454-oNBTyr before (blue) and after addition of tool compound (red).
NMR spectra of OMePhe mutant proteins
1H-13C and 1H-15N HSQC spectra were recorded for each FAS-TE mutant labeled with 13C/15N OMePhe (Figure 4). With the exception of Tyr-2343-OMePhe, a single 1H-13C cross peak was observed for the methoxy group in a narrow spectral window from 3.55 to 3.82 ppm in the proton and from 54.8 to 56.0 ppm in the carbon dimension. There was no cross peak observed for Tyr-2343-OMePhe presumably because of conformational exchange. Upon addition of the tool compound, a sharp cross peak for the methoxy peak of Tyr-2343-OMePhe appears at approximately 2.45 ppm (1H) and 53 ppm (13C). Less dramatic chemical shift changes are apparent for Leu-2222-OMePhe, Tyr-2347-OMePhe, Tyr-2351-OMePhe, Gln-2373-OMePhe, Phe-2375-OMePhe, and Tyr-2454-OMePhe. Interestingly, Tyr-2307-OMePhe does not show any chemical shift changes despite its position at the center of the active site, adjacent to the catalytic residue Ser-2308 (see Figure 7 and 8). This suggests that the bulky methoxy group inhibits binding. Interestingly, for some of the proteins (Leu-2222-OMePhe, Gln-2373-OMePhe, Phe-2375-OMePhe and Tyr-2454-OMePhe) as much as 20% cannot be converted to a complex even in the presence of a four-fold molar excess of the tool compound (data not shown). This is possibly the result of limited solubility of the tool compound.
Figure 4.

1H-13C NMR spectra of FAS-TE mutants with 13C/15N -labeled OMePhe incorporated at 11 different positions: 1H-13C HSQC spectra before (blue) and after addition of tool compound (red). With the exception of the compound complex of Tyr-2343-OMePhe, all methoxy cross peaks of the unnatural amino acid have chemical shifts between 56.5 and 54.0 ppm for carbon and 3.85 and 3.35 ppm for protons. For Tyr-2343-OMePhe all observable peaks are shown. These include natural abundance peaks of TCEP, residual glycerol from concentration devices, residual TRIS from the purification buffer and other small molecule components of the NMR buffer. These peaks are also observed for all other mutants for which only the boxed region in the Tyr-2343-OMePhe mutant spectrum is shown.
Figure 7.
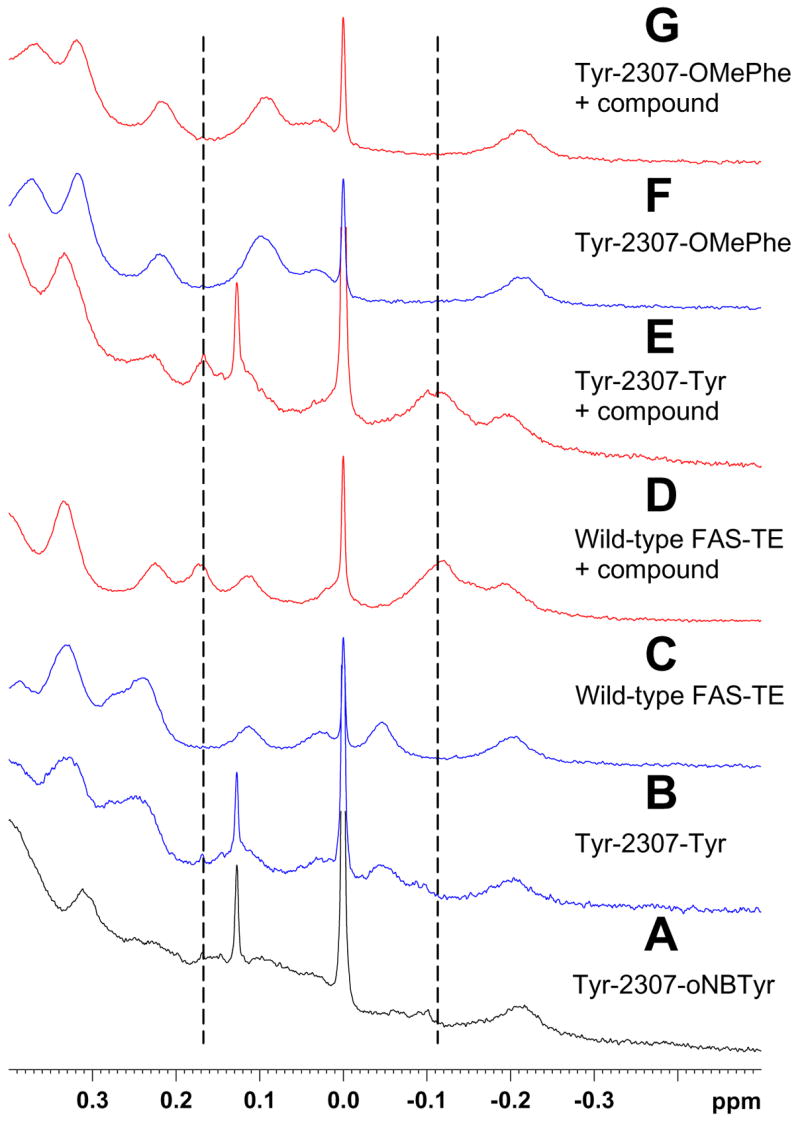
Site-directed labeling of active site residue Tyr-2307. After UV cleavage, 15N-oNBTyr incorporated at Tyr-2307 does in contrast to OMePhe and OCF3Phe not inhibit tool compound binding. Methyl region of 1H NMR spectra of Tyr-2307-oNBTyr before (A) and after UV cleavage (B), of wild-type protein before (C) and after addition of tool compound (D), of Tyr-2307-oNBTyr after UV cleavage and addition of tool compound (E), and of Tyr-2307-OMePhe before (F) and after addition of tool compound (G). The position of methyl resonances characteristic of the complex are indicated by dashed lines.
Figure 8.

Chemical shift mapping of tool compound 17 binding to FAS-TE. A) Combined chemical shift data. Chemicals shift changes ΔCS of 19F resonances (OCF3Phe; purple) were scaled relative to 1H by the gyromagnetic ratio while 1H-13C (OMePhe; violet) and 1H-15N (oNBTyr; pale yellow) chemical shifts were combined using the expression ΔCS = [(Δ1H)2 + (0.252*Δ13C)2]0.5 and ΔCS = [(Δ 1H)2 + (0.102* Δ 15N)2]0.5, respectively. OMePhe and OCF3Phe at Tyr-2307 block binding (see text). For oNBTyr mutants only a limited set of chemical shift changes could be compiled because of conformational exchange (see text). B) Structure of the covalent orlistat complex (2PX6.pdb).30 The average chemical shift changes induced by the binding of 17 are calculated for each unnatural amino acid and color-coded for each position: Δ CS < 0.1 ppm, blue; 0.1 to 0.2 ppm, salmon; > 0.2 ppm, red. Disordered loops are indicated by dashed lines. The active site residues Ser-2308, Asp-2338 and His-2481 are shown in magenta.
The observations for Tyr-2343-OMePhe suggest that parts of FAS-TE undergo conformational exchange on the millisecond time scale. Tool compound binding apparently reduces conformational exchange but only partially. 1H-15N HSQC measurements of the same samples support this interpretation as an amide resonance peak was observed for only six of the 11 mutants even in the presence of four-fold excess of the tool compound (Supplemental Figure S9). 1H-15N cross peaks were observed for Leu-2222-OMePhe, Thr-2255-OMePhe, Tyr-2347-OMePhe, His-2408-OMePhe, Thr-2450-OMePhe and Tyr-2454-OMePhe; with the exception of Leu-2222 and Tyr-2347, these are mutants that show no or only small chemical shift changes upon compound binding in the 1H-13C HSQC data (Figure 4). Because of their superior relaxation behavior, methyl groups are generally more readily observable in large proteins than backbone amide resonances.45,46 This seems to be the case for the OMePhe mutants of FAS-TE as well.
NMR spectra of OCF3-Phe mutant proteins
1D 19F-NMR spectra were recorded for each OCF3Phe FAS-TE mutant (Figure 5). In each case, a single peak was observed within 0.9 ppm of a resonance line recorded for a 0.5 mM solution of the unnatural amino acid, OCF3Phe dissolved in the same buffer and identical conditions. The width of the fluorine resonance line varied for each position again suggesting conformational exchange. The chemical shift of fluorine resonances is highly sensitive to changes in the environment5–7 and the line widths are a sensitive monitor of conformational exchange and reflect conformational fluctuations in the protein.6,7,47
Figure 5.

19F NMR spectra of FAS-TE mutants with OCF3Phe incorporated at 11 different positions. Spectra in blue are of the uncomplexed protein while the spectra in red were recorded after the addition of tool compound. Significant line broadened presumably because of conformational exchange is observed for some but not all residues. For comparison a spectrum of a 0.5 mM sample of OCF3Phe is shown.
Addition of tool compound results in significant chemical shift changes for residues near the binding site. In some cases, for example, for Tyr-2343-OCF3Phe, Tyr-2351-OCF3Phe, His-2408-OCF3Phe and Tyr-2454-OCF3Phe compound addition results in sharpening of the fluorine line; for Gln-2373-OCF3Phe and Phe-2375-OCF3Phe the opposite is observed. As with Tyr-2307-OMePhe, no changes are observed for Tyr-2307-OCF3Phe suggesting again that the addition of the bulky OCF3 (or OMe) group to this near active site residue blocks binding. The small chemical shift changes to Thr-2450-OCF3Phe and Thr-2255-OCF3Phe may be a solvent effect as the tool compound is added as DMSO stock (<0.5% final DMSO concentration).
NMR spectra of oNBTyr mutant proteins
1H-15N HSQC spectra were recorded for each FAS-TE mutant labeled with 15N-oNBTyr (Figure 6) before and after photo-cleavage by UV light and after addition of tool compound. As expected from the 1H-15N HSQC data of OMePhe mutants (Supplemental Figure S9), cross peaks are observed for only a subset of mutants, namely Leu-2222-oNBTyr, Tyr-2351-oNBTyr, Gln-2373-oNBTyr, Phe-2375-oNBTyr, His-2408-oNBTyr, Thr-2450-oNBTyr and Tyr-2454-oNBTyr. UV cleavage of the photo-caging group results in modest chemical shift changes for four of these mutants while the signal for the three others disappears presumably because of conformational exchange (Figure 6). For the five mutants that are tyrosine in wild-type FASTE, only Tyr-2307 is observed in the UV-cleaved, uncomplexed form. Its chemical shift corresponds to an observable cross peak in the spectrum of wild-type apo-protein selectively labeled with 15N-tyrosine (Figure 6B). In the latter spectrum, only nine of the 16 expected cross peaks are observed indicating that seven tyrosines undergo conformational exchange or rapid exchange with bulk solvent. After the addition of tool compound 15 of the 16 tyrosines (Figure 4D) are observed with various intensities and line widths suggesting that line broadening and absence of signals in the uncomplexed protein is indeed caused by conformational exchange (the last tyrosine is part of the presumably unstructured expression tag). The comparison with the individually 15N-oNBTyr labeled mutant proteins allows the assignment of five of the tyrosine amide resonances.
Figure 6.

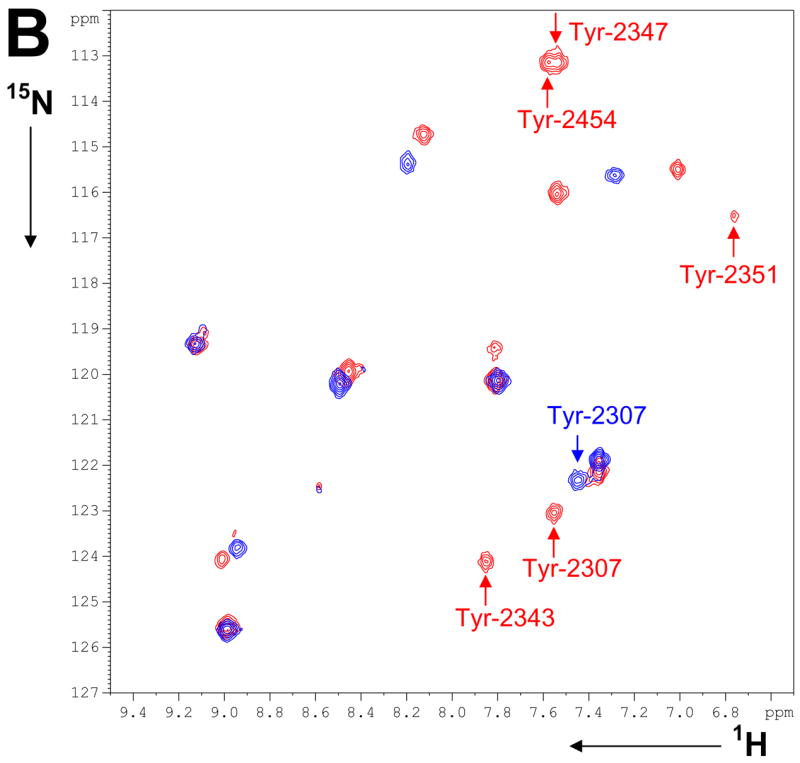
1H-15N NMR spectra of FAS-TE mutants with 15N-labeled oNBTyr incorporated at 11 different positions: A) 1H-15N HSQC spectra before (black) and after UV-cleavage (blue), and after addition of tool compound (red). B) 1H-15N HSQC spectra of wild-type FAS-TE selectively labeled with 15N-tyrosine before (blue) and after addition of tool compound (red). Spectra of individually 15N-oNBTyr labeled mutants facilitated signal assignments as indicated by residue numbers.
Figure 6B.
As shown above, the data for OMePhe and OCF3Phe indicate that unnatural amino acid incorporation at Tyr-2307, next to the active site residue Ser-2308, inhibits binding of the tool compound presumably because of steric interference by the added OCH3 or OCF3 group. UV photo-cleavage of Tyr-2307-oNBTyr re-establishes binding (Figure 6A) because the natural binding surface is regenerated as can be clearly seen from the comparison of 1D 1H spectra of wild-type and mutant proteins (Figure 7).
Mapping the binding site of a tool compound
The tool compound binds tightly to all FAS-TE mutants (except for Tyr-2307-OMePhe and Tyr-2307-OCF3Phe). Binding occurs in the slow binding regime for all 1H, 13C, 15N and 19F resonances. The chemical shift changes induced by compound addition map the binding site of the ligand to the active site of FAS-TE (Figure 8). Data obtained from 1H-13C, 1H-15N and 19F NMR spectra are all consistent in that His-2408, Thr-2450 and Thr-2255 mutants are not affected by binding. The most pronounced chemical shift changes occur at residue Tyr-2343 that is apparently disordered in the uncomplexed proteins (by NMR but not in the crystal structure of the orlistat complex30). Increasingly smaller chemical shift changes are observed for neighboring Tyr-2347 and Tyr-2351 mutants. Compound binding most likely stabilizes this flexible region of the protein as suggested by sharper fluorine resonance lines at residues Tyr-2343, Tyr-2351 and Tyr-2454 (Figure 5). Modest chemical shift changes are also observed for Gln-2373 and Phe-2375 mutants at the top surface of the binding pocket. At the far right site of the pocket, among the largest chemical shift changes are observed for Leu-2222 mutants while Thr-2255 and His-2408 mutant resonances are not shifted by compound binding. On the other end, unnatural amino acids at residue Thr-2450 and Tyr-2454 are also not affected and consistent with the overall features of the orlistat complex.30
Discussion
Improving incorporation efficiency
NMR studies require milligram quantities of isotopically labeled protein. In our previous proof-of-concept experiment, we incorporated 15N-labeled OMePhe using a mutant M. jannaschii tyrosyl tRNA/tRNA synthetase pair into myoglobin.19 Approximately 0.5 mg of protein was produced from 0.5 L of E. coli culture in defined minimal media (GMML) using ~100 mg of the custom synthesized unnatural amino acid to yield a single 55 μM NMR sample
Initial incorporation tests in rich rather than defined minimal media with a well-expressing FASTE construct indicated that most of the protein produced was truncated at the TAG incorporation codon (Supplemental Figure S1). This is in contrast to findings in GMML media where yields of full-length mutant protein can reach up to 70% of that of wild-type for some proteins.34 At the higher expression levels achieved in rich media the amount of amber tRNA aminoacylated with the unnatural amino acid is apparently insufficient. To overcome this limitation, we modified the incorporation plasmid and added a copy of the orthogonal RS under the control of an inducible promoter (Figure 2). Simultaneous overexpression of the target protein and of the RS dramatically reduced truncation at the TAG incorporation codon (Supplemental Figure S1). This modification of the plasmid appears to be beneficial for the incorporation of at least three different unnatural amino acids: OMePhe, OCF3Phe and oNBTyr. Using standard shake flask cultures and rich media, single NMR samples from 50 ml of E. coli culture were obtained for most mutants using only 8 mg of oNBTyr, 12.5 mg of OCF3Phe, or 25 mg of OMePhe (see details below). For well expressing protein such as FAS-TE, the current system allows for the economical production of protein NMR samples in rich growth media and as such should be widely applicable.
Selecting a new tRNA synthetase for a fluorinated unnatural amino acid
As discussed above, fluorinated unnatural amino acids and OCF3Phe in particular represent attractive NMR labels. Consequently an expression system for incorporating OCF3Phe into proteins was established. Initially, we attempted to express proteins with OCF3Phe using the existing OMePhe RS and its amber tRNA,13 but the two amino acids are sufficiently different that this RS does not facilitate incorporation of OCF3Phe into proteins. In hindsight, this is not surprising as a CF3 group should be ~70% bulkier than CH3 (C-F bond length 1.39 Å vs. C-H 1.09 Å; van der Waals radii of 1.35 Å for F vs. 1.2 Å for H), and may be more hydrophobic as suggested by partitioning studies of hexafluoroleucine and leucine48 (also see ref. 42 and references therein).
Using three libraries that randomize positions Tyr32, Leu65, Asp158, Leu162 and either His70 and Gln155 (L1) or Phe108 and Gln109 (L2)34 or His70, Phe108, Gln109, Gln155 and Ile159 (L3D)35 several tRNA synthetases specific for OCF3Phe were selected. His70 and Gln155 were conserved probably because they contact the ring and not the para substitution. As in other evolved tRNA synthetases, residues Tyr32 and Asp158 are mutated to remove H-bonds to the hydroxyl group of natural tyrosine. In the OMePhe RS, the mutations Tyr32Gln, Asp158Ala, and Leu162Pro increase the size of the binding pocket to accommodate the additional methyl group. Comparing the newly evolved OCF3Phe RS to the OMePhe RS suggests that the OCF3 group requires more space in the active site, as the OCF3Phe RS repeats the Asp158Ala mutation but also has other mutations (Tyr32Ala, Leu65Ser) that further reduce the bulk of side chains near the OCF3 group (based on the structure of the OMePhe RS).49 Interestingly position 109 is mutated from a hydrophilic Gln to a hydrophobic residue of variable size, possibly because of the suggested higher hydrophobicity of the OCF3 group (relative to OCH3). Finally, residues 108 and 162, which point outward in the OMePhe structure, seem to be less important in the case of OCF3Phe, as they are mutated to a wide variety of side chains in the selection winners.
The fidelity of amino acid incorporation of the newly evolved RS used in conjunction with the OMePhe tRNA13 was evaluated by mass spectrometric methods38 and found to be better than 99.5%. This suggests that it should be possible to evolve specific tRNA synthetases for other fluorinated, especially trifluoro-substituted amino acids. These unnatural amino acids do not have to be enantiomerically pure. Racemic mixtures of OCF3Phe (this study) and of several other unnatural amino acids acids50 have been used successfully to evolve selective RS mutants that retain their specificity for L-amino acids presumably because the residues randomized in the selection library are surrounding the end of the amino acid side chain rather than interfering with backbone recognition. For FAS-TE, selective incorporation of L-OCF3Phe is suggested by unperturbed 1D NMR spectra although DL-OCF3Phe was used in the incorporation experiment (Figure S6, see below).
Incorporation of three unnatural amino acids at 11 different sites
Our results indicate that all three unnatural amino acids (supplied as L-amino acids for OMePhe and oNBTyr and as DL-racemic mixture for OCF3Phe) can be incorporated at all 11 sites in a 33 kDa protein with good yields (Table 1) and with little effect on the structure of the mutant proteins (Figure 3, Supplemental Figures S4, S5, S6, S7 and S8). As with single-site mutations using natural amino acids, unnatural amino acid incorporations can interfere with protein structure and stability. In the current study, care was taken to introduce the unnatural amino acids at solvent exposed sites. Subsequently, with the exception of incorporation at Tyr-2307 adjacent to the active site, no effect on the ability of FAS-TE mutants to bind a tool compound in the active site was observed (see below). However, the observed variations in expression yields may reflect destabilizing effects on some of the mutations or context dependent variations in incorporation efficiencies, and warrant future investigations.
Monitoring conformational exchanges
At 33 kDa, FAS-TE is a sizable protein and normally deuterium labeling is employed in order to minimize spin diffusion and increase sensitivity and resolution.51 With the exception of Tyr-2343-OMePhe, all of the methoxy groups of the 11 OMePhe mutants are observed (Figure 4). Solvent exposed methoxy groups may be less sensitive to exchanges processes because of smaller chemical shift differences between the exchanging forms, and may be more readily observed because of the generally favorable relaxation properties of methyl groups. The small 1H-1H dipole-dipole couplings of the methoxy protons to the ring protons may be beneficial as well. For Tyr-2343-OMePhe conformational exchange most likely causes line broadening as addition of the tool compound gives rise to a significantly shifted resonance line. This interpretation is supported by 1H-15N data obtained with 13C/15N-OMePhe mutants (Figure S9), 15N-oNBTyr labeled mutant proteins and wild-type protein selectively labeled with 15N-tyrosine (Figure 6 and S8): Before addition of the tool compound amide signals of seven tyrosines including those of Tyr-2343, Tyr-2347, Tyr-2351 and Tyr-2454 are exchange broadened. Additional evidence for conformational exchange is also provided by 19F NMR data with OCF3Phe mutants. 19F NMR of fluorinated analogs of natural amino acids have long been known to be very sensitive to side chain dynamics.6,7,47 Line broadening is evident for incorporation at the same FAS-TE location for different unnatural amino acids. Resonance lines that are most affected correspond to residues that have high B-factors or missing electron densities in the available crystal structures (residues 2325 to 2330, 2344 to 2356, 2452 to 2457, and 2487 are missing in the published X-ray structure of apo-FAS-TE, 1XKT.pdb,32 and similarly in the orlistat complex, 2PX6.pdb30), suggesting conformational heterogeneity for these regions of the protein. This suggests that the NMR observations are indeed the result of conformational exchange processes in the natural protein and not the result of the unnatural amino acid incorporation into mutant FAS-TE. These observations in turn indicate that unnatural amino acids can be used to probe protein structure and dynamics at specific sites in large proteins.
Chemical shift mapping of a small molecule binding site in FAS-TE
Although different in absolute value, trends and magnitude of the chemical shift changes induced by tool compound binding in OMePhe, OCF3Phe and oNBTyr mutants agree and identify the active site of FAS-TE as the binding pocket (Figure 8). Data obtained from 1H-13C, 1H-15N and 19F NMR spectra are all consistent in that residues His-2408, Thr-2450 and Thr-2255 mutants are not affected by binding. When compared to the crystal structure of the FAS-TE complex with orlistat,30 Thr-2255 and His-2408 are at least 12 Å from the closest orlistat atom while the distance to Thr-2450 is 16 Å. Similarly, mutants of Thr-2450 exhibits minimal chemical shift changes as this residue is more than 13 Å from any orlistat atom. These observations indicate that the tool compound must bind in the active site not unlike orlistat. The similarities between the orlistat complexes and the complex of the tool compound are further supported by large chemical shift changes for Leu-2222 mutants. Leu-2222, therefore likely marks the extent of the compound binding pocket on the right side. The residue with the most pronounced chemical shift changes for its mutants, Tyr-2343 forms close contacts with the hexanoyl tail of orlistat in its hydrolyzed form and with both hexanoyl and the beginning of the palmitic core in the covalent orlistat complex (Figure 8). Tyr-2343, together with Tyr-2347 and Tyr-2351 are part of an alpha-helix that is observed in only one of two asymmetric units of an unpublished crystal structure (G. Spraggon, B. Bursulaya et al., unpublished data) and is absent in all published structures. Based on the sharper resonance lines, it is possible that tool compound binding stabilizes this helix but a more detail analysis must await a co-crystal structure or more detailed NMR studies. Because of the inherent flexibility of this part of the binding pocket we have not attempted to model binding of the tool compound but all preliminary evidence suggests a compound localization similar to that of orlistat.
The initial observations for Tyr-2307 mutants are misleading because introducing a bulky CH3 or CF3 group at the end of the unnatural amino acids instead of the natural tyrosine side chain apparently interferes with binding. UV cleavage of oNBTyr incorporated at Tyr-2307 restores binding (Figure 7) suggesting that the binding surface was perturbed in the Tyr-2307-OMePhe and Tyr-2307-OCF3Phe mutant proteins. As with any type of mutation there is the need to carefully consider the sites of change because altering critical residues to unnatural amino acids may impact protein function. Clearly photocaged unnatural amino acids constitute an attractive and unique labeling strategy as, with for example oNBTyr, a single NMR label can be introduced at a selected tyrosine residue without perturbing the primary protein sequence.
Overall, the current study illustrates that robustness of our improved in vivo incorporation protocol. The three unnatural NMR-active amino acids can now potentially be employed to applications ranging from screening for site-selective binders to mapping small-molecule target interactions in the absence of a crystal structure. Most interesting will be applications with very large proteins with thousands of residues for which obtaining signal assignments by traditional approaches is a truly formidable challenge. A recent test incorporation of NMR-acitve unnatural amino acids into fully deuterated FAS-TE is a first step toward such applications.
Conclusions
Three different NMR-active unnatural amino acids were incorporated with high efficiency at different sites in a 33 kDa protein of pharmaceutical interest without adversely effecting protein structure and ligand binding. As expected, 13C-labeled methyl groups introduced with the unnatural amino acid, because of their relaxation behavior, proved to be particularly advantageous for studies of a large protein. As illustrated here, unnatural amino acid labeling can be used to probe conformational exchange processes and small molecule ligand binding. More specifically, tool compound binding induces conformational rearrangements near the active site of FAS-TE and stabilizes part of the binding surface that is flexible in the X-ray structure of the orlistat complex. Although all mutations were well tolerated, introducing OMePhe or OCF3Phe at one active site residue inhibited binding of a tool compound. The same site was probed with a photo-caged oNBTyr that offers the advantage of introducing NMR labels at specific tyrosine residues without altering the protein sequence and therefore binding surface. Together with oNBCys (ref. 52 and M. Jahnz, D. Summerer, B.H. Geierstanger, P.G. Schultz, unpublished results) and o-nitroveratryl-Ser,53 the concept of photo-caged unnatural amino acid incorporation opens new and exciting avenues for site-specific labeling and should therefore enable NMR studies of large proteins that can currently not be studies by traditional methods.
Supplementary Material
Supporting Information Available: Sequence information of all newly selected tRNA synthetases, SDS gels of incorporation tests, gel and MS data of OCF3Phe incorporation, and additional NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme I.
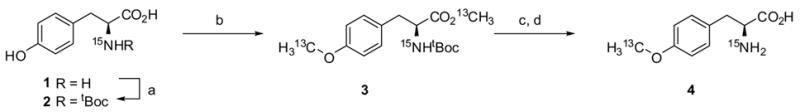
Conditions: (a) (tBuOCO)2O, NEt3, H2O/1,4-dioxane (1:1 v/v), 23°C, 16 hrs, 92% ; (b) 13CH3I, K2CO3, DMF, 0 to 23°C, 97%; (c) TFA/DCM (1:4 v/v), 23°C; (d) NaOH, H2O/MeOH (1:1 v/v), 23°C, 41% (2 steps).
Scheme II.
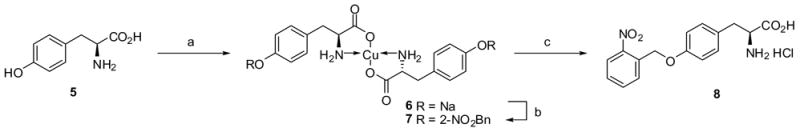
Conditions: (a) CuSO45H2O, NaOH, H2O, 23°C ; (b) 2-nitrobenzyl chloride, NaOH, H2O/MeOH (1:1 v/v), DMF, 23°C, 46% (2 steps); (c) 1M HCl, 23°C, 83%.
Scheme III.

Conditions: (a) 3, 4-(methylenedioxy)phenylmagnesium bromide, THF/toluene (1:1 v/v), −78 to 23°C, 98% ; (b) 1M HCl, THF, 70°C; (c) trifluoromethanesulfonyl azide, CuSO4, K2CO3, H2O/MeOH, 0 to 23°C, 42% (2 steps); (d) 4-nitrobenzene-1-sulfonyl chloride, pyridine, cat. DMAP, DCM, 23°C, 55%; (e) cyclohexanamine, NMP, 80°C; (f) 2,4-difluorobenzoyl chloride, DIEA, DCM, 23°C, 65%; (g) H2, 5% Pd/C, MeOH, 23°C; (h) n-butylisocyanate, DIEA, DCM, 23°C, 28% (2 steps).
Acknowledgments
We thank Jianqiao Lin for help with protein expression and purification for some of the oNBTyr mutants, Genevieve Welch for help with Figure 2, and Dr. Scott Lesley for helpful discussions and comments. This work was in part supported by grants from the National Institutes of Health (GM62159 to P.G.S).
References
- 1.Wuthrich K. Nat Struct Biol. 1998;5:492–495. doi: 10.1038/728. [DOI] [PubMed] [Google Scholar]
- 2.Mittermaier A, Kay LE. Science. 2006;312:224–8. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 3.Tugarinov V, Hwang PM, Kay LE. Annu Rev Biochem. 2004;73:107–146. doi: 10.1146/annurev.biochem.73.011303.074004. [DOI] [PubMed] [Google Scholar]
- 4.Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. Methods Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 5.Gerig JT. Prog Nucl Magn Reson Spectroscopy. 1994;26:293–370. [Google Scholar]
- 6.Frieden C, Hoeltzli SD, Bann JG. Methods Enzymol. 2004;380:400–15. doi: 10.1016/S0076-6879(04)80018-1. [DOI] [PubMed] [Google Scholar]
- 7.Danielson MA, Falke JJ. Annu Rev Biophys Biomol Struct. 1996;25:163–95. doi: 10.1146/annurev.bb.25.060196.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gakh YG, Gakh AA, Gronenborn AM. Magn Reson Chem. 2000;38:551–558. [Google Scholar]
- 9.Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Mei Ono A, Guntert P. Nature. 2006;440:52–7. doi: 10.1038/nature04525. [DOI] [PubMed] [Google Scholar]
- 10.Tugarinov V, Kay LE. ChemBioChem. 2005;6:1567–77. doi: 10.1002/cbic.200500110. [DOI] [PubMed] [Google Scholar]
- 11.Gardner KH, Rosen MK, Kay LE. Biochemistry. 1997;36:1389–401. doi: 10.1021/bi9624806. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Xie J, Schultz PG. Annu Rev Biophys Biomol Struct. 2006;35:225–49. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 13.Ryu Y, Schultz PG. Nat Methods. 2006;3:263–5. doi: 10.1038/nmeth864. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Schultz PG. Chem Biol. 2001;8:883–90. doi: 10.1016/s1074-5521(01)00063-1. [DOI] [PubMed] [Google Scholar]
- 15.Xie J, Schultz PG. Nat Rev Mol Cell Biol. 2006;7:775–82. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- 16.Xie J, Schultz PG. Curr Opin Chem Biol. 2005;9:548–54. doi: 10.1016/j.cbpa.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Xie J, Schultz PG. Methods. 2005;36:227–38. doi: 10.1016/j.ymeth.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Liu W, Brock A, Chen S, Chen S, Schultz PG. Nat Methods. 2007;4:239–244. doi: 10.1038/nmeth1016. [DOI] [PubMed] [Google Scholar]
- 19.Deiters A, Geierstanger BH, Schultz PG. ChemBioChem. 2005;6:55–8. doi: 10.1002/cbic.200400319. [DOI] [PubMed] [Google Scholar]
- 20.Hajduk PJ, Mack JC, Olejniczak ET, Park C, Dandliker PJ, Beutel BA. J Am Chem Soc. 2004;126:2390–8. doi: 10.1021/ja039480v. [DOI] [PubMed] [Google Scholar]
- 21.Skrynnikov NR, Mulder FAA, Hon B, Dahlquist FW, Kay LE. J Am Chem Soc. 2001;123:4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- 22.Jackson JC, Hammill JT, Mehl RA. J Am Chem Soc. 2007;129:1160–6. doi: 10.1021/ja064661t. [DOI] [PubMed] [Google Scholar]
- 23.Deiters A, Groff D, Ryu Y, Xie J, Schultz PG. Angew Chem Int Ed Engl. 2006;45:2728–31. doi: 10.1002/anie.200600264. [DOI] [PubMed] [Google Scholar]
- 24.Jayakumar A, Tai MH, Huang WY, al-Feel W, Hsu M, Abu-Elheiga L, Chirala SS, Wakil SJ. Proc Natl Acad Sci USA. 1995;92:8695–9. doi: 10.1073/pnas.92.19.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White SW, Zheng J, Zhang YM. Rock Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 26.Kuhajda FP. Cancer Res. 2006;66:5977–80. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 27.Ronnett GV, Kleman AM, Kim EK, Landree LE, Tu Y. Obesity. 2006;14(Suppl 5):201S–7S. doi: 10.1038/oby.2006.309. [DOI] [PubMed] [Google Scholar]
- 28.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Cancer Res. 2004;64:2070–5. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 29.Knowles LM, Axelrod F, Browne CD, Smith JW. J Biol Chem. 2004;279:30540–5. doi: 10.1074/jbc.M405061200. [DOI] [PubMed] [Google Scholar]
- 30.Pemble CW, Johnson LC, Kridel SJ, Lowther WT. Nat Struct Mol Biol. 2007;14:704–9. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- 31.Klock HE, Koesema EJ, Knuth MW, Lesley SA. Proteins. 2008;71:982–94. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 32.Chakravarty B, Gu Z, Chirala SS, Wakil SJ, Quiocho FA. Proc Nat Acad Sci USA. 2004;101:15567–72. doi: 10.1073/pnas.0406901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown CM, Stockwell PA, Trotman CN, Tate WP. Nucleic Acids Res. 1990;18:6339–45. doi: 10.1093/nar/18.21.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 35.Schultz KC, Supekova L, Ryu YH, Xie JM, Perera R, Schultz PG. J Am Chem Soc. 2006;128:13984–13985. doi: 10.1021/ja0636690. [DOI] [PubMed] [Google Scholar]
- 36.Wang L, Zhang Z, Brock A, Schultz PG. Proc Natl Acad Sci USA. 2003;100:56–61. doi: 10.1073/pnas.0234824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michnick SW, Rosen MK, Wandless TJ, Karplus M, Schreiber SL. Science. 1991;252:836–9. doi: 10.1126/science.1709301. [DOI] [PubMed] [Google Scholar]
- 38.Chen S, Schultz PG, Brock A. J Mol Biol. 2007;371:112–22. doi: 10.1016/j.jmb.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 39.Davis AL, Keeler J, Laue ED, Moskau D. J Magn Reson (1969) 1992;98:207–216. [Google Scholar]
- 40.Mori S, Abeygunawardana C, Johnson MO, Vanzijl PCM. J Magn Reson Series B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 41.Bodanzky N, Bodanzky A. The Practice of Peptide Synthesis. 2. Springer Verlag; Berlin-Heidelberg: 1994. [Google Scholar]
- 42.Budisa N, Pipitone O, Siwanowicz I, Rubini M, Pal PP, Holak TA, Gelmi ML. Chem Biodivers. 2004;1:1465–75. doi: 10.1002/cbdv.200490107. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi T, Nureki O, Ishitani R, Yaremchuk A, Tukalo M, Cusack S, Sakamoto K, Yokoyama S. Nat Struct Biol. 2003;10:425–32. doi: 10.1038/nsb934. [DOI] [PubMed] [Google Scholar]
- 44.Alper PB, Hung SC, Wong CH. Tetrahedron Lett. 1996;37:6029–6032. [Google Scholar]
- 45.Kay LE, Bull TE, Nicholson LK, Griesinger C, Schwalbe H, Bax A, Torchia DA. J Magn Reson. 1992;100:538–558. [Google Scholar]
- 46.Kay LE, Bull TE, Nicholson LK, Griesinger C, Schwalbe H, Bax A, Torchia DA. J Magn Reson Series A. 1994;111:231–232. [Google Scholar]
- 47.Bai P, Luo L, Peng ZY. Biochemistry. 2000;39:372–380. doi: 10.1021/bi992056f. [DOI] [PubMed] [Google Scholar]
- 48.Lee KH, Lee HY, Slutsky MM, Anderson JT, Marsh EN. Biochemistry. 2004;43:16277–84. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Wang L, Schultz PG, Wilson IA. Protein Sci. 2005;14:1340–9. doi: 10.1110/ps.041239305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santoro SW, Wang L, Herberich B, King DS, Schultz PG. Nat Biotech. 2002;20:1044–8. doi: 10.1038/nbt742. [DOI] [PubMed] [Google Scholar]
- 51.Gardner KH, Kay LE. Annu Rev Biophys Biomol Struct. 1998;27:357–406. doi: 10.1146/annurev.biophys.27.1.357. [DOI] [PubMed] [Google Scholar]
- 52.Wu N, Deiters A, Cropp TA, King D, Schultz PG. J Am Chem Soc. 2004;126:14306–7. doi: 10.1021/ja040175z. [DOI] [PubMed] [Google Scholar]
- 53.Lemke EA, Summerer D, Geierstanger BH, Brittain SM, Schultz PG. Nat Chem Biol. 2007;3:769–72. doi: 10.1038/nchembio.2007.44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Sequence information of all newly selected tRNA synthetases, SDS gels of incorporation tests, gel and MS data of OCF3Phe incorporation, and additional NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.